

ABSTRACT
The mechanisms by which hepatitis B virus (HBV) establishes and maintains chronic hepatitis B infection (CHB) are poorly defined. Innate immune responses play an important role in reducing HBV replication and pathogenesis. HBV has developed numerous mechanisms to escape these responses, including the production of the secreted hepatitis B e antigen (HBeAg), which has been shown to regulate antiviral toll-like receptor (TLR) and interleukin-1 (IL-1) signaling. IL-18 is a related cytokine that inhibits HBV replication in hepatoma cell lines and in the liver through the induction of gamma interferon (IFN-γ) by NK cells and T cells. We hypothesized that HBV or HBV proteins inhibit IFN-γ expression by NK cells as an accessory immunomodulatory function. We show that HBeAg protein inhibits the NF-κB pathway and thereby downregulates NK cell IFN-γ expression. Additionally, IFN-γ expression was significantly inhibited by exposure to serum from individuals with HBeAg-positive but not HBeAg-negative chronic HBV infection. Further, we show that the HBeAg protein suppresses IL-18-mediated NF-κB signaling in NK and hepatoma cells via modulation of the NF-κB pathway. Together, these findings show that the HBeAg inhibits IL-18 signaling and IFN-γ expression, which may play an important role in the establishment and/or maintenance of persistent HBV infection.
IMPORTANCE It is becoming increasingly apparent that NK cells play a role in the establishment and/or maintenance of chronic hepatitis B infection. The secreted HBeAg is an important regulator of innate and adaptive immune responses. We now show that the HBeAg downregulates NK cell-mediated IFN-γ production and IL-18 signaling, which may contribute to the establishment of infection and/or viral persistence. Our findings build on previous studies showing that the HBeAg also suppresses the TLR and IL-1 signaling pathways, suggesting that this viral protein is a key regulator of antiviral innate immune responses.
INTRODUCTION
The mechanisms by which hepatitis B virus (HBV) establishes and maintains persistent infection are not fully understood. It has become increasingly apparent that innate immune response via the effector functions of a range of cell types, including Kupffer cells, natural killer (NK) cells, and hepatocytes, play an important role in controlling HBV infection (1–3). Our group has previously shown that stimulation of the interleukin-1 (IL-1) and toll-like receptor 2 (TLR2) signaling pathways inhibits HBV replication in vitro (4). In turn, we and others have shown that the hepatitis B e antigen (HBeAg; p17) downregulates antiviral TLR2- and IL-1β-mediated responses (5–7). HBeAg is secreted as a nonparticulate form of the hepatitis B virus (HBV) nucleocapsid protein (hepatitis B core antigen [HBcAg]; p21), which is processed from larger precore polyproteins (p25 and p22) (8). Although not required for HBV replication, the precore protein and HBeAg are critical for the establishment of persistent infection. The HBV precore protein and HBeAg are important regulators of innate and adaptive immune responses that contribute to the establishment and/or maintenance of persistent infection.
IL-18 is a proinflammatory cytokine synthesized and secreted by mononuclear cells, including Kupffer cells. In the presence of the necessary costimulatory ligands, such as IL-12 (9), IL-18 stimulates IFN-γ production by NK cells, T cells, dendritic cells (DCs), and B cells. IL-18 signaling is activated following the interaction of two receptors: the alpha-receptor, IL-18R1, and the beta-receptor, AcPL, both of which dimerize following ligand binding to the alpha component, initiating signal transduction by AcPL (10). Murine studies have shown that IL-18 (11) inhibits HBV replication through induction of IFN-γ (12, 13), which directly inhibits the HBV life cycle at the pre- and posttranslational level. In vitro studies have recently shown that, similar to IL-1 (4), overexpression of IL-18 inhibits HBV replication in a hepatoma cell line (14), although the mechanism for this inhibition is unclear, as hepatocytes do not produce IFN-γ.
