

ABSTRACT
Modified vaccinia virus Ankara (MVA) serves as a versatile platform in vaccine development. This highly attenuated orthopoxvirus, which cannot replicate in mammalian cells, triggers strong innate immune responses, including cell migration. Previously, we have shown that induction of chemokine (C-C motif) ligand 2 (CCL2) by MVA is necessary for the recruitment of monocytes and T cells, but not neutrophils, to the lung. Here, we identified neutrophil-attracting chemokines produced by MVA-infected primary murine lung fibroblasts and murine bone marrow-derived macrophages. We demonstrate that MVA, but not vaccinia virus (VACV) strain WR, induces chemokine expression, which is independent of Toll-like receptor 2 (TLR2) signaling. Additionally, we show that both chemokine (C-C motif) receptor 1 (CCR1) and chemokine (C-X-C motif) receptor 2 (CXCR2) are involved in MVA-induced neutrophil chemotaxis in vitro. Finally, intranasal infection of Ccr1−/− mice with MVA, as well as application of the CCR1 antagonist J-113863, revealed a role for CCR1 in leukocyte recruitment, including neutrophils, into the lung.
IMPORTANCE Rapid attraction of leukocytes to the site of inoculation is unique to MVA in comparison to other VACV strains. The findings here extend current knowledge about the regulation of MVA-induced leukocyte migration, particularly regarding neutrophils, which could potentially be exploited to improve other VACV strains currently in development as oncolytic viruses and viral vectors. Additionally, the data presented here indicate that the inflammatory response may vary depending on the cell type infected by MVA, highlighting the importance of the site of vaccine application. Moreover, the rapid recruitment of neutrophils and other leukocytes can directly contribute to the induction of adaptive immune responses elicited by MVA inoculation. Thus, a better understanding of leukocyte migration upon MVA infection is particularly relevant for further development and use of MVA-based vaccines and vectors.
INTRODUCTION
Coordinated cell activation and migration are critical for an optimal host immune response during viral infection. However, many viruses, including poxviruses, have evolved several distinct strategies to interfere with that system (1, 2). Ideally, immediately after a viral infection signals from infected cells and resident sentinel cells trigger an immune response that results in hierarchical recruitment of leukocytes (3). Neutrophils are one of the first cell types to arrive at the site of infection, and their role in antiviral immune responses is receiving renewed attention (4, 5).
Vaccinia virus (VACV) induces activation and functional priming of neutrophils (6), which phagocytize and inactivate VACV by an antibody-dependent mechanism (7, 8). Systemic myxoma virus infection induces neutrophil recruitment to the liver microvasculature, where adherent neutrophils form large aggregates with platelets, releasing neutrophil extracellular traps that help to prevent infection of adjacent cells (9). Recent studies have indicated that the role of neutrophils during poxvirus infection may extend beyond direct antiviral activity, as during infection with modified vaccinia virus Ankara (MVA), neutrophils transport antigen from the dermis to the bone marrow, where a distinct subset of virus-specific CD8+ T cells is induced (10). This reverse transmigration of neutrophils was mediated by chemokine (C-C motif) receptor 1 (CCR1), a chemokine receptor that seemingly fulfills a number of nonredundant functions in host defense (11). CCR1 is widely expressed on hematopoietic and nonhematopoietic cells of both the innate and adaptive immune system and is particularly important for the development of the inflammatory response (12).
Among VACV strains, MVA is unique in its ability to induce chemokine production and cell migration (13, 14). Recruitment of monocytes and lymphocytes to the lungs after MVA infection is driven by chemokine (C-C motif) ligand 2 (CCL2), a ligand for CCR2. However, recruitment of neutrophils to the lungs of Ccl2−/− mice after intranasal infection is almost unaffected (14). There are several cell types in the lung that may potentially produce neutrophil-attracting chemokines, including macrophages and fibroblasts (15, 16). Alveolar macrophages are an important sentinel cell in the lung (17), and fibroblasts constitute some 35 to 40% of cells in the lung interstitium (18). Interestingly, nasal polyps, which are sites of chronic inflammation, were found to comprise a large proportion of fibroblasts (19). Therefore, we investigated the potential of VACV-infected macrophages and lung fibroblasts to induce neutrophil-attracting chemokines and the contribution of their cognate receptors to neutrophil migration.
MATERIALS AND METHODS
Viruses.
MVA (cloned isolate F6), recombinant MVA expressing the green fluorescent protein (MVA-GFP) under the control of the VACV P7.5 early/late promoter (20), and VACV strain Western Reserve (WR), which was originally provided by Bernard Moss (NIH, Bethesda, MD), were propagated in chicken embryo fibroblasts (CEF). Virus stocks were purified by sucrose density centrifugation and titrated using standard methodology (21). All virus stocks were checked for mycoplasma contamination prior to use. Supernatants collected from infected cells were treated with 800 mJ of UV (250 nm; 4.5 min) in a Stratalinker UV Cross-linker 1800 to inactivate any remaining input virus and stored at −80°C until use. To analyze virus growth, cells were infected at a multiplicity of infection (MOI) of 0.05. After 1 h of incubation, the inoculum was removed, and the cells were washed 3 times with phosphate-buffered saline (PBS) before the addition of the appropriate medium containing 2% fetal calf serum (FCS). At the indicated time points, cells were harvested by scraping into medium and frozen until use. Viral titers were determined by the 50% tissue culture infectious dose (TCID50) using the Reed-Muench methodology (22) on the chicken embryo fibroblast line DF1, which was maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with stable glutamine and 10% FCS (Biochrom AG, Berlin, Germany).
Mice.
Male and female C57BL/6NCrl mice were obtained from Charles River (Sulzfeld, Germany) and used as age- and sex-matched wild-type (WT) controls. Ccr1−/− mice (12) were backcrossed for 12 generations to C57BL/6NCrl, and Tlr2−/− mice were back-crossed for 9 generations to the C57BL/6 background (23). The mice were housed under specific-pathogen-free conditions in a temperature- and light-controlled room (21 to 23°C; 55% ± 3% relative humidity; 12-h light–12-h dark cycle) and were fed a standard rodent diet with sterilized water ad libitum. All animal procedures were in accordance with the institutional Animal Welfare Committee, as well as the regulations in the German Animal Welfare Act for the protection of laboratory animals. All experimentations were approved by the Government of Upper Bavaria, Munich, Germany.
Flow cytometric analysis.
Bronchoalveolar lavage (BAL) of mice was carried out as described previously (14). Cells obtained from BAL fluid were stained using CD11b-fluorescein isothiocyanate (FITC) (M1/70), CD3-phycoerythrin (PE) (145-2C11), CD8α-PeCy7 (53-6.7), Ly6G-allophycocyanin (APC) (HK1.4), and Ly6C-APC-Cy7 (1A8) (BioLegend, London, United Kingdom) and analyzed on a FACSCanto II (BD Biosciences, San Jose, CA) or on a MACSQuant VYB flow cytometer (Miltenyi Biotec GmbH, Bergisch-Gladbach, Germany). For analysis, neutrophils were gated as CD11b+ Ly6G+ cells, inflammatory monocytes as CD11b+ Ly6G− Ly6Chi cells, and lymphocytes as CD3+ CD8+ or CD3+ CD8− cells. For the analysis of lymphocytes, gating from forward/side scatter was used to exclude granulocytes and monocytes. Dead cells were excluded by staining with 7-aminoactinomycin D (7-AAD) or a Zombie Aqua Fixable Viability Kit (BioLegend).
