

ABSTRACT
Porcine reproductive and respiratory syndrome virus (PRRSV) is a highly infectious pathogen that causes severe diseases in pigs and great economic losses to the swine industry worldwide. Type I interferons (IFNs) play a crucial role in antiviral immunity. In the present study, we demonstrated that infection with the highly pathogenic PRRSV strain JXwn06 antagonized type I IFN expression induced by poly(I·C) in both porcine alveolar macrophages (PAMs) and blood monocyte-derived macrophages (BMo). Subsequently, we showed that the inhibition of poly(I·C)-induced IFN-β production by PRRSV was dependent on the blocking of NF-κB signaling pathways. By screening PRRSV nonstructural and structural proteins, we demonstrated that nonstructural protein 4 (nsp4), a viral 3C-like serine protease, significantly suppressed IFN-β expression. Moreover, we verified that nsp4 inhibited NF-κB activation induced by signaling molecules, including RIG-I, VISA, TRIF, and IKKβ. nsp4 was shown to target the NF-κB essential modulator (NEMO) at the E349-S350 site to mediate its cleavage. Importantly, nsp4 mutants with defective protease activity abolished its ability to cleave NEMO and inhibit IFN-β production. These findings might have implications for our understanding of PRRSV pathogenesis and its mechanisms for evading the host immune response.
IMPORTANCE Porcine reproductive and respiratory syndrome virus (PRRSV) is a major agent of respiratory diseases in pigs. Like many other viruses, PRRSV has evolved a variety of strategies to evade host antiviral innate immunity for survival and propagation. In this study, we show that PRRSV nsp4 is a novel antagonist of the NF-κB signaling pathway, which is responsible for regulating the expression of type I interferons and other crucial cytokines. We then investigated the underlying mechanism used by nsp4 to suppress NF-κB-mediated IFN-β production. We found that nsp4 interfered with the NF-κB signaling pathway through the cleavage of NEMO (a key regulator of NF-κB signaling) at the E349-S350 site, leading to the downregulation of IFN-β production induced by poly(I·C). The data presented here may help us to better understand PRRSV pathogenesis.
INTRODUCTION
Porcine reproductive and respiratory syndrome (PRRS) is one of the most important infectious diseases in the swine industry worldwide (1, 2). The causative pathogen is porcine reproductive and respiratory syndrome virus (PRRSV), which is an enveloped, single-stranded RNA virus and belongs to the genus Arterivirus, family Arteriviridae, order Nidovirales (3). In 2006, a highly pathogenic PRRSV (HP-PRRSV) associated with porcine high-fever syndrome (PHFS) was reported in China and caused tremendous economic losses in China and Vietnam (4, 5). This strain was identified to have a discontinuous 30-amino-acid depletion in nonstructural protein 2 (nsp2) (5). The PRRSV genome includes at least 10 open reading frames (ORFs) and encodes 8 structural proteins and at least 15 nonstructural proteins. The nonstructural proteins are encoded as polyprotein precursors from two open reading frames, ORF1a and ORF1ab. The precursors are thought to be cleaved into 15 nonstructural proteins (nsp1α, nsp1β, nsp2 to -6, nsp2TF, nsp7a, nsp7b, and nsp8 to -12) by four virus-encoded proteases upon infection (6–8). In addition to their protease activities in viral polyprotein processing, the viral proteases may act as “multitaskers” by interacting with key factors in innate immune response.
Type I interferons (IFNs) are crucial antiviral cytokines that represent one of the first lines of host defenses against virus infection (9, 10). The expression of IFNs is regulated by intracellular signaling cascades that are activated by pattern recognition receptors (PRRs) sensing specific pathogen-associated molecular patterns (PAMPs) (11–13). The endosome-lysosome PRR Toll-like receptor 3 (TLR3) recognizes viral nucleic acid and recruits the adaptor molecule TIR-containing adaptor-inducing IFN-β (TRIF) (14). TRIF binds TRAF6 and then activates transforming growth factor β (TGF-β)-activated kinase-1 (TAK1) and TAK1-binding proteins (TABs), resulting in the phosphorylation of NF-κB essential modulator (NEMO) and the activation of an IKK complex. IκBα is phosphorylated and proteasomally degraded, leading to the release and translocation of the transcription factor NF-κB heterodimer (p65/p50) to the nucleus to regulate the expression of IFN-β and inflammatory cytokines (15). In parallel, IRF3 is activated through TRIF/TRAF3/TANK to TBK1 signaling (16, 17). Alternative cytosol PRRs, encoded by retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5), signal the production of IFNs via the adaptor VISA/MAVS/IPS-1 (18–21).
To replicate and spread successfully, viruses have evolved various strategies to evade host defenses. Previous studies reported that IFN-α was undetectable when PRRSV-infected macrophages were superinfected with swine transmissible gastroenteritis virus (TGEV), a known inducer of type I IFNs (22). PRRSV was also found to suppress IFN production activated by double-stranded RNA (dsRNA) (23, 24). PRRSV proteins, including nsp1, nsp2, nsp11, and N, have been identified and characterized as IFN antagonists (25, 26). nsp1 is considered a multifunctional protein regulating type I IFN responses (23, 27–29), and nsp11 and N protein have been shown to suppress IFN-β induction by antagonizing IRF3 activation (30, 31). In a report by Beura et al. (23), the authors showed that PRRSV nsp4 protein might inhibit IFN-β promoter activity activated by IRF3 overexpression.
In the present study, we found evidence that IFN-β induction is impaired by HP-PRRSV by downregulation of the NF-κB signaling pathway in infected cells. Subsequently, we observed that nsp4 protein could suppress IFN-β expression by mediating the cleavage of NEMO, a significant subunit of the IκB kinase complex. Furthermore, we found that nsp4 specifically targeted NEMO at the site of E349-S350, cleaving off the zinc finger domain from the protein. nsp4 mutants with defective protease activity abolished its ability to cleave NEMO and suppress IFN-β production.
MATERIALS AND METHODS
Cell lines and viruses.
HeLa cells were cultured in Dulbecco's minimum essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin. CRL-2843 cells, a porcine alveolar macrophage cell line, were maintained in RPMI 1640 medium supplemented with 10% FBS and penicillin-streptomycin. Porcine alveolar macrophages (PAMs) were obtained by postmortem lung lavage of 8-week-old specific-pathogen-free (SPF) pigs and maintained in RPMI 1640 medium with 10% heat-inactivated FBS and penicillin-streptomycin. Blood monocyte-derived macrophages (BMo) were prepared as previously described by Wang et al. (32). All the cells were cultured and maintained at 37°C with 5% CO2.
HP-PRRSV strain JXwn06 (GenBank accession no. EF641008.1) was isolated from a pig farm with an atypical PRRS outbreak in Jiangxi Province, China, in 2006. Virus preparation was propagated and titrated on PAMs. Briefly, 5 × 104 PAMs were prepared in a 96-well plate and then infected with serially 10-fold-diluted PRRSV. Virus infection was determined 72 h postinfection using immunofluorescent staining for the PRRSV N protein. The viral titer was determined by the Reed-Muench method and expressed as 50% tissue culture infective doses (TCID50)/ml. Virus was stored at −80°C until used.
Plasmids.
Genes encoding viral proteins were amplified from the JXwn06 genome and cloned into pcDNA3.1-myc-his (N-terminally tagged). All construction vectors were confirmed by sequencing (compared with the PRRSV strain JXwn06 sequence; GenBank accession number EF641008.1). The PRRSV nsp4 expression plasmid and mutants were constructed and cloned into the pCMV-Myc as described elsewhere (33). A porcine IFN-β–luciferase reporter plasmid was constructed by cloning porcine IFN-β promoter (from nucleotide [nt] −281 to +59; the +1 position refers to the transcription start site of the porcine IFN-β gene [GenBank accession no. M86762.1]) into the vector pGL3-Basic (Promega). Transcription factor NF-κB responding elements were synthesized and annealed to form double-strand DNA and then separately cloned into pGL3-basic (34). The plasmids pRK-Flag-RIG-I, pRK-Flag-VISA, pRK-Flag-TRIF, and pRK-Flag-IKKβ have been described previously (35, 36). cDNA encoding porcine NEMO was amplified using reverse transcription-PCR (RT-PCR) from total RNAs extracted from porcine alveolar macrophages (PAMs) and cloned into pRK5-Flag. The NEMO mutants, including the E331, E334, E339, E349, E354, and E362 amino acid substitution mutants, were constructed by overlap extension PCR using Pfu DNA polymerase. All the primers are listed in Tables 1 and 2.