NK cells are lymphocytes that eliminate virus-infected cells by both direct cytolysis and the production of several antiviral cytokines, including IFN-γ. HBV infection stimulates NK cells, most likely via the activation of DCs and macrophages that produce IL-12, IL-18, and chemokines, including CXCR3 (15). NK cells are present in the liver and the periphery, with the majority of intrahepatic NK cells having a CD56bright phenotype, whereas CD56dim NK cells are found predominantly in the periphery. It is generally believed that CD56dim NK cells produce less IFN-γ and are more cytotoxic than CD56bright cells. Despite this, a recent study has shown that a large proportion of the IFN-γ-producing NK cells in the setting of chronic hepatitis B (CHB) belong to the CD56dim subset (16). Indeed, it has been shown that IFN-γ expression by NK cells is lower in CHB patients than in uninfected controls, and IFN-γ expression is restored by antiviral therapy that reduces HBV replication (16). This implicates a role for either HBV itself or cellular factors upregulated by HBV replication in impairing IFN-γ production by NK cells and contributing to viral persistence. This reduction in IFN-γ production may be due in part to the upregulation of immunosuppressive cytokines, such as IL-10, which suppress IFN-γ expression by NK cells (18). The blocking of these cytokines restores the capacity of NK cells in both the liver and periphery to produce IFN-γ (18). Interestingly, Schlaak and colleagues have shown that IFN-γ responses were lower following stimulation of peripheral blood mononuclear cells (PBMCs) from chronically infected HBeAg-positive patients than from HBeAg-negative patients (19). This suggests that molecules upregulated during HBeAg-positive CHB or the HBeAg itself contribute to IFN-γ suppression.
Therefore, we have investigated whether the HBeAg protein impairs IL-18-mediated IFN-γ expression by NK cells. We show that IFN-γ production by NK cells within the periphery of healthy subjects is downregulated following treatment with HBeAg protein but not the closely related HBV core protein. Additionally, we found that IFN-γ production by NK cells was significantly inhibited by exposure to serum from individuals with HBeAg-positive but not HBeAg-negative chronic HBV infection. We further investigated whether the HBeAg and related proteins altered IL-18 signaling itself, using in vitro NK and hepatoma cell culture models. We show that HBeAg precursor proteins suppress IL-18-mediated NF-κB signaling in NK92 and Huh7 hepatoma cells, whereas the related core protein did not. Together, these findings show that the HBV precore protein and HBeAg regulate IFN-γ production and IL-18 signaling and suggest that this regulation plays a role in the establishment and/or maintenance of persistent HBV infection.
MATERIALS AND METHODS
Whole-blood assay for in vitro analysis of IL-18-mediated IFN-γ expression.
Whole-blood samples (100 μl) from healthy control volunteers were incubated with 10 ng/ml of IL-18 (MBL, Nagoya, Japan) and 10 ng/ml IL-12 (MBL, Nagoya, Japan) in the presence of 100 μl serum from patients with CHB or chronic HCV infection, a healthy control serum, or RPMI 1640 (control) (Life Technologies, CA) at a final dilution of 1:2 unless otherwise stated. All samples were incubated in a 96-well plate for 24 h at 37°C, 5% CO2, similar to previously described methods (16). After 18 h of stimulation, 10 μg/ml of brefeldin A (Sigma-Aldrich, Sydney, Australia) was added to each well and incubated for the remaining 6 h at 37°C, 5% CO2. Following this, samples were incubated with Live/Dead fixable aqua dead cell stain (Life Technologies) at a 1/200 dilution for 30 min at room temperature in the dark. Subsequently, samples were further incubated with anti-CD3 peridinin chlorophyll protein (PerCP) (SP34-2; Becton, Dickinson [BD], North Ryde, Australia), anti-CD14 phycoerythrin (PE)-Cy7 (M5E2; BD, North Ryde, Australia), anti-CD56 allophycocyanin (APC) (B159; BD, North Ryde, Australia), and anti-IL-18R1-fluorescein isothiocyanate (FITC) (Ab93546; Abcam, Sapphire Bioscience, Waterloo, Australia) for an additional 