Isolation, cultivation, and infection of primary MLFs.
The lungs of PBS-perfused mice were removed; rinsed in PBS; placed in DMEM-Ham's F-12 (1:1) medium (Biochrom) supplemented with 20% FCS, 15 mM HEPES, 100 U/ml penicillin, and 100 μg/ml streptomycin (murine lung fibroblast [MLF] growth medium); and cut into small segments (approximately 1 mm3). The lung segments were digested for 2 h in MLF growth medium containing 5 mg/ml collagenase I (Biochrom) at 37°C with agitation at 400 rpm. The digested lung segments were then pushed through a 70-μm nylon filter (BD Biosciences). The homogenized lung tissue was then washed, resuspended in MLF growth medium, and placed in a 10-cm cell culture-treated petri dish. The cells were identified as described previously (24) and split at a 1:3 ratio when 80% confluent; otherwise, the medium was refreshed every 2 or 3 days. For infection, 1 × 106 MLFs were seeded into each well of 6-well plates and left overnight to attach. The following day, 2 ml of DMEM-Ham's F-12 containing 15 mM HEPES and 0.5% FCS was added to each well, and the cells were infected with virus at the indicated MOI.
Generation and infection of BMDM.
Bone marrow (BM) cells were obtained from the femurs and tibiae of C57BL/6 mice. Red blood cells were lysed using a mouse erythrocyte-lysing kit (R&D Systems Inc., Minneapolis, MN). The cells were then cultivated in very-low-endotoxin (VLE)-RPMI 1640 medium supplemented with 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 20% conditioned medium from L929 cells. The fibroblasts were removed by collecting nonadherent cells after the first 24 h. The medium was replaced after 4 days with VLE-RPMI 1640 medium and supplemented with 10% FCS, standard antibiotics, and 10% L929 conditioned medium. Two days later, 1.5 × 106 cells were reseeded to 6-well plates (Sarstedt, Nümbrecht, Germany) in VLE-RPMI 1640 medium supplemented with 10% FCS and incubated overnight before infection. The purity and differentiation of bone marrow-derived macrophages (BMDM) was verified by microscopy and by surface expression of CD11b, accompanied by upregulation of Toll-like receptor 2 (TLR2) and loss of Gr-1 expression. Cultures of BMDM were typically determined to be ≥98% pure.
Isolation of primary neutrophils.
Bone marrow cells from C57BL/6 mice were prepared as described above. Subsequently, neutrophils were obtained by negative selection in a magnetic sorting column using a neutrophil isolation kit (Miltenyi Biotec GmbH) according to the manufacturer's instructions. After isolation, the neutrophils were resuspended at a concentration of 5 × 106 cells/ml in Hanks balanced salt solution (HBSS) (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) and used immediately.
Protein array.
Cell culture supernatants were screened with a RayBio murine cytokine array III (RayBiotech, Norcross, GA) according to the manufacturer's instructions. Chemiluminescence signals were detected by a ChemiDoc MP System and analyzed using Image Lab software (Bio-Rad, Hercules, CA).
ELISA.
Enzyme-linked immunosorbent assays (ELISAs) for CCL5 and CCL9 were performed using DuoSet antibody kits (R&D Systems Inc.) and were carried out according to the manufacturer's instructions.
Reverse transcription (RT)-PCR.
Total RNA was isolated using an RNeasy Plus Kit (Qiagen, Hilden, Germany) 6 h postinfection (p.i.) and converted to cDNA using Omniscript reverse transcriptase (Qiagen). PCR was carried out as described previously (25). Primer pairs for amplification of murine GAPDH (glyceraldehyde-3-phosphate dehydrogenase), specific murine chemokines, and the VACV E3L gene were designed using Primer3 software (26). The sequences of oligonucleotides and PCR product sizes are listed in Table 1. All oligonucleotides were synthesized by Eurofins Genomics (Ebersberg, Germany). PCR products were run on a 1.5% agarose gel and stained with GelRed (Biotium, Hayward, CA), and the identity of each PCR product was verified by sequencing (Eurofins Genomics). Pictures of gels were acquired with a ChemiDoc MP System and analyzed using Image Lab software (Bio-Rad).
TABLE 1.
Summary of oligonucleotides used in RT-PCR
| Target gene | Primera | Oligonucleotide sequence (5′–3′) | Product size (bp) |
|---|---|---|---|
| Murine GAPDH | mGAPDH-F | GAC AAC TCA CTC AAG ATT GTC AG | 540 |
| mGAPDH-R | GTA GCC GTA TTC ATT GTC ATA CC | ||
| Murine CCL5 | mCCL5-F | CCT CAC CAT CAT CCT CAC TG | 302 |
| mCCL5-R | AAT GAC AGG GAA GCT ATA CAG G | ||
| Murine CCL9 | mCCL9-F | CTA TAA CTC ACG GAT TCA GTG TTC | 550 |
| mCCL9-R | TTA TAG ATA GTT TGA GCT GCC ATC | ||
| Murine CXCL1 | mCXCL1-F | ATT CAC CTC AAG AAC ATC CAG | 690 |
| mCXCL1-R | CCA GGA GAA ACA GGG TTA AAG | ||
| Murine CXCL2 | mCXCL2-F | TTG ACT TCA AGA ACA TCC AGA G | 754 |
| mCXCL2-R | CAT TAC ATT GAA ATC CAG CAT C | ||
| VACV E3L | E3L-F | GTT CTG ACG CAG AGA TTG TG | 406 |
| E3L-R | TTA CTA GGC CCC ACT GAT TC |
F, forward; R, reverse.
Quantitative real-time RT-PCR.
RNA isolation from MLFs and synthesis of cDNA were carried out as described above. For in vivo analysis, lungs of mice were snap-frozen in liquid nitrogen and subsequently homogenized. Then, RNA extraction and synthesis of cDNA were performed as described above. Expression levels of selected transcripts were determined in duplicate by quantitative PCR using a Roche LightCycler 480 II and SYBR green I master mix. A single 20-μl PCR mixture contained 10 μl of the 2× SYBR green master mix, 6 pmol of forward and reverse primers, 0.6 units Taq polymerase, and 2 μl of the 1:10-diluted cDNA. No-template controls (NTC) were included for each target to ensure that all reagents used to set up the reactions were free of contaminating foreign DNA. The following cycle conditions were used: 1 cycle of 5 min at 95°C and 45 cycles of 15 s at 95°C, 15 s at 60°C, and 20 s at 72°C. During the first 8 cycles, the annealing temperature was decreased in decrements of 1°C from 67°C to 60°C. Each PCR run was terminated with a melting-curve analysis. Samples were denatured for 10 s at 95°C, reannealed for 1 min at 60°C, and then denatured again by slowly heating the samples from 60°C to 97°C with a ramp-up rate of 0.11°C/s. Primer pairs were either from qPrimerDepot (27) or designed with the software Primer-BLAST (28) and were cDNA specific. The annealing temperature was always set to approximately 60°C, and the amplicon size range was between 80 and 150 bp. Relative quantification was performed by the ΔΔCT method (29) using 18S rRNA as a reference. Threshold cycle (CT) values were determined with the LightCycler 480 software using the second derivative maximum method. The resulting relative expression values were multiplied by 108.