TABLE 1.
Primers used for cloning of PRRSV nonstructural and structural genes (from PRRSV strain JXwn06)
| Primer | Sequence (5′–3′) |
|---|---|
| nsp1-F | GCGATATCATGTCTGGGATACTTGATCG |
| nsp1-R | CCGCTCGAGTCAACCGTACCACTTATGACTG |
| nsp2-F | GGGGTACCATGGCCGGAAAGAGAGCAAGG |
| nsp2-R | GCTCTAGATCATCCCCCTGAAGGCTTGGAAAT |
| nsp3-F | CGGGATCCATGGGCCCACACCTCATTGCTGCCTT |
| nsp3-R | CCGCTCGAGTCACTCAAGGAGGGACCCGAGCT |
| nsp4-F | GGGGTACCATGGGCGCTTTCAGAACTCAAAAGC |
| nsp4-R | GCTCTAGATCATTCCAGTTCGGGTTTGGCAGC |
| nsp5-F | GGGGTACCATGGGAGGCCTTTCCACAGTTCAAC |
| nsp5-R | GCTCTAGATCACTCGGCAAAGTATCGCAAGAAG |
| nsp6-F | GGGGTACCATGGGAAAGTTGAGGGAAGGGGTGTCGCAATCCTGCG |
| nsp6-R | GCTCTAGATCACTCATGACTCATCCCGCAGGATTGCGACACCCCTT |
| nsp7-F | GGGGTACCATGTCGCTGACTGGTGCCCTC |
| nsp7-R | GCTCTAGATCATTCCCACTGAGCTCTTCTATT |
| nsp8-F | GGGGTACCATGGCCGCCAAGCTTTCCGTG |
| nsp8-R | GCTCTAGATCAGCAGTTTAAACACTGCTCCTTAG |
| nsp9-F | GGGGTACCATGGCCGCCAAGCTTTCCGTG |
| nsp9-R | GCTCTAGATCACTCATGATTGGACCTGAGTT |
| nsp10-F | CGGGATCCATGGGGAAGAAGTCCAGAATGTGC |
| nsp10-R | CCGCTCGAGTCATTCCAGGTCTGCGCAAATAGC |
| nsp11-F | CGGGATCCATGGAAGGGTCGAGCTCCCCG |
| nsp11-R | CCGCTCGAGTCATTCAAGTTGAAAATAGGCCGTC |
| nsp12-F | GGGGTACCATGGGCCGCCATTTTACCTGGT |
| nsp12-R | GCTCTAGATCATCAATTCAGGCCTAAAGTTGG |
| GP2a-F | CGCGGATCCATGAAATGGGGTCTATGCAAAG |
| GP2a-R | CCGCTCGAGTCACCATGAGTTCAAAAGAAAAG |
| GP2b-F | CGCGGATCCATGGGGTCTATGCAAAGCCT |
| GP2b-R | CCGCTCGAGTCATAAGATCTTCTGTAATTGC |
| GP3-F | CGCGGATCCATGGCTAATAGCTGTACATTCC |
| GP3-R | CCGCTCGAGCTATCGCCGTGCGGCACTG |
| GP4-F | CGCGGATCCATGGCTGCGTCCTTTCTTT |
| GP4-R | CCGCTCGAGTCAAATTGCCAGTAGGATGG |
| GP5-F | CGCGGATCCATGTTGGGGAAGTGCTTGACC |
| GP5-R | CCGCTCGAGCTAGAGACGACCCCATTGTTC |
| GP5a-F | CGCGGATCCATGTTTAAGTATGTTGGGG |
| GP5a-R | CCGCTCGAGTCACATAGCGTTAAGTTAT |
| M-F | CGCGGATCCATGGGGTCGTCTCTAGACG |
| M-R | CCGCTCGAGTTATTTGGCATATTTAACAAGGTT |
| N-F | CGCGGATCCATGCCAAATAACAACGGCAAG |
| N-R | CCGCTCGAGTCATGCTGAGGGTGATGCT |
| nsp11′12-F | CTAGTCTAGAGGGTCGAGCTCCCCGCTCC |
| nsp11′12-R | CCCAAGCTTCTAATTCAGGCCTAAAGTTGGTTC |
TABLE 2.
Primers used for NEMO and IFN-β promoter cloning
| Primer | Sequence (5′–3′) |
|---|---|
| Sus-NEMO-F | GTGCTCTAGAACTTGTTGGATGAGCAGGACCC |
| Sus-NEMO-R | CCCAAGCTTGCCTACTCGATACACTCCATGAC |
| E331A-F | GCAGCTGGCTGAGAGGAAGGAGCTCCTGCAGGCACAGCTGGAGC |
| E331A-R | CTGTACTCCCTCTGCAGCTGCTCCAGCTGTGCCTGCAGGAGC |
| E334A-F | GAGAGGAAGGAGCTCCTGCAGGAGCAGCTGGCACAGCTGCAGAG |
| E334A-R | CAGCCGGCTGTACTCCCTCTGCAGCTGTGCCAGCTGCTC |
| E339A-F | GCAGGAGCAGCTGGAGCAGCTGCAGAGGGCATACAGCCGG |
| E339A-R | CTGGCAGCTGGTCTTCAGCCGGCTGTATGCCCTCTGCAG |
| E349A-F | GAGGGAGTACAGCCGGCTGAAGACCAGCTGCCAGGCATCGGCCAG |
| E349A-R | GTGCCGCTTCCTCATGTCTTCGATCCTGGCCGATGCCTGGCAGC |
| E354A-F | GACCAGCTGCCAGGAATCGGCCAGGATCGCAGACATGAGG |
| E354A-R | GAGACCTCGACGTGCCGCTTCCTCATGTCTGCGATCCTGG |
| E362A-F | GGCCAGGATCGAAGACATGAGGAAGCGGCACGTCGCAGTCTCCCAG |
| E362A-R | TGGGGCGGGGGGCAAGGTGGGCTGGGAGACTGCGACGTGCC |
| IFNβ promoter-F | GGGGTACCTTTTTTTTGCACTTACAGCATAT |
| IFNβ promoter-R | CCCAAGCTTGTTACTGGCTCCACTACTC |
Antibodies and reagents.
Antibodies against IκBα, p-IκBα, p65, and NEMO (also known as IKKγ) were purchased from Cell Signaling Technology. Anti-α-tubulin antibody was purchased from MBL International Corporation. Anti-Flag and anti-β-actin antibodies were purchased from Sigma. Antibodies against c-Myc and histone 3 were purchased from Santa Cruz. Goat anti-mouse or anti-rabbit IgG secondary antibodies were also purchased from Santa Cruz. The antisera for nsp4 and GP5 were prepared by our lab. Poly(I·C) was purchased from InvivoGen.
RNA isolation and quantitative real-time PCR (qPCR).
Total RNAs from cells were extracted with TRIzol (Invitrogen) following the manufacturer's instructions. Moloney murine leukemia virus (M-MLV) reverse transcriptase was used for reverse transcription according to the manufacturer's protocol (TaKaRa). Quantitative RT-PCR (qPCR) analysis was performed using a FastSYBR mixture (Cwbiotech) on a ViiA7 real-time PCR system (Applied Biosystems). Gene-specific primers for porcine IFN-β were 5′AGCACTGGCTGGAATGAAAC3′ (forward) and 5′TCCAGGATTGTCTCCAGGTC3′ (reverse), and primers for porcine IFN-α were 5′CTGCTGCCTGGAATGAGAGCC3′ (forward) and 5′TGACACAGGCTTCCAGGTCCC3′ (reverse), as described in a previous study (37). The type I IFN expression was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (5′CCTTCCGTGTCCCTACTGCCAAC3′ [forward] and 5′GACGCCTGCTTCACCACCTTCT3′ [reverse]) and presented as the change (n-fold) in induction relative to the control.