30 min at room temperature in the dark. Red blood cells then were lysed using OptiLyse C (Beckman Coulter, Gladesville, Australia) at room temperature for 10 min and washed. White blood cells were permeabilized using 1× BD fluorescence-activated cell sorting (FACS) permeabilizing solution 2 (BD, North Ryde, Australia) for 10 min and washed. Finally, samples were incubated at room temperature for 1 h with anti-IFN-γ-AF700 (B27; BD, North Ryde, Australia) in the dark. Cells then were washed and fixed with 1% paraformaldehyde (Sigma-Aldrich, Sydney, Australia). Acquisition was performed on an LSRII flow cytometer (BD) with 106 lymphocyte events collected and analyzed using FlowJo, version 9.2 (TreeStar, Ashland, Oregon). IFN-γ production in NK CD56bright and NK CD56dim cells was measured as (i) the percentage of NK cells expressing IFN-γ and (ii) IFN-γ mean fluorescence intensity (MFI). The specificity of the assay for IL-12/IL-18-driven IFN-γ production was assessed by blocking with antibodies to IL-18R1 (ab93546; Abcam, Sapphire Bioscience, Waterloo, Australia) and AcPL (ab47269; Abcam, Sapphire Bioscience, Waterloo, Australia) receptors and/or stimulation with 10 ng/ml lipopolysaccharide (LPS; Sigma-Aldrich, Sydney, Australia).
HBV proteins.
One hundred μl of whole blood from uninfected controls was stimulated with 100 μl of supernatant obtained from cells stably transformed with genes expressing the HBV HBeAg (p17) or core protein (p21) or untransformed controls (referred to as HBeAg, core, or control conditioned medium, respectively) (6, 20). The conditioned medium was collected 9 days after seeding cells, and the HBeAg titer was determined by the Abbot Architect procedure (21) and typically measured 600 Paul Erlich (PE) IU/ml. Since the conditioned medium was diluted 1:1 with serum in our assay, each well typically contained 300 PE IU/ml of HBeAg. This is biologically relevant, with clinical studies showing this to be within the second quartile of HBeAg titers (21). Core protein expression in the HBcAg media was confirmed by Western blotting (not shown) and contained no HBeAg (20). Whole blood also was stimulated with HBV virion-positive conditioned medium from AD38 cells, which are HepG2-derived hepatoblastoma cells stably transformed with HBeAg-positive infectious cDNA (22). The supernatant from these cells had an HBV viral load of 3.2 × 107 IU/ml and also was weakly HBeAg positive, with HBeAg levels typically registering 20 PE IU/ml. The effect of each of these conditioned media on IFN-γ signaling was assessed by flow cytometry as described above.
NK cell culture assay for analysis of IL-18-mediated IFN-γ expression.
The assay described above also was performed using NK-92 cells (ATCC CRL-2407), which are an IL-2-dependent PBMC-derived NK cell line. NK-92 cells were grown as recommended by the ATCC in alpha-minimal essential medium (Sigma) supplemented with 10% fetal calf serum (FCS; HyClone), 10% horse serum (Invitrogen), and 50 IU/ml IL-2 (Peprotech). Cells were seeded into 96-well plates, and 24 h postseeding they were treated with 100 μl HBeAg, core, or control conditioned medium. Cells were harvested 24 h posttreatment, and the effect of each of these conditioned media on IFN-γ signaling was assessed by flow cytometry as described above.
Patient samples.
Peripheral blood samples were obtained from 13 patients with chronic HBeAg-positive hepatitis B, 20 patients with chronic HBeAg-negative hepatitis, 10 patients with chronic hepatitis C virus (HCV) infection (HCV RNA, 3.2 × 106 ± 2.4 × 106 IU/ml), and 12 uninfected controls. All patients were treatment naive, HIV negative, and (with the exception of the HCV controls) HCV negative. HBV viral load was measured by the Versant HBV DNA 3.0 assay (linear range, 2 × 103 to 1 × 108 IU/ml; Bayer, Tarrytown, NY), and the HBV genotype and HBeAg and HBsAg levels in serum were determined and measured as described previously (21). The 13 HBeAg-positive subjects had a mean HBV viral load of 1.47 × 109 IU/ml and serum HBeAg level of 2,254 PE IU/ml. For the 20 HBeAg-negative subjects, the mean HBV viral load was 2.2 × 103 IU/ml (Table 1).