Chemotaxis assay.
Chemotaxis assays were performed as previously described (30). Briefly, freshly isolated neutrophils were resuspended at a concentration of 5 × 106 cells/ml in HBSS and allowed to migrate for 40 min in a 96-well Multi-Screen-MIC plate (3-μm pore size; Merck Millipore, Billerica, MA). Where indicated, neutrophils were pretreated with 10 nM the CCR1 antagonist J-113863 (CCR1a) or the chemokine (C-X-C motif) receptor 2 (CXCR2) antagonist SB265610 (CXCR2a). Concentrations of the antagonists were applied according to the manufacturer's instructions (R&D Systems Inc.); otherwise, cells were incubated with the equivalent volume of solvent for 5 min prior to running the assay. The number of transmigrating cells in the bottom chamber was determined using a MACSQuant VYB flow cytometer (Miltenyi Biotec).
Statistical analysis.
All data were assembled using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA); significance was determined by a nonparametric Mann-Whitney U test, with a P value of <0.05 deemed to be statistically significant.
RESULTS
MVA infects but does not replicate in primary murine lung fibroblasts and bone marrow-derived macrophages.
Macrophages and fibroblasts are potential targets for viral infection in the lung that may contribute to the development of local immune responses by production of inflammatory mediators (31–33), making them potentially important sentinel cells. To assess the infectivity of these cell types, monolayers of MLFs and BMDM were infected with a recombinant MVA-GFP at different MOIs and measured after 16 h by flow cytometry. These experiments demonstrated that both MLFs and BMDM are readily infected with MVA and are permissive for VACV gene expression and that the level of infection correlates with the MOI applied (Fig. 1A). As expected, MVA was unable to replicate in MLFs or BMDM, whereas VACV WR productively infected MLFs, but not BMDM (Fig. 1B).
FIG 1.

MVA infects but does not replicate in MLFs and BMDM. (A) MLFs and BMDM were infected with MVA-GFP at the indicated MOIs, incubated for 16 h, and analyzed by flow cytometry. (B) Virus replication was determined as described in Materials and Methods. DF1 cells served as a positive control.
MVA induces chemokine production in MLFs and BMDM.
To examine the potential of lung fibroblasts to contribute to the development of the innate immune response, we investigated chemokine production by MVA-infected primary MLFs. Culture supernatants from mock- and MVA-infected primary MLFs and BMDM were screened by a protein array to identify chemokines that may play a role in migration of neutrophils during MVA infection. The analysis revealed that CCL5, CCL9, CXCL1, and CXCL2 were upregulated in MVA-infected primary MLFs. We also detected induction of CCL2 protein (Fig. 2B), which has been reported previously for MVA-infected monocytes (14). As with MVA-infected MLFs, we detected an increase of CCL9 and CXCL2 in supernatants from MVA-infected BMDM. In contrast, CCL5 and CXCL1 were not induced (Fig. 2C). Additionally, we observed an increase of CCL12 in MVA-infected BMDM. CCL12 is a murine ligand for CCR2 that has no direct human counterpart (34).
FIG 2.

Detection of differentially expressed cytokines in cell culture supernatants of MLFs and BMDM. (A) Scheme of the spotted primary antibodies on the RayBio Mouse Cytokine Antibody Array 3. (B) Images after chemiluminescence detection of cytokines in cell culture supernatants from MVA-infected and mock-infected MLFs. Cell culture supernatants were harvested 16 h p.i. (C) Images after chemiluminescence detection of cytokines in cell culture supernatants from MVA-infected and mock-infected BMDM. Cell culture supernatants were harvested 24 h p.i. The tables show upregulated chemokines. The ratios indicated were calculated by using the intensities of the corresponding protein spots after background (Neg) correction and normalization of the intensities according to the mean intensities of the positive controls (Pos).
MVA-infected, but not VACV WR-infected, primary MLFs produce neutrophil-attracting chemokines.
MVA is a highly attenuated VACV strain that, unlike other strains, induces strong chemokine production in monocytes and triggers early recruitment of leukocytes to the lung (14). Therefore, we compared chemokine induction by MVA in primary MLFs to that of the replication-competent VACV WR.
Analysis of specific mRNAs in MVA-infected primary MLFs by RT-PCR showed induction of Ccl5, Cxcl1, and Cxcl2 by MVA and constitutive expression of Ccl9 (Fig. 3A). As expected VACV WR infection did not induce Ccl5 or Cxcl2 in primary MLFs and triggered only a slight upregulation of Cxcl1. Both CCL5 and CCL9 bind to CCR1, which is expressed on neutrophils and is involved in cell migration (12). As CCR1 is activated by a number of other chemokines from the C-C motif subfamily, which are not detectable by the protein array used, expression of other potential CCR1 binding chemokines was examined by quantitative real-time RT-PCR. We confirmed the induction of Ccl5 mRNA by MVA infection and the constitutive expression of Ccl9. Moreover, we also detected constitutive expression of Ccl6 and Ccl8 that was not affected by MVA infection (Fig. 3B). The induction of CCL5 at the protein level was confirmed by ELISA, which also showed MVA-induced, but not VACV WR-induced, CCL5 production in MLFs. In accordance with the results of the protein array, CCL9 protein was constitutively produced by uninfected MLFs and significantly increased by MVA infection. Infection of MLFs with VACV WR did not increase the concentration of CCL9 protein (Fig. 3C).
FIG 3.

MVA-infected, but not WR-infected, MLFs produce neutrophil-attracting chemokines. (A) MLFs were infected with MVA or WR at the indicated MOI, and specific mRNA was detected by RT-PCR 6 h p.i. (B) Total RNA from MLFs infected at an MOI of 0.5 with MVA or WR was analyzed by quantitative real-time RT-PCR for specific CCR1 ligands. The error bars indicate standard deviations (SD). LPS, lipopolysaccharide. (C) Concentrations of CCL5 (n = 3) and CCL9 (n = 5) in culture supernatants of MLFs infected at an MOI of 0.5 with MVA or WR were determined by ELISA after 16 h of infection. The data are means and standard errors of the mean (SEM). *, P < 0.05.
MVA induces CCL5 expression in BMDM independently of TLR2 signaling.
Previously, it was reported that MVA signals via TLR2 (35) and that TLR2 induces CXCL1 and CXCL2 in macrophages at the transcriptional level (36). Examination of mRNA from BMDM infected with MVA for 6 h showed that Cxcl1 and Cxcl2 mRNA levels were as low as in mock-infected cells. Even if BMDM were infected with MVA at an MOI of 2 or 4, transcription of Cxcl1 and Cxcl2 was not induced (M. H. Lehmann, unpublished data). However, in contrast to the results from the protein array, Ccl5 mRNA was strongly induced in MVA-infected BMDM from WT and Tlr2−/− mice. As expected, the TLR2 ligand P3CSK4, used as a positive-control stimulus, induced Cxcl1, Cxcl2, and Ccl5 mRNAs in WT but not in Tlr2−/− cells (Fig. 4A). Induction of Ccl5 expression in MVA-infected BMDM was confirmed at the protein level by ELISA. As expected, this marked increase of the CCL5 protein concentration in supernatants of MVA-infected BMDM was not seen in VACV WR-infected BMDM (Fig. 4B).