Luciferase reporter assays.
HeLa and CRL-2843 cells were seeded in 24-well plates at a cell density of 4 × 104 cells per well. At 14 to 16 h after plating, cells were transfected with a control plasmid or plasmids expressing viral nonstructural and structural proteins along with pGL3-IFN-β-luc, pGL3-NF-κB-luc, and pRL-TK using Lipofectamine LTX and Plus reagents (Invitrogen). pRL-TK plasmid was used as a control for transfection efficiency. The total amount of DNA was kept constant by adding vector control plasmid. At 24 h after transfection, cells were treated with poly(I·C) for 8 h or left untreated. Cell extracts were prepared and analyzed for firefly and Renilla luciferase activities using the dual-luciferase reporter assay kit (Promega) according to the manufacturer's instructions.
Western blot analysis.
Cells were harvested and lysed in radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime) with 100 U proteinase inhibitor (Cwbiotech) and 20 μM NaF. Nuclear- and cytoplasmic-protein samples were extracted with a nuclear and cytosol fractionation kit (Beyotime). Protein levels in each sample were quantified with a bicinchoninic acid (BCA) assay kit (Pierce Biotechnology, Inc.). Similar amounts of proteins from each extract were resolved by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). Membranes were blocked with 5% nonfat milk in phosphate-buffered saline with 0.05% Tween 20 (PBST) and incubated for 2 h at room temperature with the antibodies at a suitable dilution as recommended (anti-IκBα, -p-IκBα, -p65, -NEMO, and -histone 3 at 1:1,000; anti-β-actin, -Flag, -α-tubulin, and -c-Myc at 1:2,000). The membranes were then incubated with the appropriate secondary antibody for 1 h at a dilution of 1:10,000. The antibodies were visualized by use of the enhanced chemiluminescence (ECL) reagent according to the manufacturer's instructions.
Statistical analysis.
All experiments were performed with at least three independent replicates. Results were analyzed by GraphPad Prism software using Student's t test. Differences in data were considered to be statistically significant if the P value was less than 0.05.
RESULTS
HP-PRRSV inhibits type I IFN expression induced by poly(I·C).
A previous study demonstrated that PRRSV (VR2332) has evolved mechanisms to subvert type I IFN responses (38). To investigate whether HP-PRRSV inhibits the production of type I IFNs, we infected porcine alveolar macrophages (PAMs) and blood monocyte-derived macrophages (BMo) with HP-PRRSV strain JXwn06 and then either stimulated them with poly(I·C) or left them untreated. At 6 h poststimulation, the mRNA levels of type I IFNs were analyzed using qPCR. As shown in Fig. 1, HP-PRRSV induced a very low level of IFN-β (Fig. 1A) and IFN-α (Fig. 1B) expressions in PAMs compared to poly(I·C), a well-known inducer of type I IFNs (it mimics the viral PAMP). And interestingly, HP-PRRSV significantly suppressed type I IFN expression to 6% (IFN-β) (Fig. 1A) and 60% (IFN-α) (Fig. 1B) of that induced by poly(I·C). Similar results were also obtained in BMo cells, and IFN-β and IFN-α mRNA levels were downregulated to 24% and 86% of that induced by poly(I·C), respectively (Fig. 1C and D). Efficient PRRSV infection in these cells was confirmed by qPCR (data not shown). These results suggest that HP-PRRSV infection antagonizes the production of type I IFNs.
FIG 1.

HP-PRRSV inhibits type I IFN expression induced by poly(I·C). (A and B) Porcine alveolar macrophages (PAMs) were mock infected or infected with HP-PRRSV (JXwn06) at a multiplicity of infection (MOI) of 0.1. (C and D) Blood monocyte-derived macrophages (BMo) were inoculated with medium alone or PRRSV at an MOI of 0.1. At 24 h postinfection, cells were treated with poly(I·C) (10 μg/ml) for 6 h or left untreated. Total RNAs were extracted from cells, and qPCR was performed to analyze the expression of IFN-β (A and C) and IFN-α (B and D). GAPDH was used as an internal control. Data are representative of three independent experiments (means and standard deviations [SD]). Statistical analysis was performed by Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Identification of PRRSV nsp4 as an inhibitor of IFN-β induction.
To examine which PRRSV proteins have effect on antagonizing the production of IFN-β, PRRSV structural and nonstructural protein genes (derived from the viral genome of JXwn06) were cloned into mammalian expression vectors. HeLa cells were cotransfected with each of the plasmids encoding these proteins and a IFN-β–luciferase reporter plasmid. At 24 h posttransfection, cells were treated with poly(I·C) for 8 h, and luciferase activities were then examined. As expected (23, 30, 31), IFN-β promoter activation was decreased to 17%, 50%, and 58% by nsp1, nsp11, and N protein compared to the control vector, respectively (Fig. 2A). Our results also showed that overexpression of nsp4 strongly suppressed poly(I·C)-stimulated IFN-β promoter activity, by 65% compared to the control vector (Fig. 2A). Also, the IFN-β promoter activity was reduced by 14%, 40%, and 56% compared to the control vector when cells were transfected with 0.1, 0.3, and 0.5 μg of nsp4, respectively, suggesting that this inhibition by nsp4 was dose dependent (Fig. 2B). Considering that nsp4 is a 3C-like protease and has a significant inhibition effect on IFN-β production, we decided to focus on this protein. To further verify whether nsp4 can block IFN-β transcriptional activation, CRL-2843 cells were transfected with nsp4 expression vector and then stimulated with extracellular poly(I·C). Cells were harvested, and IFN-β expression was measured by qPCR. Results revealed that nsp4 inhibited the IFN-β expression by 7%, 16%, and 27% at doses of 0.2, 0.5, and 1 μg, respectively (Fig. 2C). Taken together, these data show that PRRSV nsp4 protein can suppress IFN-β expression induced by poly(I·C).
FIG 2.

Inhibition of poly(I·C)-induced activation of the IFN-β promoter by PRRSV proteins. (A) PRRSV nonstructural and structural protein expression vectors and pcDNA3.1 were cotransfected into HeLa cells with pGL3-IFN-β-Luc and pRL-TK, a control for transfection efficiency. Twenty-four hours later, cells were transfected with poly(I·C) for 8 h, and cell lysates were analyzed for luciferase activities. (B) HeLa cells were cotransfected with pGL3-IFN-β-Luc, pRL-TK, and nsp4 expression plasmids at different doses (0.1, 0.3, or 0.5 μg). Cells were transfected with poly(I·C) for 8 h, and luciferase activities were measured. (C) CRL-2843 cells were transfected with control vector or nsp4 expression plasmid. Cells were treated with or without poly(I·C) (10 μg/ml) for 6 h, and IFN-β expression was analyzed using qPCR. Data are means and SD from three independent experiments. Differences were evaluated by Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
PRRSV nsp4 blocks the NF-κB signaling pathway.
To further verify whether and how nsp4 inhibits IFN-β induction and determine at which step the inhibition occurs, IFN-β promoter and NF-κB responsive promoter activities were evaluated. CRL-2843 cells were transfected with IFN-β–luciferase reporter plasmid along with the nsp4 expression plasmid. At 24 h posttransfection, cells were stimulated with poly(I·C), and IFN-β–luciferase activity was examined. As shown in Fig. 3A, IFN-β promoter activity induced by poly(I·C) was reduced by 10%, 39%, and 62% when transfected with 0.1, 0.3, and 0.5 μg of nsp4 (compared to that of control vector), respectively. NF-κB-responsive promoter activity induced by poly(I·C) was similarly affected by nsp4 expression, with a downregulation of 22%, 52%, and 68% (compared to that of the control vector) when nsp4 was used at 0.1, 0.3, and 0.5 μg, respectively (Fig. 3B).
FIG 3.