TABLE 1.
Clinical demographics of HBV-infected patients who supplied the serum used in the whole-blood assay
| Parameter | Value according to HBeAg status |
P valuea | |
|---|---|---|---|
| Positive (n = 13) | Negative (n = 20) | ||
| Age (yr) | 29 ± 6 | 44 ± 14 | <0.011 |
| Male gender (%) | 54 | 65 | NS |
| HBV genotype (proportion) | B (58%), C (25%), D (8%) | B (69%), C (25%), D (6%) | NS |
| HBV DNA (IU/ml) | 1.47 × 109 ± 2.7 × 109 | 2.2 × 106 ± 4.9 × 106 | NS |
| HBeAg (PE IU/ml) | 2,254 × 103 ± 3.6 × 103 | 0 | <0.011 |
| HBsAg (IU/liter) | 2.0 × 104 ± 2.6 × 104 | 5.9 × 103 ± 6.9 × 103 | NS |
| ALT (IU/liter) | 117 ± 76 | 68 ± 59 | <0.032 |
NS, not significant.
All studies were approved by the St. Vincent's Hospital Melbourne human research ethics committee (HREC), HREC numbers 093/04, 005/11, 045/11, and 043/11.
Cytokine arrays on serum.
The levels of IL-10 (no. 561514), IFN-γ (561515), and IL-12p70 (561518) in serum were determined using human enhanced-sensitivity flex set assays (BD, Sydney, Australia). These assays detect specific cytokines in the range of 274 to 200,000 fg/ml. Briefly, 50 μl of serum was mixed with 20 μl of mixed capture beads, incubated for 2 h, and then washed and treated with enhanced-sensitivity detection reagent (no. 561521) according to the manufacturer's instructions. Samples were acquired on a BD FACSCanto II cytometer.
Plasmids and transfection for in vitro analysis of IL-18-mediated NF-κB signaling.
Expression of mammalian expression plasmids encoding variants of the HBV precore gene (p25 and p22) or core gene (p21) was confirmed by Western blotting and quantified by the Abbot Architect procedure (21). The replication and protein expression of greater-than-genome-length (1.3 mer) infectious cDNAs encoding wild-type (wt) HBV or the G1896A precore stop codon mutation have been described previously by our group (4, 23).
NF-κB luciferase reporter assay.
The effect of the HBV precore protein on IL-18-mediated cell signaling in Huh7 cells was analyzed using a previously described luciferase reporter assay (24) adapted for Huh7 cells. It was not possible to perform these studies in NK-92 cells, as they were not permissive to transfection with the appropriate expression constructs (not shown). Briefly, Huh7 cells were seeded into 24-well plates and transfected (Fugene, Roche, IN) 24 h postseeding with 100 ng pAcPL and 100 ng of HBV HBeAg precursor construct p25, p22, or p21 (core) or plasmids encoding infectious wt HBV or G1896A precore mutant HBV cDNA. Cells also were cotransfected with 188 ng of pNF-κB and 62 ng of TK renilla plasmid (transfection control). A cytomegalovirus (CMV)-driven empty vector plasmid was included to ensure the same amount of DNA was added per transfection. Cells were stimulated with 60 ng/ml IL-18 (MBL) 24 h posttransfection and harvested in passive lysis buffer (Promega, Sydney, Australia). The level of NF-κB and TK renilla-driven luciferase expression was measured using an Optima luminometer (BMG Labtech, Mornington, Australia) according to the manufacturer's instructions.
PhosFlow assay.