FIG 4.
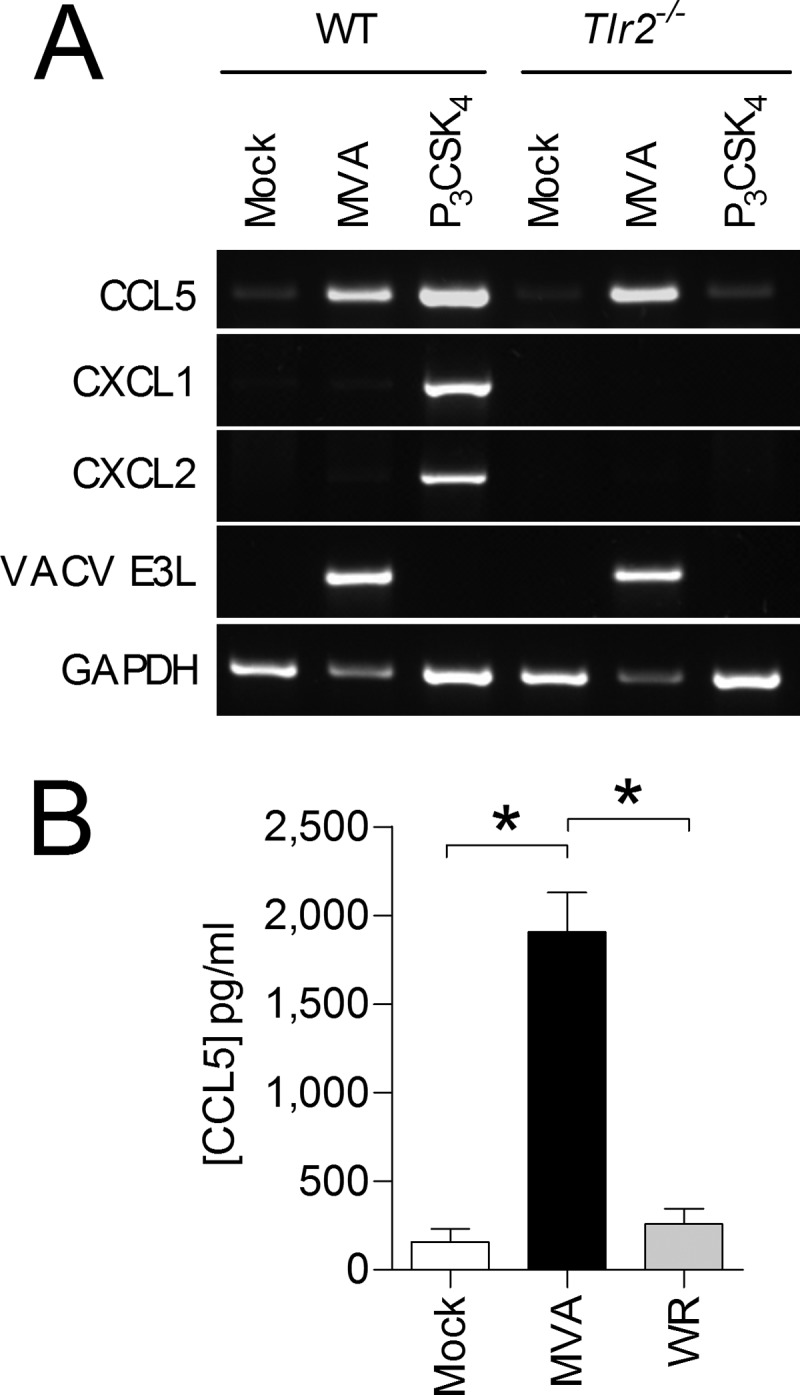
MVA-infected, but not WR-infected, BMDM induce expression of CCL5. (A) WT and Tlr2−/− BMDM were infected with MVA at an MOI of 1 and incubated for 6 h. Specific mRNAs were detected by RT-PCR. One of three independent experiments is shown. (B) BMDM were infected with MVA or WR at an MOI of 1 and incubated for 16 h. Cellular supernatants were analyzed by means of a CCL5-specific ELISA. The data are means and SEM from two independent experiments. *, P < 0.05.
CCR1 and CXCR2 are required for efficient migration of neutrophils in vitro.
To investigate whether MVA-infected MLFs and BMDM produce biologically active neutrophil-attracting agents, in vitro chemotaxis assays using primary mouse neutrophils were performed. UV-treated cell culture supernatants from MVA-infected, but not VACV WR-infected, MLFs and BMDM induced chemotaxis of neutrophils, which is in accordance with the observed capabilities of these strains to induce the production of neutrophil-attracting chemokines in vitro. Supernatants from MVA-infected MLFs were notably more efficient at inducing neutrophil chemotaxis than BMDM supernatants, with approximately 5-fold induction compared to 3-fold induction, respectively (Fig. 5). As demonstrated, MVA-infected MLFs and BMDM produce chemokines that act on chemokine receptors CCR1 and CXCR2. Both receptors are known to play key roles in neutrophil mobilization and migration (37–40). Therefore, the relevance of these two chemokine receptors in MVA-induced neutrophil chemotaxis was assessed by way of specific non-peptide receptor antagonists. Inhibition of CXCR2 reduced neutrophil migration toward supernatants from MVA-infected MLFs and BMDM to background levels. While treatment of neutrophils with the CCR1 antagonist significantly reduced the induction of neutrophil chemotaxis in response to supernatants from MVA-infected MLFs (Fig. 5A), the effect was less pronounced in supernatants from MVA-infected BMDM (Fig. 5B).
FIG 5.

CCR1 and CXCR2 mediate chemotaxis of neutrophils toward supernatants from MVA-infected MLFs and BMDM. MLFs (A) and BMDM (B) were infected with MVA or WR at an MOI of 1. Cell culture supernatants were harvested after 16 h and tested for the ability to induce chemotaxis of primary neutrophils. Where indicated (+), neutrophils were pretreated for 5 min with 10 nM the CCR1 antagonist J-113863 (CCR1a) or 10 nM the CXCR2 antagonist SB265610 (CXCR2a) prior to the running of the assay; all other samples were treated with an equivalent amount of solvent. The data are means and SEM (n = 4). *, P < 0.05.
MVA induces CXCL2 and CCL5 in murine lungs.
Neutrophils are found in high numbers in the lung 24 h after intranasal infection with MVA (14). It is reasonable to expect that the factor attracting them has to be produced prior to their recruitment. Assuming that such a factor is induced at the transcriptional level by MVA, we decided to examine the mRNA levels of Ccr1 and Cxcr2 chemokine ligands in murine lungs 8 h after infection. Quantitative real-time RT-PCR from the lungs of infected mice showed induction of Cxcl1 and Cxcl2 mRNAs (Fig. 6A).