PRRSV nsp4 blocks the NF-κB signaling pathway. (A and B) nsp4 expression plasmids at different doses (0.1, 0.3, and 0.5 μg) were cotransfected into CRL-2843 cells with pGL3-IFN-β-Luc (A) or pGL3-NF-κB-Luc (B) and pRL-TK. pRL-TK was used as an internal control of transfection efficiency. Twenty-four hours after transfection, cells were treated with or without poly(I·C) (10 μg/ml) for 8 h and analyzed for luciferase activities. (C and D) CRL-2843 cells were transfected with control vector or nsp4 expression plasmid at different doses (0.2, 0.5, or 1 μg). Twenty-four hours later, cells were stimulated with or without poly(I·C) (10 μg/ml) for 2 h. (C) IκBα and p-IκBα protein levels were analyzed using Western blotting, and β-actin was used as a loading control. (D) Total cell lysates were separated into nuclear-protein (N.P) and cytoplasmic-protein (C.P) fractions for detecting the distribution of p65. Histone 3 and α-tubulin were used as nuclear and cytoplasmic controls, respectively. Data are representative of three independent experiments (means and SD). Statistical analysis was performed by Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To investigate the mechanism of nsp4-mediated inhibition of NF-κB activation, we assessed several hallmark steps of the NF-κB activation process, such as IκBα phosphorylation and p65 nuclear translocation, upon poly(I·C) treatment. As expected, the level of phosphorylated IκBα in the control cells was remarkably increased when they were treated with poly(I·C). However, poly(I·C)-induced IκBα phosphorylation was significantly decreased by nsp4 (Fig. 3C). Consistent with this observation, p65 translocation to the nucleus was also affected (Fig. 3D). These results indicate that nsp4 may inhibit NF-κB activation by targeting the process upstream of IκBα phosphorylation.
We next examined whether IκBα was phosphorylated and degraded during the course of PRRSV infection. PRRSV-infected PAMs were treated with poly(I·C) and then collected for Western blot analysis using a phospho-specific antibody for p-IκBα and an antibody for IκBα. As expected, poly(I·C) treatment increased the level of phosphorylated IκBα, leading to its degradation. However, in PRRSV-infected cells, IκBα phosphorylation and degradation were suppressed (Fig. 4A). Subsequently, p65 nuclear translocation was also affected by PRRSV. As shown in Fig. 4B, the level of p65 present in the nuclear fraction increased upon poly(I·C) treatment, while the nuclear translocation of p65 was decreased in the presence of PRRSV. These data suggest that PRRSV, as well as nsp4, interferes with the poly(I·C)-activated NF-κB signaling pathway by reducing IκBα phosphorylation and p65 translocation to the nucleus.
FIG 4.

HP-PRRSV suppresses the NF-κB signaling pathway. (A and B) PAMs were mock infected or infected with HP-PRRSV (JXwn06) at an MOI of 0.1. Twenty-four hours later, cells were stimulated with or without poly(I·C) (10 μg/ml) for 2 h. (A) Cell lysates were subjected to Western blotting to examine the levels of IκBα, p-IκBα, GP5, and β-actin. (B) Cells were lysed, and then nuclear-protein (N.P) and cytoplasmic-protein (C.P) fractions were separated for detecting the distribution of p65. Histone 3 and α-tubulin were used as nuclear and cytoplasmic controls, respectively.
nsp4 may target IKK signaling molecules.
It is well established that IFN-β production is initiated by the activation of Toll-like receptor 3 (TLR3) with extracellular dsRNA or the DEx(D/H) box RNA helicases, RIG-I and MDA-5, with intracellular RNA. We then examined at which step the nsp4 protein mediates the inhibition of IFN-β expression. HeLa cells were cotransfected with a plasmid expressing RIG-I, VISA, or TRIF protein, a plasmid expressing nsp4 protein, and an IFN-β–luciferase reporter plasmid. As expected, all the signaling components (RIG-I, VISA, and TRIF) induced IFN-β promoter activity. However, their activities were downregulated to 62%, 20%, and 40% by nsp4, respectively (Fig. 5A to C). Similarly, NF-κB-responsive promoter activity was constitutively stimulated in cells transfected with either a RIG-I, VISA, or TRIF construct, while the luciferase activity was suppressed about 27%, 65%, and 51% in the presence of nsp4, respectively (Fig. 5E to G). These results suggest that nsp4 inhibits TLR3 and that RIG-I mediated the induction of IFN-β and NF-κB.
FIG 5.

Effects of nsp4 on RIG-I, VISA, TRIF, and IKKβ. (A to D) Expression vectors for the signaling molecules RIG-I (A), VISA (B), TRIF (C), and IKKβ (D) and the control vector pRK5-flag were cotransfected into HeLa cells with pGL3-IFN-β-Luc, pRL-TK, and nsp4 expression plasmids. pRL-TK was used as an internal control. Twenty-four hours later, cells were harvested for luciferase activity analysis. (E to H) Experiments were performed as for panels A to D with the pGL3-NF-κB-Luc vector. Data are means and SD from three independent experiments. Statistical analysis was performed by Student's t test. **, P < 0.01; ***, P < 0.001.
IKKβ is an essential component of IκB kinase (IKK) complex, which phosphorylates IκBα and contributes to its degradation, leading to p65/p50 heterodimer nuclear translocation and the activation of NF-κB signaling pathway (39). Thus, we tested the effect of nsp4 on the IKKβ-mediated IFN-β and NF-κB activations. Our results revealed that IFN-β and NF-κB activation induced by IKKβ overexpression was suppressed about 36% and 75% by nsp4, respectively (Fig. 5D and H). Taken together, our observations imply that PRRSV nsp4 may interfere with IKK signaling molecules upstream of IκBα to block NF-κB activation.
The 3C-like serine protease catalytic activity is essential for nsp4 to inhibit IFN-β production.
PRRSV nsp4 is a member of a relatively rare group of proteolytic enzymes, the 3C-like serine proteases, which contains the canonical catalytic triad of His39-Asp64-Ser118 and three domains located at amino acids 1 to 69, 89 to 153, and 157 to 199, respectively (33). To determine whether the serine protease activity was essential for nsp4 to block IFN-β production, we constructed a series of nsp4 mutants for mutational analysis (Fig. 6A). We next sought to determine whether nsp4 protease activity was affected in these mutants. The fusion protein nsp11′12 was cloned into a mammalian expression vector with an N-terminal Flag tag, which is used as a substrate because the 3C-like protease can cleave the junction in the nsp11′12 fusion protein in the canonical cleavage sequence (E/G). HeLa cells were transfected with nsp11′12 fusion protein and nsp4 plasmids, and 24 h later, Western blot analysis was performed. As shown in Fig. 6B, only wild-type nsp4 was capable of cleaving the substrate. The mutation of nsp4 caused a complete abolishment of nsp4 cleavage activity, demonstrating that the catalytic triad of His39-Asp64-Ser118 and three domains are essential for nsp4 protease activity.
FIG 6.

The serine protease catalytic activity is essential for nsp4 to inhibit IFN-β production. (A) Schematic diagram representing the PRRSV nsp4 protein serial mutant constructs. (B) A Flag-tagged nsp11′12 fusion protein expression plasmid was cotransfected into HeLa cells with plasmids encoding nsp4 mutants. Cell lysates were analyzed by Western blotting with antibodies specific for Flag and c-Myc. β-Actin was used as a loading control. (C to F) HeLa cells (C and D) or CRL-2843 cells (E and F) were cotransfected with pGL3-IFN-β-Luc (C and E) or pGL3-NF-κB-Luc (D and F), pRL-TK, and nsp4 mutant expression plasmids. Twenty-four hours later, cells were stimulated with or without poly(I·C) (10 μg/ml). Luciferase assays were performed 8 h after the stimulation. Data are representative of three independent experiments (means and SD). Statistical analysis was performed by Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We then investigated the effect of these nsp4 mutants on IFN-β promoter and NF-κB-responsive promoter activations. Each of the mutant vectors was cotransfected into HeLa cells with an IFN-β– or NF-κB–luciferase reporter vector (Fig. 6C and D). The luciferase activities were measured after poly(I·C) treatment. Our results showed that nsp4 overexpression led to the suppression of IFN-β promoter and NF-κB-responsive promoter activities to 27% and 38%, respectively. However, 3A, N69, and C157 completely abolished the inhibitory ability of nsp4, and the luciferase activities of the IFN-β promoter and NF-κB-responsive promoter were similar to the control level. The MID mutant maintained part of the inhibitory ability, and IFN-β and NF-κB induction was downregulated to about 54% and 66%, respectively. Similar results were obtained in porcine CRL-2843 cells (Fig. 6E and F). These data suggest that the 3C-like serine protease catalytic activity is critical for nsp4 to inhibit IFN-β promoter and NF-κB-responsive promoter activations.