The ability of the HBeAg to suppress IL-18/IL-12-mediated NF-κB signaling was investigated using a PhosFlow assay. Cells were seeded into 96-well plates (1 million cells per well), and 24 h postseeding they were treated with 100 μl HBeAg, core, or control conditioned medium. Twenty hours later cells were stimulated for 15 min with 20 ng/ml IL-12/IL-18 and then immediately fixed by the addition of an equal volume of prewarmed (to room temperature) PhosFlow fix buffer I (BD) to the cell suspension according to the manufacturer's guidelines. Cells were washed twice with BD PhosFlow perm/wash buffer I and were added to tubes containing PE mouse anti-NF-κB p65 (pS529) (no. 558423) or appropriate isotype control antibodies and incubated for 30 min at room temperature in the dark. Cells were washed with BD PhosFlow perm/wash buffer I and resuspended for immediate flow-cytometric analysis with the BD FACSCanto II cytometer.
Statistical analyses.
To determine whether any virological or patient parameters shown in Table 1 were associated with IFN-γ expression, analysis was performed using GraphPad version 5.03 for Windows (GraphPad Software Inc., La Jolla, CA). Data initially were assessed for normality and log transformed where necessary. Normally distributed, continuous data were reported as means ± standard errors of means. Nonparametric data were reported as median (range) values. Categorical data were reported as numbers (percentages). Exploratory analysis was conducted using student t tests, chi-square tests for equal proportion, or nonparametric tests where appropriate. Multivariate analysis for continuous, normally distributed variables was conducted using generalized linear modeling, and for binomially distributed variables it was performed using multiple logistic regressions. The log-rank test was used for comparison between groups. In all