FIG 6.

MVA infection induces production of neutrophil-attracting chemokines in vivo. Mice were i.n. infected with 1 × 107 PFU of MVA or the equivalent volume of PBS. (A) Analysis of chemokine expression in snap-frozen whole lungs by quantitative real-time RT-PCR 8 h p.i. Determination of relative expression values was performed as described in Materials and Methods. (B) The concentration of CXCL2 in BAL fluid was determined 24 h p.i. by multiplex bead-based technology. (C) The concentration of CCL5 in BAL fluid was determined by ELISA at the indicated time points. The dotted line intersecting the y axis indicates the lower limit of detection. The data are means and SEM (n = 3). *, P < 0.05.
An increased CXCL1 protein concentration in the lung after infection with MVA has been reported previously (14). Here, induction of CXCL2 was confirmed by a Bio-Plex mouse cytokine assay, which demonstrated clear upregulation of CXCL2 protein in the lungs of MVA-infected mice (Fig. 6B). We did not detect any induction or upregulation of Ccl5, Ccl6, Ccl7, Ccl8, or Ccl9 mRNA in the lungs of infected mice 8 h p.i. However, CCL5 protein was detected by ELISA 24 h and 48 h after MVA infection (Fig. 6C).
CCR1 is required for efficient recruitment of leukocytes to the lung.
CCR1 is known to play an important role in neutrophil recruitment (40), but with regard to MVA infection, it was shown recently that early recruitment of neutrophils to the dermis was not affected in Ccr1−/− mice (10). The results from our in vitro chemotaxis assays indicate a role for CCR1 in neutrophil migration during MVA infection. Interestingly, the effects of the CCR1 antagonist J-113863 were different depending on the cellular origin of the supernatants used to trigger chemotaxis of primary neutrophils. Therefore, one might hypothesize that the role of CCR1 in neutrophil recruitment may vary depending on the particular tissues or organs.
Since we were interested in studying neutrophil recruitment to the lung, mice were intranasally (i.n.) infected with MVA and treated with the well-characterized CCR1 antagonist J-113863 (41, 42) or an equivalent amount of vehicle. BAL was performed 16 h after infection, and cells in the BAL fluid were analyzed with flow cytometry. Indeed, neutrophil recruitment in MVA-infected mice treated with the CCR1 antagonist was reduced to levels similar to those of the mock-infected controls (Fig. 7), demonstrating that CCR1 is required for the recruitment of neutrophils to the lung during the early phase of MVA infection.
FIG 7.

Neutrophil recruitment to the lungs of MVA-infected mice is blocked by a CCR1 antagonist. C57BL/6 mice were i.n. infected with 1 × 107 PFU of MVA or an equal volume of PBS as a control and then injected i.p. with 10 mg/kg of body weight of the CCR1 antagonist (CCR1a) J-113863 or an equivalent amount of vehicle. Cells in the lung were recovered at 16 h p.i. by BAL and analyzed by flow cytometry. (A) Representative dot plots from each group showing infiltration of CD11b+ Ly6G+ neutrophils. PMN, polymorphonuclear leukocytes. (B) Summary of the analysis. For groups of MVA-infected mice, n = 9; for groups of PBS-dosed mice, n = 5. ***, P < 0.001.
Additionally Ccr1-deficient mice were i.n. infected with MVA, and leukocyte recruitment to the lungs was monitored at 48 h p.i., a time point allowing the detection of several types of important leukocytes (14). Compared to WT control mice, MVA-induced leukocyte recruitment was impaired in Ccr1-deficient mice. In particular, we observed significant reductions in the number of infiltrating CD11b+ Ly6G+ neutrophils and CD11b+ LC6Chi inflammatory monocytes (Fig. 8), corresponding well to our in vitro results. Moreover, we observed a significant reduction in the number of recruited CD3+ lymphocytes, including CD8+ T cells.
FIG 8.

MVA-induced leukocyte recruitment to the lungs is impaired in Ccr1-deficient mice. C57BL/6 WT and Ccr1−/− mice were i.n. infected with 1 × 107 PFU of MVA. An equal volume of PBS was used as a control. Cells in the lung were recovered at 48 h p.i. by BAL and analyzed by flow cytometry. Neutrophils (PMN) were gated as CD11b+ Ly6C+ Ly6G+, inflammatory monocytes (IM) were gated as CD11b+ Ly6Chi, and lymphocytes were gated according to expression of CD3 and CD8. (A) Representative dot plots from each group. The gates show the positions of relevant cell types. (B) Summary of the analysis. For groups of MVA-infected mice, n ≥ 8; for groups of PBS-dosed mice, n ≥ 5. The bars represent the mean numbers of gated cell populations, and the error bars indicate SEM. *, P < 0.05; ***, P < 0.001.
DISCUSSION
Recombinant poxviruses are increasingly used in clinical studies for the treatment of infectious diseases and cancer (43, 44). However, little is known about the mechanisms underlying the induction of the host immune response by these viruses, which may be a critical factor contributing to the success or failure of a therapy. Gaining a better understanding of the mechanisms of MVA-induced leukocyte migration may assist in the design of improved and more effective vaccines.
In contrast to other conventional VACV strains, MVA triggers robust innate immune responses, including induction of inflammatory cytokines and leukocyte migration (13, 45–47). In previous experiments, we have shown that the chemokine CCL2, which binds to CCR2, plays an important role in the recruitment of monocytes and lymphocytes to the site of MVA infection. However, MVA-induced neutrophil migration was not affected in Ccl2−/− mice (14). In this report, we (i) identify neutrophil-attracting chemokines, which are produced in primary cells and in the lung after infection with MVA; (ii) show the importance of both CCR1 and CXCR2 for MVA-induced neutrophil migration in vitro; and (iii) demonstrate the contribution of CCR1 to MVA-triggered leukocyte migration in vivo.
Our study of chemokine expression during MVA infection demonstrated that MVA-infected primary MLFs and BMDM produce the neutrophil-attracting chemokines CCL5 and CXCL2, whereas CXCL1 was detected only in MLFs. Chemotaxis assays indicated a critical role for CXCR2 in MVA-induced neutrophil migration in vitro. Notably, supernatants from MLFs were more efficient in inducing neutrophil chemotaxis than supernatants from BMDM. The observed differences between the two cell types in their abilities to induce chemotaxis of neutrophils could be due to the lack of CXCL1 induction in BMDM. The failure of the protein array to detect CCL5 in supernatants from BMDM could hint at another possibility. Macrophage proteases can cleave CCL5, resulting in amino-terminally truncated forms that are biologically less active (48). Proteolytic processing of CCL5 might alter an epitope that is important for detection by the protein array but not by the CCL5-specific ELISA. Proteolytic enzymes are readily isolated from the lungs of patients (49), and their activity can have important consequences for the immune response. Processing of CCL5 alters its receptor specificity, with cleaved CCL5 binding preferentially to CCR5 (50) while inhibiting CCR1-mediated effects at the same time (51, 52). Despite increased amounts of CCL5 protein in MVA-infected BMDM supernatants, it is possible that CCL5 may be proteolytically processed and therefore a weaker agonist for CCR1. This might explain why BMDM supernatants are less efficient than MLF supernatants in inducing neutrophil chemotaxis and appear to be less affected by blockade of CCR1.