NEMO is the target of PRRSV nsp4.
The data presented above suggested that nsp4 might target IKK signaling molecules to inhibit the NF-κB signaling pathway. To investigate which molecule(s) nsp4 may target to interfere with NF-κB activation, HeLa cells were transfected with nsp4 plasmid and its possible targets, including IKKα, IKKβ, and NEMO (IKKγ). At 24 h after transfection, cell lysates were processed to assess nsp4-mediated cleavage by Western blotting. As shown in Fig. 7A, a reduction in NEMO abundance was observed when nsp4 was coexpressed, and this was accompanied by the appearance of a smaller band (∼40 kDa) reactive with anti-Flag antibody. Moreover, the degree of NEMO cleavage induced by nsp4 was dose dependent (Fig. 7A). However, no cleavage of IKKα and IKKβ by nsp4 was observed (data not shown).
FIG 7.

NEMO is the target of PRRSV nsp4. (A and B) Increasing amounts of nsp4 expression plasmids (A) or nsp4 mutant expression plasmids (B) were cotransfected into HeLa cells with N-terminally Flag-tagged porcine NEMO expression plasmid. Twenty-four hours later, cells were harvested, and Western blot analysis for NEMO and nsp4 expression was performed by incubating with antibodies against Flag or c-Myc, respectively. (C) CRL-2843 cells were transfected with control vector or nsp4 mutant expression plasmids. Western blot analysis was performed 36 h after the transfection. (D) PAMs were mock infected or infected with PRRSV (MOI of 0.1) for 36 h and inoculated with or without poly(I·C) (10 μg/ml). Cells were harvested at 2 h postinoculation, and the expression of NEMO and nsp4 was determined by Western blotting, with β-actin as the reference control.
To investigate whether the cleavage of NEMO is dependent on 3C-like serine protease catalytic activity of nsp4, cells were cotransfected with NEMO and wild-type or mutant nsp4 vectors individually. Our data showed that only wild-type nsp4 cleaved NEMO and produced a smaller band of 40 kDa, whereas all of the mutants lost their ability to induce NEMO cleavage (Fig. 7B). To further confirm whether PRRSV nsp4 can cleave NEMO, we evaluated the effect of nsp4 on endogenous NEMO. As shown in Fig. 7C, transfection with nsp4 reduced the level of NEMO strikingly, while nsp4 mutants without protease activity lost their ability to decrease endogenous NEMO.
Next, we analyzed the impact of PRRSV infection on endogenous NEMO. PAMs were mock infected or infected with PRRSV for 36 h and then either stimulated with poly(I·C) or left untreated. Cells were harvested, and the expression of NEMO was examined by Western blotting. Our results showed that the level of NEMO was significantly decreased in cells infected with PRRSV compared to mock-infected cells (Fig. 7D).
The E349-S350 pair on NEMO is necessary for nsp4-mediated cleavage.
We next examined which residue(s) within NEMO could be cleaved by nsp4. Previous studies have suggested that 3C-like serine protease preferentially cleaves glutamic acid-glycine (E-G) bonds in both the viral polyproteins and cellular targets but may also exhibit proteolytic activity against glutamic acid-serine (E-S) or glutamic acid-alanine (E-A) bonds (40). As NEMO cleavage produced an ∼40-kDa (N-terminal) product, we inferred that a cleavage site(s) may exist between amino acids 330 and 370 (Fig. 8A). To examine this, we constructed a series of mutants with site-directed mutations within NEMO at residues that may serve as nsp4 cleavage sites (E331, E334, E339, E349, E354, and E362). As illustrated in Fig. 8B and C, wild-type NEMO was cleaved when nsp4 was coexpressed, producing a smaller 40-kDa cleavage protein. Of these mutants, only one (the E349A mutant) was resistant to nsp4-mediated cleavage. In contrast, the E331, E334, E339, E354, and E362 mutants were cleaved in the presence of nsp4. These results indicate that the E349-S350 pair of NEMO is a unique cleavage site targeted by nsp4.
FIG 8.

E349-S350 pair is the site of nsp4-mediated cleavage of NEMO. (A) Primary sequences of amino acids 330 to 370 within porcine NEMO. In this region, glutamic acid was replaced with alanine. (B and C) Flag-tagged wild-type NEMO or NEMO mutants were cotransfected into HeLa cells with an nsp4 expression vector or control vector. Cells were collected 24 h posttransfection, and Western blot analysis for NEMO mutant and nsp4 expression was performed by incubating with antibodies against Flag and c-Myc, respectively. β-Actin was used as a loading control.
Overall, these data provide evidence that PRRSV nsp4 is an IFN-β antagonist by mediating the cleavage of NEMO to inhibit NF-κB activation, leading to the suppression of IFN-β production (Fig. 9).
FIG 9.
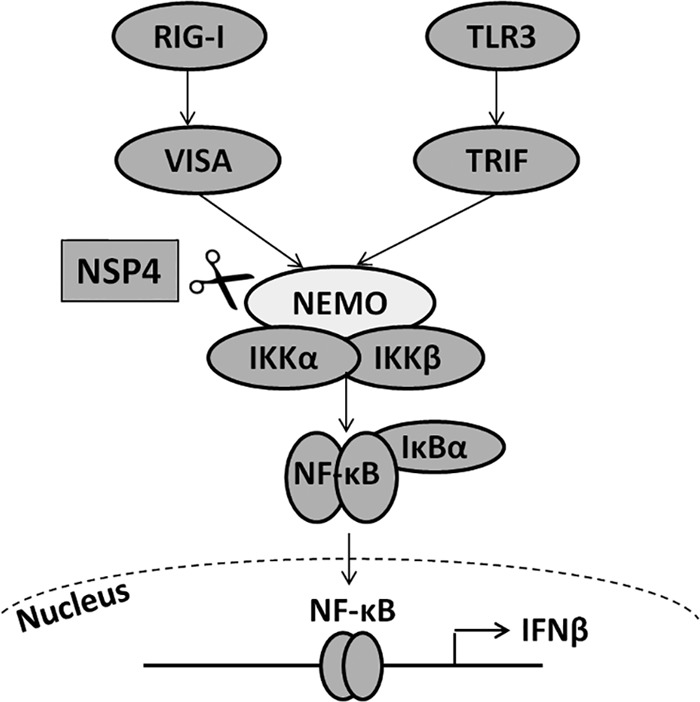
Model showing that nsp4 suppresses the NF-κB signaling pathway by mediating the cleavage of NEMO, leading to the downregulation of IFN-β expression.
DISCUSSION
In the present study, we demonstrated that HP-PRRSV, as well as its nonstructural protein nsp4, has the ability to inhibit IFN-β production. Our data showed that HP-PRRSV interfered with dsRNA-induced type I IFN expression in porcine alveolar macrophages and blood monocyte-derived macrophages by blocking the NF-κB signaling pathway. We further identified PRRSV nsp4 as one of the interferon antagonists. Notably, nsp4 targeted the E349-S350 site of NEMO and mediated its proteolytic cleavage, leading to the suppression of the NF-κB signaling pathway.