cases a two-sided P value of less than or equal to 0.05 was considered statistically significant. Data shown in Fig. 3 and 4 (and Fig. S1 in the supplemental material) were analyzed using SPSS software, version 18 (Chicago, IL, USA). Mann-Whitney U tests were used to analyze data shown in Fig. 3A. A two-way repeated-measures analysis of variance (ANOVA) was used to analyze data shown in Fig. 3B. Data shown in Fig. 4 and 5 were analyzed using a Spearman test and a Friedman test, followed by Wilcoxon signed-rank tests with Bonferroni adjustment to the alpha values. In these analyses, 2 tests were used, leading to a new alpha level of 0.025. For cytokine analysis, results were analyzed using Kruskal-Wallis equality-of-proportions rank tests and two-sample Wilcoxon rank-sum (Mann-Whitney) tests. For PhosFlow analysis and results presented in Fig. 5, significance was determined using Student's t test assuming unequal variance (GraphPad Prism 5).
FIG 3.
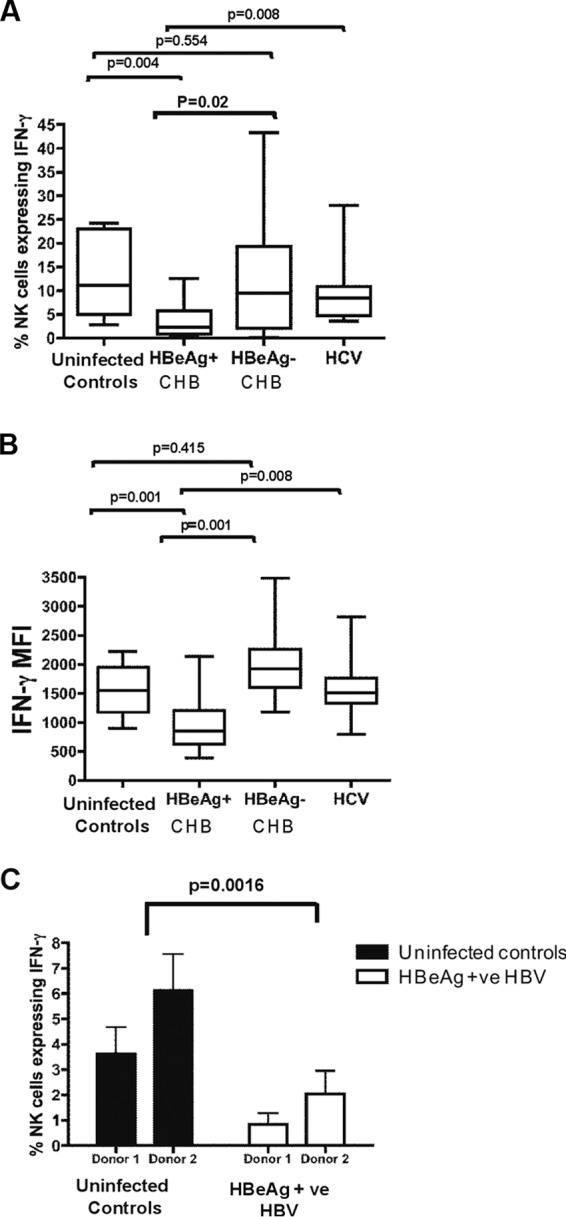
Whole blood from a single uninfected control donor was incubated with either uninfected control serum (n = 10), chronic HBeAg-positive serum (n = 10), chronic HBeAg-negative serum (n = 20), or chronic HCV serum (n = 10) and stimulated with IL-18/IL-12. (A) Percentage of NK cells expressing IFN-γ. (B) Mean fluorescence intensity of IFN-γ-producing NK cells. (C) Whole blood from 4 different uninfected control donors was incubated with either uninfected control serum (n = 2) or HBeAg-positive HBV serum (n = 2). HBeAg-positive serum significantly reduced IFN-γ expression in NK cells from all uninfected donors. The line on each graph depicts the median for each of three independent experiments.
FIG 4.
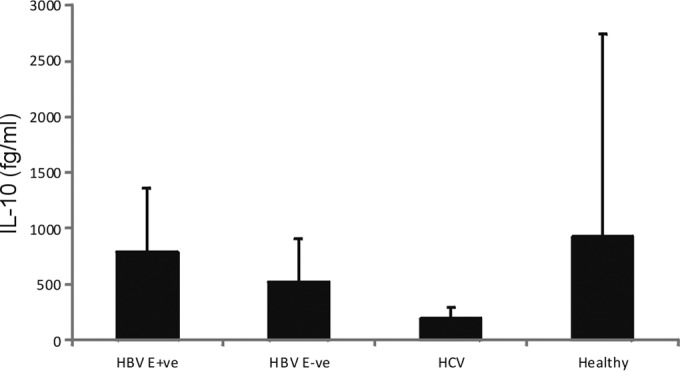
IL-10 levels in serum of patients used in the whole-blood assay, detected using the enhanced-sensitivity BD array.
FIG 5.