Using a murine intranasal-infection model, we demonstrated that MVA induces the expression of chemokines that act on CCR1 and CXCR2, which are known to mediate migration of neutrophils. As the role of CXCR2 in neutrophil migration is relatively well understood, we focused on CCR1. Similar to our in vitro findings, neutrophil recruitment was significantly reduced, but not abolished, in Ccr1−/− mice at 48 h postinfection. On the other hand, treatment of mice with a CCR1 antagonist nearly completely prevented neutrophil recruitment at 16 h postinfection. However, J-113863, the CCR1 antagonist used, has been shown to interfere with tumor necrosis factor alpha (TNF-α) (41), which is also involved in neutrophil recruitment (53); therefore, the off-target effects of J-113863 may potentiate the effects of CCR1 inhibition. Based on our results, it is tempting to speculate that CCR1 and CXCR2 ligands may interact in a synergistic manner to enable optimal recruitment of neutrophils. Indeed such cross-regulation between CCR1 and CXCR2 has been shown previously in vitro (54), and CXCL2 and CCL3 have been shown to act in a cascade that induces neutrophil recruitment by triggering the release of TNF-α and leukotriene B4 (55). In this context, the two VACV CC chemokine binding proteins, A41 (56, 57) and the 35-kDa viral CC chemokine inhibitor (58–60), may play an important role in poxvirus immune evasion, as inhibition of CCR1 ligands, such as CCL5, may be sufficient to affect neutrophil recruitment. However, not all VACV strains, including VACV WR, express the 35-kDa C-C chemokine inhibitor (61), yet many are still able to block chemokine induction and leukocyte recruitment. VACV, including the WR strain, encodes many viral immune evasion proteins (2) that are absent in MVA. Therefore, prevention of chemokine production and early neutrophil recruitment by VACV strain WR probably occurs at the level of host gene expression.
Reduced numbers of infiltrating leukocytes could potentially have important consequences for the development of the antiviral immune response. The reduction of T lymphocytes, particularly CD8+ cytotoxic T cells, in MVA-infected Ccr1−/− mice indicates that the adaptive immune response may also be affected. This is not entirely unexpected, as CCR1 appears to be expressed on distinct subsets of T lymphocytes and is important for T cell migration to the lymph nodes (62). In addition CCR1 was also shown to play a role in an antigen transport mechanism whereby antigen-loaded neutrophils migrate to the bone marrow, where a distinct subset of CD8+ T cells is induced (10).
In summary, we have demonstrated that CCR1 is important for MVA-triggered leukocyte migration in vivo, particularly with regard to neutrophil and inflammatory monocyte recruitment. Additionally we showed that MVA-infected, but not VACV WR-infected, cells produce neutrophil-attracting chemokines, which is independent of TLR2 signaling. These findings extend the knowledge about the induction of the host immune response by MVA, which is important for its application as a viral vector.
ACKNOWLEDGMENTS
We thank Katharina Heinzelmann and Oliver Eickelberg (Comprehensive Pneumology Center, Helmholtz Zentrum, Munich, Germany) for their help with the preparation of primary murine lung fibroblasts.
This study was partly supported by the European Commission FP7 project VECTORIE (contract no. 261466) and by the German Centre for Infection Research (DZIF) (TTU 07.803). Lino E. Torres-Dominguez was supported by a grant from the German Academic Exchange Service (DAAD).
Footnotes
Published ahead of print 9 July 2014
REFERENCES
- 1.Alcami A. 2003. Viral mimicry of cytokines, chemokines and their receptors. Nat. Rev. Immunol. 3:36–50. 10.1038/nri980 [DOI] [PubMed] [Google Scholar]
- 2.Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SW, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J. Gen. Virol. 94:2367–2392. 10.1099/vir.0.055921-0 [DOI] [PubMed] [Google Scholar]
- 3.Melchjorsen J, Sorensen LN, Paludan SR. 2003. Expression and function of chemokines during viral infections: from molecular mechanisms to in vivo function. J. Leukoc. Biol. 74:331–343. 10.1189/jlb.1102577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mocsai A. 2013. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J. Exp. Med. 210:1283–1299. 10.1084/jem.20122220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drescher B, Bai F. 2013. Neutrophil in viral infections, friend or foe? Virus Res. 171:1–7. 10.1016/j.virusres.2012.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forster R, Wolf G, Mayr A. 1994. Highly attenuated poxviruses induce functional priming of neutrophils in vitro. Arch. Virol. 136:219–226. 10.1007/BF01538831 [DOI] [PubMed] [Google Scholar]
- 7.West BC, Eschete ML, Cox ME, King JW. 1987. Neutrophil uptake of vaccinia virus in vitro. J. Infect. Dis. 156:597–606. 10.1093/infdis/156.4.597 [DOI] [PubMed] [Google Scholar]
- 8.Jones JF. 1982. Interactions between human neutrophils and vaccinia virus: induction of oxidative metabolism and virus inactivation. Pediatr. Res. 16:525–529. 10.1203/00006450-198207000-00005 [DOI] [PubMed] [Google Scholar]
- 9.Jenne CN, Wong CH, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, McFadden G, Kubes P. 2013. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 13:169–180. 10.1016/j.chom.2013.01.005 [DOI] [PubMed] [Google Scholar]
- 10.Duffy D, Perrin H, Abadie V, Benhabiles N, Boissonnas A, Liard C, Descours B, Reboulleau D, Bonduelle O, Verrier B, Van Rooijen N, Combadiere C, Combadiere B. 2012. Neutrophils transport antigen from the dermis to the bone marrow, initiating a source of memory CD8+ T cells. Immunity 37:917–929. 10.1016/j.immuni.2012.07.015 [DOI] [PubMed] [Google Scholar]
- 11.Domachowske JB, Bonville CA, Gao JL, Murphy PM, Easton AJ, Rosenberg HF. 2000. The chemokine macrophage-inflammatory protein-1 alpha and its receptor CCR1 control pulmonary inflammation and antiviral host defense in paramyxovirus infection. J. Immunol. 165:2677–2682. 10.4049/jimmunol.165.5.2677 [DOI] [PubMed] [Google Scholar]