Innate immune responses are the first line of host defense to fight virus infection. The critical role of cytokines and chemokines produced by host cells is not only to control virus infection in the early phase but also to facilitate viral clearance by the adaptive immune system (41, 42). Type I IFN responses have been identified as one of the most significant cytokines for resistance to virus infection. It has been reported that PRRSV is sensitive to IFNs (22). Recombinant porcine interferons or synthetic double-stranded RNAs could inhibit PRRSV replication and protect pigs from PRRSV infection (43, 44). However, PRRSV infection triggers poor type I IFN responses. There are two possible explanations. A simple one is that type I IFN is not induced during PRRSV infection. Alternatively, PRRSV evolves mechanisms to counteract the production of type I IFNs. Previous studies have determined that various PRRSV strains can inhibit the production of type I IFNs induced by dsRNA (23, 24, 38). In the present study, we showed that the HP-PRRSV strain significantly blocked poly(I·C)-induced IFN-β production (Fig. 1A and C), indicating that HP-PRRSV is a poor inducer of type I IFNs. Nan et al. found that a novel PRRSV isolate (A2MC2) induced production of type I IFNs and appeared to have an undetectable inhibitory effect on the ability of IFN-α to induce an antiviral response (45), suggesting that different PRRSV strains have distinct influences on interactions with type I IFN responses. Supporting the above assumption, we found that the HP-PRRSV isolate exhibited more inhibition of IFN-β activation in PAMs than the typical type 2 PRRSV isolate VR2332 (data not shown). Interestingly, HP-PRRSV infection causes much more severe symptoms, including high fever, high morbidity, and high mortality in pigs of all ages, than VR2332 infection (46). In consideration of the broad range of effects of type I IFNs on different aspects of immunity, including dendritic-cell (DC) maturation, T-cell activation, and inhibition of viral propagation, these observations implicate that the inhibition of the type I interferon responses by PRRSV may be involved in its pathogenesis (47–49).
We then investigated the ability of HP-PRRSV proteins to inhibit IFN-β production in HeLa cells. Using reporter assays, we found that at least four proteins had the ability to suppress IFN-β promoter activity induced by poly(I·C): one structural protein (N) and three nonstructural proteins (nsp1, nsp4, and nsp11) (Fig. 2A). nsp1, nsp11, and N protein have been identified as IFN-β antagonists, and their mechanisms have partially been studied at the molecular level (25, 26). To our surprise, we did not observe the inhibitory effect of HP-PRRSV nsp2 on IFN-β promoter activity induced by poly(I·C), which is in contrast to reports by others (50, 51). The possible explanation is that the nsp2 sequence is variable among different strains. Interestingly, we also found that HP-PRRSV JXwn06 nsp4 had a greater inhibitory effect on IFN-β induction than the typical less-pathogenic PRRSV strain CH-1a nsp4 (data not shown). Thus, it is reasonable to speculate that HP-PRRSV nsp4 might contribute to PRRSV pathogenesis and evasion of the host immune response much more than the less pathogenic strains. However, these observations need to be further investigated. Nevertheless, nsp4 in combination with other proteins, including nsp1, nsp11, N protein, and possibly other proteins, may play an important role in PRRSV pathogenesis by interfering with type I interferon production.
Here, we demonstrated that nsp4 blocked phosphorylation of IκBα and retained p65 in the cytoplasm. NF-κB plays a critical role in the regulation of innate and adaptive immunity. It is also a key regulator of cell survival, adhesion, proliferation and cell death (apoptosis) (52). To establish persistent infection in host cells, viruses have evolved various strategies to activate or inhibit the NF-κB signaling pathway (53). Activation of NF-κB can be either an antiapoptotic response for viruses to survive and propagate or a proapoptotic response as a mechanism for agents to release mature particles and improve the efficiency of infection (54). Therefore, some viruses, such as Epstein-Barr virus (EBV), Kaposi's sarcoma-associated herpesvirus (KSHV), and hepatitis C virus (HCV), have developed strategies to manipulate NF-κB to strengthen their propagation and transmission (55, 56). On the other hand, NF-κB plays an important role in the regulation of IFN-β production. Thus, many agents target NF-κB to evade innate immune responses. For instance, the measles virus V protein prevents NF-κB activation by binding to p65 and retaining the factor in the cytoplasm; the severe acute respiratory syndrome coronavirus (SARS-CoV) papain-like protease (PLP) blocks the NF-κB signaling pathway via stabilization of the NF-κB inhibitor, IκBα; foot-and-mouth disease virus (FMDV) Lpro regulates the activity of NF-κB by degradation of nuclear p65/RalA (57–59). In our study, we found a mechanism by which PRRSV downregulated NF-κB activation.
PRRSV nsp4 is a multifunctional protein in the polypeptide protein process and in viral replication which belongs to the member of a relatively rare group of proteolytic enzymes, the chymotrypsin-like serine proteases (3CLSP) (60). Crystal structures reveal that nsp4 contains three domains. Domains I and II form the typical chymotrypsin-like two-β-barrel fold, and the C-terminal domain III is dispensable for proteolytic activity. A canonical catalytic triad that is composed of Ser118, His39, and Asp64 is located in the open cleft between the two-β-barrel domains (61). To understand whether the 3C-like protease activity is essential for nsp4 to prevent IFN-β induction, nsp4 site mutant (Ser118, His39, and Asp64) and deletion mutants (domain I, II, or III) were constructed. We found that all of the mutants lost their ability to inhibit IFN-β promoter and NF-κB-responsive promoter activation, except that the domain II deletion mutant retained part of the inhibitory ability. The limited inhibitory ability of the MID mutant is independent of the 3C-like protease activity, because it is incapable of cleaving the reported substrate, nsp11′12 fusion protein. We also found that the MID mutant could not cleave NEMO. A possible explanation could be that the MID mutant may associate with NEMO and impose a physical barrier that disrupts the function of NEMO required to activate NF-κB. Further studies are needed to investigate the mechanism underlying this observation.
Our results indicated that PRRSV nsp4 interfered with the NF-κB signaling pathway by targeting the NF-κB essential modifier (NEMO; also known as IKKγ), a regulatory subunit of the IKK complex. NEMO is required for signaling in the canonical NF-κB pathway, and NEMO-deficient cells exhibit a more severe and broader loss of NF-κB activation than IKKβ knockout cells, suggesting that targeting NEMO could be more effective for viruses to subvert NF-κB pathway (62, 63). Supporting this idea, nsp4 was shown to mediate the cleavage NEMO but had no such effect on IKKα and IKKβ (data not shown). Human NEMO is a 48-kDa protein containing N-terminal two-coiled-coil domains, a leucine zipper, and a C-terminal Zn finger-like domain (64). There are a high degree of sequence conservation and the same functional domains between porcine NEMO and its human counterpart (65). Previous publications have reported that the C-terminal region of NEMO mediates the activation of IKK and interaction with upstream signaling adapters and that the N terminus is responsible for the interaction with IKKs (66, 67). We observed that nsp4 specifically targeted the E349-S350 pair within porcine NEMO, unlike the foot-and-mouth disease virus (FMDV) 3Cpro, which cleaves NEMO at the Q383-R384 site (68). Thus, nsp4 cleaves the C-terminal Zn finger-like domain, which plays a significant role as a bridge in recruiting upstream signaling molecules to activate the NF-κB signaling pathway.
In conclusion, we identified that HP-PRRSV infection could interfere with type I IFN expression induced by poly(I·C) in infected cells. Our data further showed that downregulation of IFNs by HP-PRRSV required suppression of NF-κB signaling pathway. In addition, PRRSV nsp4 was proved to have the ability to inhibit IFN-β production by blocking NF-κB activation in HeLa cells and CRL-2843 cells. Importantly, we found that nsp4 targeted NEMO at the E349-S350 site, cleaving off the C-terminal ZF-like domain from the protein. These findings may provide some insights into the mechanisms by which PRRSV evades host innate immunity.
ACKNOWLEDGMENTS
We thank Hongbing Shu (Wuhan University) for providing the plasmids pRK-Flag-RIG-I, pRK-Flag-VISA, and pRK-Flag-TRIF.
This work was supported by the State Key Laboratory of Agrobiotechnology (grant 2013SKLAB1-5 and 2014SKLAB1-3), China Agricultural University, China, Chinese Universities Scientific Fund (grant 2014JD074), and the Research Fund for the Doctoral Program of Higher Education of China (grant 20130008110028).