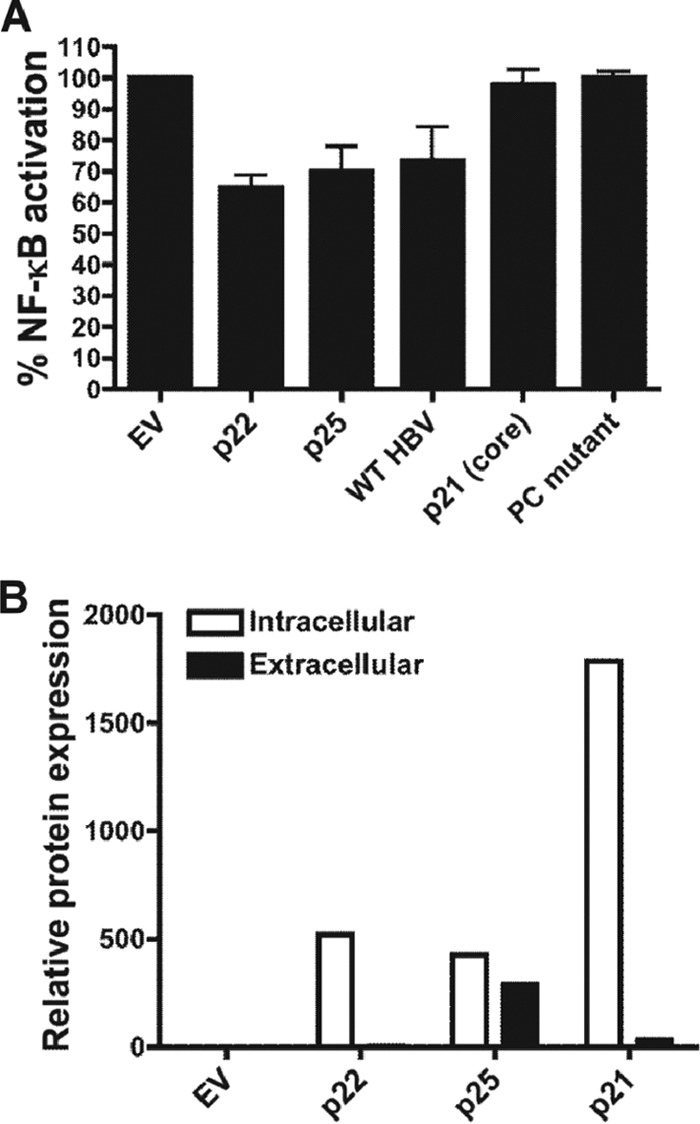
Downregulation of IL-18-stimulated NF-κB-driven luciferase expression in Huh7 cells. (A) Luciferase expression corrected to IL-18-stimulated Huh7 cells transfected with empty vector following transfection of cells with CMV-driven expression plasmids expressing the HBeAg precursor (p22 or p25), the HBV core protein (p21), or infectious cDNAs encoding wt HBV or G1896A precore mutant HBV. Samples statistically different from empty vector controls are indicated by an asterisk (P < 0.05). (B) Relative protein expression of CMV-driven reporter plasmids in transfected Huh7 cells, as determined by quantitative serology (21).
RESULTS
Interrogation of IL-18-mediated IFN-γ expression by NK cells.
We measured IL-18-mediated IFN-γ production by NK cells using a previously described whole-blood assay (16). Briefly, healthy donor whole blood was stimulated with IL-18 and IL-12 to stimulate robust IFN-γ expression from NK cells (Fig. 1A and B). This resulted in a mean of 12.79% (±9.95%) of CD14− CD3− CD56+ NK cells expressing IFN-γ in the presence of healthy donor serum. The specificity of IL-18-mediated IFN-γ expression was demonstrated by blockade of the IL-18 receptor using antibodies to either component of the receptor (IL-18R1 or AcPL), which effectively inhibited NK cell IFN-γ expression (Fig. 1B).
FIG 1.

IL-18/IL-12 stimulation of NK cells in whole blood. (A) Intracellular IFN-γ expression by NK cells analyzed by gating on viable CD14− CD3− CD56+ lymphocytes. The first panel shows cells sorted into lymphocytes based on forward (FSC-A, x axis) and side scatter (SSC-A, y axis). The second panel shows the NK cell population analyzed. (B) Addition of either anti-IL-18R1 or anti-AcPL antibody prior to stimulation significantly reduced IFN-γ production.
HBeAg protein downregulates IFN-γ production by NK cells.