- 12.Gao JL, Wynn TA, Chang Y, Lee EJ, Broxmeyer HE, Cooper S, Tiffany HL, Westphal H, Kwon-Chung J, Murphy PM. 1997. Impaired host defense, hematopoiesis, granulomatous inflammation and type 1-type 2 cytokine balance in mice lacking CC chemokine receptor 1. J. Exp. Med. 185:1959–1968. 10.1084/jem.185.11.1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guerra S, Najera JL, Gonzalez JM, Lopez-Fernandez LA, Climent N, Gatell JM, Gallart T, Esteban M. 2007. Distinct gene expression profiling after infection of immature human monocyte-derived dendritic cells by the attenuated poxvirus vectors MVA and NYVAC. J. Virol. 81:8707–8721. 10.1128/JVI.00444-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lehmann MH, Kastenmuller W, Kandemir JD, Brandt F, Suezer Y, Sutter G. 2009. Modified vaccinia virus Ankara triggers chemotaxis of monocytes and early respiratory immigration of leukocytes by induction of CCL2 expression. J. Virol. 83:2540–2552. 10.1128/JVI.01884-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki T, Chow CW, Downey GP. 2008. Role of innate immune cells and their products in lung immunopathology. Int. J. Biochem. Cell Biol. 40:1348–1361. 10.1016/j.biocel.2008.01.003 [DOI] [PubMed] [Google Scholar]
- 16.Mukaida N. 2003. Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 284:L566–L577 [DOI] [PubMed] [Google Scholar]
- 17.Fels AO, Cohn ZA. 1986. The alveolar macrophage. J. Appl. Physiol. 60:353–369 [DOI] [PubMed] [Google Scholar]
- 18.Crapo JD, Barry BE, Gehr P, Bachofen M, Weibel ER. 1982. Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 126:332–337 [DOI] [PubMed] [Google Scholar]
- 19.Finotto S, Dolovich J, Denburg JA, Jordana M, Marshall JS. 1994. Functional heterogeneity of mast cells isolated from different microenvironments within nasal polyp tissue. Clin. Exp. Immunol. 95:343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Staib C, Lowel M, Erfle V, Sutter G. 2003. Improved host range selection for recombinant modified vaccinia virus Ankara. Biotechniques 34:694–696 698:700 [DOI] [PubMed] [Google Scholar]
- 21.Kremer M, Volz A, Kreijtz JH, Fux R, Lehmann MH, Sutter G. 2012. Easy and efficient protocols for working with recombinant vaccinia virus MVA. Methods Mol. Biol. 890:59–92. 10.1007/978-1-61779-876-4_4 [DOI] [PubMed] [Google Scholar]
- 22.Reed LJ, Muench H. 1937. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27:493–497 [Google Scholar]
- 23.Spiller S, Dreher S, Meng G, Grabiec A, Thomas W, Hartung T, Pfeffer K, Hochrein H, Brade H, Bessler W, Wagner H, Kirschning CJ. 2007. Cellular recognition of trimyristoylated peptide or enterobacterial lipopolysaccharide via both TLR2 and TLR4. J. Biol. Chem. 282:13190–13198. 10.1074/jbc.M610340200 [DOI] [PubMed] [Google Scholar]
- 24.Konigshoff M, Wilhelm A, Jahn A, Sedding D, Amarie OV, Eul B, Seeger W, Fink L, Gunther A, Eickelberg O, Rose F. 2007. The angiotensin II receptor 2 is expressed and mediates angiotensin II signaling in lung fibrosis. Am. J. Respir. Cell Mol. Biol. 37:640–650. 10.1165/rcmb.2006-0379TR [DOI] [PubMed] [Google Scholar]
- 25.Lehmann MH, Weber J, Gastmann O, Sigusch HH. 2002. Pseudogene-free amplification of human GAPDH cDNA. Biotechniques 33:766, 769–770. [DOI] [PubMed] [Google Scholar]
- 26.Untergrasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. 2012. Primer3-new capabilities and interfaces. Nucleic Acids Res. 40:e115. 10.1093/nar/gks596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cui W, Taub DD, Gardner K. 2007. qPrimerDepot: a primer database for quantitative real time PCR. Nucleic Acids Res. 35:D805–D809. 10.1093/nar/gkl767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. 2012. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13:134. 10.1186/1471-2105-13-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 30.Lehmann MH, Masanetz S, Kramer S, Erfle V. 2006. HIV-1 Nef upregulates CCL2/MCP-1 expression in astrocytes in a myristoylation- and calmodulin-dependent manner. J. Cell Sci. 119:4520–4530. 10.1242/jcs.03231 [DOI] [PubMed] [Google Scholar]
- 31.Rolfe MW, Kunkel SL, Standiford TJ, Orringer MB, Phan SH, Evanoff HL, Burdick MD, Strieter RM. 1992. Expression and regulation of human pulmonary fibroblast-derived monocyte chemotactic peptide-1. Am. J. Physiol. 263:L536–L545 [DOI] [PubMed] [Google Scholar]
- 32.Ghildyal R, Dagher H, Donninger H, de Silva D, Li X, Freezer NJ, Wilson JW, Bardin PG. 2005. Rhinovirus infects primary human airway fibroblasts and induces a neutrophil chemokine and a permeability factor. J. Med. Virol. 75:608–615. 10.1002/jmv.20315 [DOI] [PubMed] [Google Scholar]
- 33.Martin TR, Frevert CW. 2005. Innate immunity in the lungs. Proc. Am. Thorac. Soc. 2:403–411. 10.1513/pats.200508-090JS [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarafi MN, Garcia-Zepeda EA, MacLean JA, Charo IF, Luster AD. 1997. Murine monocyte chemoattractant protein (MCP)-5: a novel CC chemokine that is a structural and functional homologue of human MCP-1. J. Exp. Med. 185:99–109. 10.1084/jem.185.1.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delaloye J, Roger T, Steiner-Tardivel QG, Le Roy D, Knaup Reymond M, Akira S, Petrilli V, Gomez CE, Perdiguero B, Tschopp J, Pantaleo G, Esteban M, Calandra T. 2009. Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS Pathog. 5:e1000480. 10.1371/journal.ppat.1000480 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.De Filippo K, Henderson RB, Laschinger M, Hogg N. 2008. Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways. J. Immunol. 180:4308–4315. 10.4049/jimmunol.180.6.4308 [DOI] [PubMed] [Google Scholar]
- 37.Burdon PC, Martin C, Rankin SM. 2005. The CXC chemokine MIP-2 stimulates neutrophil mobilization from the rat bone marrow in a CD49d-dependent manner. Blood 105:2543–2548. 10.1182/blood-2004-08-3193 [DOI] [PubMed] [Google Scholar]
- 38.Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, Wood WI, Moore MW. 1994. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science 265:682–684. 10.1126/science.8036519 [DOI] [PubMed] [Google Scholar]