Footnotes
Published ahead of print 9 July 2014
REFERENCES
- 1.Albina E. 1997. Epidemiology of porcine reproductive and respiratory syndrome (PRRS): an overview. Vet. Microbiol. 55:309–316. 10.1016/S0378-1135(96)01322-3 [DOI] [PubMed] [Google Scholar]
- 2.Li Y, Wang X, Bo K, Wang X, Tang B, Yang B, Jiang W, Jiang P. 2007. Emergence of a highly pathogenic porcine reproductive and respiratory syndrome virus in the mid-eastern region of China. Vet. J. 174:577–584. 10.1016/j.tvjl.2007.07.032 [DOI] [PubMed] [Google Scholar]
- 3.Cavanagh D. 1997. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch. Virol. 142:629–633 [PubMed] [Google Scholar]
- 4.Huang YW, Meng XJ. 2010. Novel strategies and approaches to develop the next generation of vaccines against porcine reproductive and respiratory syndrome virus (PRRSV). Virus Res. 154:141–149. 10.1016/j.virusres.2010.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tian K, Yu X, Zhao T, Feng Y, Cao Z, Wang C, Hu Y, Chen X, Hu D, Tian X, Liu D, Zhang S, Deng X, Ding Y, Yang L, Zhang Y, Xiao H, Qiao M, Wang B, Hou L, Wang X, Yang X, Kang L, Sun M, Jin P, Wang S, Kitamura Y, Yan J, Gao GF. 2007. Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS One 2:e526. 10.1371/journal.pone.0000526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dokland T. 2010. The structural biology of PRRSV. Virus Res. 154:86–97. 10.1016/j.virusres.2010.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fang Y, Treffers EE, Li Y, Tas A, Sun Z, van der Meer Y, de Ru AH, van Veelen PA, Atkins JF, Snijder EJ, Firth AE. 2012. Efficient −2 frameshifting by mammalian ribosomes to synthesize an additional arterivirus protein. Proc. Natl. Acad. Sci. U. S. A. 109:E2920–E2928. 10.1073/pnas.1211145109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei C, Huang Z, Sun L, Xie J, Chen Y, Zhang M, Zhang C, Qi H, Qi W, Ning Z, Yuan L, Wang H, Zhang L, Zhang G. 2013. Expression and antibody preparation of GP5a gene of porcine reproductive and respiratory syndrome virus. Indian J. Microbiol. 53:370–375. 10.1007/s12088-013-0368-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381. 10.1016/j.immuni.2006.08.007 [DOI] [PubMed] [Google Scholar]
- 10.Bowie AG, Unterholzner L. 2008. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8:911–922. 10.1038/nri2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801. 10.1016/j.cell.2006.02.015 [DOI] [PubMed] [Google Scholar]
- 12.Meylan E, Tschopp J. 2006. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol. Cell 22:561–569. 10.1016/j.molcel.2006.05.012 [DOI] [PubMed] [Google Scholar]
- 13.Kawai T, Akira S. 2007. SnapShot: pattern-recognition receptors. Cell 129:1024. 10.1016/j.cell.2007.05.017 [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. 2003. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301:640–643. 10.1126/science.1087262 [DOI] [PubMed] [Google Scholar]
- 15.West AP, Koblansky AA, Ghosh S. 2006. Recognition and signaling by toll-like receptors. Annu. Rev. Cell Dev. Biol. 22:409–437. 10.1146/annurev.cellbio.21.122303.115827 [DOI] [PubMed] [Google Scholar]
- 16.Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, Mann M, Karin M. 2006. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439:204–207. 10.1038/nature04369 [DOI] [PubMed] [Google Scholar]
- 17.Gaestel M, Kotlyarov A, Kracht M. 2009. Targeting innate immunity protein kinase signalling in inflammation. Nat. Rev. Drug Discov. 8:480–499. 10.1038/nrd2829 [DOI] [PubMed] [Google Scholar]
- 18.Loo YM, Gale M., Jr 2011. Immune signaling by RIG-I-like receptors. Immunity 34:680–692. 10.1016/j.immuni.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988. 10.1038/ni1243 [DOI] [PubMed] [Google Scholar]
- 20.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682. 10.1016/j.cell.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 21.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19:727–740. 10.1016/j.molcel.2005.08.014 [DOI] [PubMed] [Google Scholar]
- 22.Albina E, Carrat C, Charley B. 1998. Interferon-alpha response to swine arterivirus (PoAV), the porcine reproductive and respiratory syndrome virus. J. Interferon Cytokine Res. 18:485–490. 10.1089/jir.1998.18.485 [DOI] [PubMed] [Google Scholar]
- 23.Beura LK, Sarkar SN, Kwon B, Subramaniam S, Jones C, Pattnaik AK, Osorio FA. 2010. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J. Virol. 84:1574–1584. 10.1128/JVI.01326-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calzada-Nova G, Schnitzlein WM, Husmann RJ, Zuckermann FA. 2011. North American porcine reproductive and respiratory syndrome viruses inhibit type I interferon production by plasmacytoid dendritic cells. J. Virol. 85:2703–2713. 10.1128/JVI.01616-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoo D, Song C, Sun Y, Du Y, Kim O, Liu HC. 2010. Modulation of host cell responses and evasion strategies for porcine reproductive and respiratory syndrome virus. Virus Res. 154:48–60. 10.1016/j.virusres.2010.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Y, Han M, Kim C, Calvert JG, Yoo D. 2012. Interplay between interferon-mediated innate immunity and porcine reproductive and respiratory syndrome virus. Viruses 4:424–446. 10.3390/v4040424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Z, Lawson S, Sun Z, Zhou X, Guan X, Christopher-Hennings J, Nelson EA, Fang Y. 2010. Identification of two auto-cleavage products of nonstructural protein 1 (nsp1) in porcine reproductive and respiratory syndrome virus infected cells: nsp1 function as interferon antagonist. Virology 398:87–97. 10.1016/j.virol.2009.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim O, Sun Y, Lai FW, Song C, Yoo D. 2010. Modulation of type I interferon induction by porcine reproductive and respiratory syndrome virus and degradation of CREB-binding protein by non-structural protein 1 in MARC-145 and HeLa cells. Virology 402:315–326. 10.1016/j.virol.2010.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beura LK, Subramaniam S, Vu HL, Kwon B, Pattnaik AK, Osorio FA. 2012. Identification of amino acid residues important for anti-IFN activity of porcine reproductive and respiratory syndrome virus non-structural protein 1. Virology 433:431–439. 10.1016/j.virol.2012.08.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi X, Wang L, Li X, Zhang G, Guo J, Zhao D, Chai S, Deng R. 2011. Endoribonuclease activities of porcine reproductive and respiratory syndrome virus nsp11 was essential for nsp11 to inhibit IFN-beta induction. Mol. Immunol. 48:1568–1572. 10.1016/j.molimm.2011.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sagong M, Lee C. 2011. Porcine reproductive and respiratory syndrome virus nucleocapsid protein modulates interferon-β production by inhibiting IRF3 activation in immortalized porcine alveolar macrophages. Arch. Virol. 156:2187–2195. 10.1007/s00705-011-1116-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Zhang H, Suo X, Zheng S, Feng WH. 2011. Increase of CD163 but not sialoadhesin on cultured peripheral blood monocytes is coordinated with enhanced susceptibility to porcine reproductive and respiratory syndrome virus infection. Vet. Immunol. Immunopathol. 141:209–220. 10.1016/j.vetimm.2011.03.001 [DOI] [PubMed] [Google Scholar]
- 33.Ma Z, Wang Y, Zhao H, Xu AT, Wang Y, Tang J, Feng WH. 2013. Porcine reproductive and respiratory syndrome virus nonstructural protein 4 induces apoptosis dependent on its 3C-like serine protease activity. PLoS One 8:e69387. 10.1371/journal.pone.0069387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q, Guo XK, Gao L, Huang C, Li N, Jia X, Liu W, Feng WH. 2014. MicroRNA-23 inhibits PRRSV replication by directly targeting PRRSV RNA and possibly by upregulating type I interferons. Virology 450–451:182–195. 10.1016/j.virol.2013.12.020 [DOI] [PubMed] [Google Scholar]
- 35.He X, Li Y, Li C, Liu LJ, Zhang XD, Liu Y, Shu HB. 2013. USP2a negatively regulates IL-1beta- and virus-induced NF-kappaB activation by deubiquitinating TRAF6. J. Mol. Cell Biol. 5:39–47. 10.1093/jmcb/mjs024 [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Li C, Xue P, Zhong B, Mao AP, Ran Y, Chen H, Wang YY, Yang F, Shu HB. 2009. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc. Natl. Acad. Sci. U. S. A. 106:7945–7950. 10.1073/pnas.0900818106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo XK, Zhang Q, Gao L, Li N, Chen XX, Feng WH. 2013. Increasing expression of microRNA 181 inhibits porcine reproductive and respiratory syndrome virus replication and has implications for controlling virus infection. J. Virol. 87:1159–1171. 10.1128/JVI.02386-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song C, Krell P, Yoo D. 2010. Nonstructural protein 1alpha subunit-based inhibition of NF-kappaB activation and suppression of interferon-beta production by porcine reproductive and respiratory syndrome virus. Virology 407:268–280. 10.1016/j.virol.2010.08.025 [DOI] [PubMed] [Google Scholar]
- 39.Perkins ND. 2007. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 8:49–62. 10.1038/nrm2083 [DOI] [PubMed] [Google Scholar]
- 40.Fang Y, Snijder EJ. 2010. The PRRSV replicase: exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus Res. 154:61–76. 10.1016/j.virusres.2010.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoebe K, Janssen E, Beutler B. 2004. The interface between innate and adaptive immunity. Nat. Immunol. 5:971–974. 10.1038/ni1004-971 [DOI] [PubMed] [Google Scholar]
- 42.Kabelitz D, Medzhitov R. 2007. Innate immunity–cross-talk with adaptive immunity through pattern recognition receptors and cytokines. Curr. Opin. Immunol. 19:1–3. 10.1016/j.coi.2006.11.018 [DOI] [PubMed] [Google Scholar]
- 43.Miller LC, Laegreid WW, Bono JL, Chitko-McKown CG, Fox JM. 2004. Interferon type I response in porcine reproductive and respiratory syndrome virus-infected MARC-145 cells. Arch. Virol. 149:2453–2463. 10.1007/s00705-004-0377-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Overend C, Mitchell R, He D, Rompato G, Grubman MJ, Garmendia AE. 2007. Recombinant swine beta interferon protects swine alveolar macrophages and MARC-145 cells from infection with porcine reproductive and respiratory syndrome virus. J. Gen. Virol. 88:925–931. 10.1099/vir.0.82585-0 [DOI] [PubMed] [Google Scholar]
- 45.Nan Y, Wang R, Shen M, Faaberg KS, Samal SK, Zhang Y-J. 2012. Induction of type I interferons by a novel porcine reproductive and respiratory syndrome virus isolate. Virology 432:261–270. 10.1016/j.virol.2012.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wernike K, Hoffmann B, Dauber M, Lange E, Schirrmeier H, Beer M. 2012. Detection and typing of highly pathogenic porcine reproductive and respiratory syndrome virus by multiplex real-time rt-PCR. PLoS One 7:e38251. 10.1371/journal.pone.0038251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Honda K, Sakaguchi S, Nakajima C, Watanabe A, Yanai H, Matsumoto M, Ohteki T, Kaisho T, Takaoka A, Akira S, Seya T, Taniguchi T. 2003. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc. Natl. Acad. Sci. U. S. A. 100:10872–10877. 10.1073/pnas.1934678100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vollstedt S, Arnold S, Schwerdel C, Franchini M, Alber G, Di Santo JP, Ackermann M, Suter M. 2004. Interplay between alpha/beta and gamma interferons with B, T, and natural killer cells in the defense against herpes simplex virus type 1. J. Virol. 78:3846–3850. 10.1128/JVI.78.8.3846-3850.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Le Bon A, Tough DF. 2002. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 14:432–436. 10.1016/S0952-7915(02)00354-0 [DOI] [PubMed] [Google Scholar]
- 50.Li H, Zheng Z, Zhou P, Zhang B, Shi Z, Hu Q, Wang H. 2010. The cysteine protease domain of porcine reproductive and respiratory syndrome virus non-structural protein 2 antagonizes interferon regulatory factor 3 activation. J. Gen. Virol. 91:2947–2958. 10.1099/vir.0.025205-0 [DOI] [PubMed] [Google Scholar]
- 51.Sun Z, Chen Z, Lawson SR, Fang Y. 2010. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J. Virol. 84:7832–7846. 10.1128/JVI.00217-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caamano J, Hunter CA. 2002. NF-kappaB family of transcription factors: central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 15:414–429. 10.1128/CMR.15.3.414-429.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santoro MG, Rossi A, Amici C. 2003. NF-kappaB and virus infection: who controls whom. EMBO J. 22:2552–2560. 10.1093/emboj/cdg267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bowie AG, Zhan J, Marshall WL. 2004. Viral appropriation of apoptotic and NF-kappaB signaling pathways. J. Cell. Biochem. 91:1099–1108. 10.1002/jcb.20026 [DOI] [PubMed] [Google Scholar]
- 55.Waris G, Livolsi A, Imbert V, Peyron JF, Siddiqui A. 2003. Hepatitis C virus NS5A and subgenomic replicon activate NF-kappaB via tyrosine phosphorylation of IkappaBalpha and its degradation by calpain protease. J. Biol. Chem. 278:40778–40787. 10.1074/jbc.M303248200 [DOI] [PubMed] [Google Scholar]
- 56.de Oliveira DE, Ballon G, Cesarman E. 2010. NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol. 18:248–257. 10.1016/j.tim.2010.04.001 [DOI] [PubMed] [Google Scholar]
- 57.Schuhmann KM, Pfaller CK, Conzelmann KK. 2011. The measles virus V protein binds to p65 (RelA) to suppress NF-kappaB activity. J. Virol. 85:3162–3171. 10.1128/JVI.02342-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. 2009. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 83:6689–6705. 10.1128/JVI.02220-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Los Santos T, Diaz-San Segundo F, Grubman MJ. 2007. Degradation of nuclear factor kappa B during foot-and-mouth disease virus infection. J. Virol. 81:12803–12815. 10.1128/JVI.01467-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Snijder EJ, Wassenaar AL, van Dinten LC, Spaan WJ, Gorbalenya AE. 1996. The arterivirus nsp4 protease is the prototype of a novel group of chymotrypsin-like enzymes, the 3C-like serine proteases. J. Biol. Chem. 271:4864–4871. 10.1074/jbc.271.9.4864 [DOI] [PubMed] [Google Scholar]
- 61.Tian X, Lu G, Gao F, Peng H, Feng Y, Ma G, Bartlam M, Tian K, Yan J, Hilgenfeld R, Gao GF. 2009. Structure and cleavage specificity of the chymotrypsin-like serine protease (3CLSP/nsp4) of porcine reproductive and respiratory syndrome virus (PRRSV). J. Mol. Biol. 392:977–993. 10.1016/j.jmb.2009.07.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israel A, Rajewsky K, Pasparakis M. 2000. NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol. Cell 5:981–992. 10.1016/S1097-2765(00)80263-4 [DOI] [PubMed] [Google Scholar]
- 63.Solt LA, Madge LA, Orange JS, May MJ. 2007. Interleukin-1-induced NF-kappaB activation is NEMO-dependent but does not require IKKbeta. J. Biol. Chem. 282:8724–8733. 10.1074/jbc.M609613200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hayden MS, Ghosh S. 2008. Shared principles in NF-kappaB signaling. Cell 132:344–362. 10.1016/j.cell.2008.01.020 [DOI] [PubMed] [Google Scholar]
- 65.Scheidereit C. 2006. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene 25:6685–6705. 10.1038/sj.onc.1209934 [DOI] [PubMed] [Google Scholar]
- 66.Makris C, Roberts JL, Karin M. 2002. The carboxyl-terminal region of IkappaB kinase gamma (IKKgamma) is required for full IKK activation. Mol. Cell. Biol. 22:6573–6581. 10.1128/MCB.22.18.6573-6581.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rothwarf DM, Zandi E, Natoli G, Karin M. 1998. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 395:297–300. 10.1038/26261 [DOI] [PubMed] [Google Scholar]
- 68.Wang D, Fang L, Li K, Zhong H, Fan J, Ouyang C, Zhang H, Duan E, Luo R, Zhang Z, Liu X, Chen H, Xiao S. 2012. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 86:9311–9322. 10.1128/JVI.00722-12 [DOI] [PMC free article] [PubMed] [Google Scholar]