- 39.Eash KJ, Greenbaum AM, Gopalan PK, Link DC. 2010. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Invest. 120:2423–2431. 10.1172/JCI41649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lionakis MS, Fischer BG, Lim JK, Swamydas M, Wan W, Richard C, Lee C, Cohen JI, Scheinberg P, Gao JL, Murphy PM. 2012. Chemokine receptor Ccr1 drives neutrophil-mediated kidney immunopathology and mortality in invasive candidiasis. PLoS Pathog. 8:e1002865. 10.1371/journal.ppat.1002865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amat M, Benjamim CF, Williams LM, Prats N, Terricabras E, Beleta J, Kunkel SL, Godessart N. 2006. Pharmacological blockade of CCR1 ameliorates murine arthritis and alters cytokine networks in vivo. Br. J. Pharmacol. 149:666–675. 10.1038/sj.bjp.0706912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gibon E, Yao Z, Rao AJ, Zwingenberger S, Batke B, Valladares R, Smith RL, Biswal S, Gambhir SS, Goodman SB. 2012. Effect of a CCR1 receptor antagonist on systemic trafficking of MSCs and polyethylene particle-associated bone loss. Biomaterials 33:3632–3638. 10.1016/j.biomaterials.2012.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boukhebza H, Bellon N, Limacher JM, Inchauspe G. 2012. Therapeutic vaccination to treat chronic infectious diseases: current clinical developments using MVA-based vaccines. Hum. Vaccin. Immunother. 8:1746–1757. 10.4161/hv.21689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JW, Gulley JL. 2012. Poxviral vectors for cancer immunotherapy. Expert Opin. Biol. Ther. 12:463–478. 10.1517/14712598.2012.668516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Price PJ, Torres-Dominguez LE, Brandmuller C, Sutter G, Lehmann MH. 2013. Modified vaccinia virus Ankara: innate immune activation and induction of cellular signalling. Vaccine 31:4231–4234. 10.1016/j.vaccine.2013.03.017 [DOI] [PubMed] [Google Scholar]
- 46.Flechsig C, Suezer Y, Kapp M, Tan SM, Loffler J, Sutter G, Einsele H, Grigoleit GU. 2011. Uptake of antigens from modified vaccinia Ankara virus-infected leukocytes enhances the immunostimulatory capacity of dendritic cells. Cytotherapy 13:739–752. 10.3109/14653249.2010.549123 [DOI] [PubMed] [Google Scholar]
- 47.Gomez CE, Perdiguero B, Jimenez V, Filali-Mouhim A, Ghneim K, Haddad EK, Quakkelaar ED, Delaloye J, Harari A, Roger T, Duhen T, Sekaly RP, Melief CJ, Calandra T, Sallusto F, Lanzavecchia A, Wagner R, Pantaleo G, Esteban M. 2012. Systems analysis of MVA-C induced immune response reveals its significance as a vaccine candidate against HIV/AIDS of clade C. PLoS One 7:e35485. 10.1371/journal.pone.0035485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim JK, Lu W, Hartley O, DeVico AL. 2006. N-terminal proteolytic processing by cathepsin G converts RANTES/CCL5 and related analogs into a truncated 4-68 variant. J. Leukoc. Biol. 80:1395–1404. 10.1189/jlb.0406290 [DOI] [PubMed] [Google Scholar]
- 49.Juillerat-Jeanneret L, Aubert JD, Leuenberger P. 1997. Peptidases in human bronchoalveolar lining fluid, macrophages, and epithelial cells: dipeptidyl (amino)peptidase IV, aminopeptidase N, and dipeptidyl (carboxy)peptidase (angiotensin-converting enzyme). J. Lab. Clin. Med. 130:603–614. 10.1016/S0022-2143(97)90110-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oravecz T, Pall M, Roderiquez G, Gorrell MD, Ditto M, Nguyen NY, Boykins R, Unsworth E, Norcross MA. 1997. Regulation of the receptor specificity and function of the chemokine RANTES (regulated on activation, normal T cell expressed and secreted) by dipeptidyl peptidase IV (CD26)-mediated cleavage. J. Exp. Med. 186:1865–1872. 10.1084/jem.186.11.1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Struyf S, De Meester I, Scharpe S, Lenaerts JP, Menten P, Wang JM, Proost P, Van Damme J. 1998. Natural truncation of RANTES abolishes signaling through the CC chemokine receptors CCR1 and CCR3, impairs its chemotactic potency and generates a CC chemokine inhibitor. Eur. J. Immunol. 28:1262–1271. [DOI] [PubMed] [Google Scholar]
- 52.Proost P, De Meester I, Schols D, Struyf S, Lambeir AM, Wuyts A, Opdenakker G, De Clercq E, Scharpe S, Van Damme J. 1998. Amino-terminal truncation of chemokines by CD26/dipeptidyl-peptidase IV. Conversion of RANTES into a potent inhibitor of monocyte chemotaxis and HIV-1-infection. J. Biol. Chem. 273:7222–7227 [DOI] [PubMed] [Google Scholar]
- 53.Ramos CD, Canetti C, Souto JT, Silva JS, Hogaboam CM, Ferreira SH, Cunha FQ. 2005. MIP-1alpha[CCL3] acting on the CCR1 receptor mediates neutrophil migration in immune inflammation via sequential release of TNF-alpha and LTB4. J. Leukoc. Biol. 78:167–177. 10.1189/jlb.0404237 [DOI] [PubMed] [Google Scholar]
- 54.Richardson RM, Pridgen BC, Haribabu B, Snyderman R. 2000. Regulation of the human chemokine receptor CCR1. Cross-regulation by CXCR1 and CXCR2. J. Biol. Chem. 275:9201–9208. 10.1074/jbc.275.13.9201 [DOI] [PubMed] [Google Scholar]
- 55.Ramos CD, Fernandes KS, Canetti C, Teixeira MM, Silva JS, Cunha FQ. 2006. Neutrophil recruitment in immunized mice depends on MIP-2 inducing the sequential release of MIP-1alpha, TNF-alpha and LTB(4). Eur. J. Immunol. 36:2025–2034. 10.1002/eji.200636057 [DOI] [PubMed] [Google Scholar]
- 56.Bahar MW, Kenyon JC, Putz MM, Abrescia NG, Pease JE, Wise EL, Stuart DI, Smith GL, Grimes JM. 2008. Structure and function of A41, a vaccinia virus chemokine binding protein. PLoS Pathog. 4:e5. 10.1371/journal.ppat.0040005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruiz-Arguello MB, Smith VP, Campanella GS, Baleux F, Arenzana-Seisdedos F, Luster AD, Alcami A. 2008. An ectromelia virus protein that interacts with chemokines through their glycosaminoglycan binding domain. J. Virol. 82:917–926. 10.1128/JVI.02111-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burns JM, Dairaghi DJ, Deitz M, Tsang M, Schall TJ. 2002. Comprehensive mapping of poxvirus vCCI chemokine-binding protein. Expanded range of ligand interactions and unusual dissociation kinetics. J. Biol. Chem. 277:2785–2789. 10.1074/jbc.M109884200 [DOI] [PubMed] [Google Scholar]
- 59.Alcami A, Symons JA, Collins PD, Williams TJ, Smith GL. 1998. Blockade of chemokine activity by a soluble chemokine binding protein from vaccinia virus. J. Immunol. 160:624–633 [PubMed] [Google Scholar]
- 60.Lalani AS, Ness TL, Singh R, Harrison JK, Seet BT, Kelvin DJ, McFadden G, Moyer RW. 1998. Functional comparisons among members of the poxvirus T1/35kDa family of soluble CC-chemokine inhibitor glycoproteins. Virology 250:173–184. 10.1006/viro.1998.9340 [DOI] [PubMed] [Google Scholar]
- 61.Graham KA, Lalani AS, Macen JL, Ness TL, Barry M, Liu LY, Lucas A, Clark-Lewis I, Moyer RW, McFadden G. 1997. The T1/35kDa family of poxvirus-secreted proteins bind chemokines and modulate leukocyte influx into virus-infected tissues. Virology 229:12–24. 10.1006/viro.1996.8423 [DOI] [PubMed] [Google Scholar]
- 62.Schaller MA, Kallal LE, Lukacs NW. 2008. A key role for CC chemokine receptor 1 in T-cell-mediated respiratory inflammation. Am. J. Pathol. 172:386–394. 10.2353/ajpath.2008.070537 [DOI] [PMC free article] [PubMed] [Google Scholar]