The HBeAg previously has been shown to downregulate innate immune signaling in a range of cell types (6, 7, 25). To determine whether the HBeAg specifically downregulated IL-18-mediated IFN-γ expression in NK cells, we stimulated whole blood from 10 healthy control blood donors with IL-18 and IL-12 in the presence of control, precore, core, or whole virus conditioned media and measured the frequency of NK cells expressing IFN-γ. IL-18 stimulation of whole blood in conjunction with HBeAg protein demonstrated significant downregulation of IFN-γ expression in NK cells compared to control media with no HBeAg (Fig. 2A [P = 0.005] and 3B [P = 0.022]). Treatment with core or with whole HBV-positive conditioned media (whole HBV, HBsAg positive, and weakly HBeAg positive) did not alter IFN-γ production by NK cells (Fig. 2A and B) in whole blood. Further analysis of NK cell phenotypes showed IFN-γ expression was higher in CD56bright cells than in CD56dim cells (Fig. 2C and D). Despite this, the HBeAg-containing conditioned media reduced IFN-γ expression similarly in both NK cell subtypes (Fig. 2C and D). To confirm this was a direct effect on NK cells, NK-92 cells were stimulated with IL-18, and IFN-γ production by NK cells was measured in the presence of condition media. To assess whether IFN-γ expression by NK cells was affected by stimuli other than IL-12/IL-18, we stimulated whole blood with LPS. LPS treatment of cells results in IFN-γ expression both directly via stimulation of TLR4 and indirectly through activation of the inflammasome, producing IL-18, which results in IFN-γ (26). Following treatment with HBeAg-containing media or HBeAg-negative media, we obtained results almost identical to those obtained following direct IL-12/IL-18 stimulation, with a significant reduction in NK cell-driven IFN-γ expression, irrespective of whether we examined the percentage of cells expressing IFN-γ (P = 0.005) (Fig. 2E) or mean fluorescence (P = 0.005) (Fig. 2F). Blocking experiments showed that anti-IL-18 receptor antibodies prevented IFN-γ expression by NK cells, showing that LPS-driven upregulation of IFN-γ most likely was mediated via activation of IL-12/IL-18 in cells such as DCs and not by direct activation of IFN-γ via the LPS receptor TLR4 in NK cells (Fig. 2G). As expected, the HBeAg also reduced IFN-γ expression in NK92 cells (Fig. 2H). Interestingly, in these cells we also observed a small but nonetheless statistically significant reduction in IFN-γ expression in cells exposed to core-expressing media compared to control media. To confirm that inhibition of IFN-γ was dose dependent on HBeAg protein, we determined the amount of HBeAg protein required to suppress IFN-γ expression following IL-12/IL-18 stimulation of whole blood. We observed that 300 PE IU/ml HBeAg suppressed NK cell IFN-γ, but this effect was ameliorated when HBeAg conditioned medium was diluted to 50 PE IU/ml (Fig. 2I). This indicates that HBeAg protein inhibits IL-18-mediated IFN-γ production by NK cells with a dose-dependent mechanism.
FIG 2.

HBeAg-specific inhibition of IFN-γ production by NK cells following stimulation with IL-18/IL-12. Whole blood (A to D) from 10 uninfected control donors or NK92 cells (H) was incubated with IL-18/IL-12 (A and B) or LPS (E and F) in the presence of conditioned media containing either HBeAg, core antigen, purified HBsAg/virions (derived from AD38 cells), or control media. (G) LPS-mediated IFN-γ expression was blocked by treatment with the IL-18R1 antibody. For whole-blood assays, the percentage of cells expressing IFN-γ is shown in panels A, C, D, E, and G, and the mean fluorescence intensity (MFI) of IFN-γ-producing cells is shown in panels B and F. The line on each graph depicts the median for each of three independent experiments. (I) IFN-γ expression in whole blood from one uninfected control donor incubated with IL-18/IL-12 in the presence of conditioned media containing serial dilutions of HBeAg, core antigen, or control media. Suppression of IFN-γ expression was dose dependent, with no significant difference from core or control media when HBeAg levels were 50 PE IU/ml or less. The line on each data set depicts the median. (J) Differences in the percentage of phosphorylated NF-κB-positive NK92 cells observed following IL-18/IL-12 stimulation and exposure to conditioned media expressing the HBeAg, core protein, or control.
We were interested next to determine a possible mechanism for this downregulation. We stimulated NK-92 cells with IL-18/IL-12 and measured NF-κB phosphorylation by flow cytometry. Treatment of NK cells with HBeAg conditioned medium significantly downregulated NF-κB phosphorylation compared to core or control conditioned medium (Fig. 2J), suggesting that HBeAg-mediated suppression of IL-12/IL-18 signaling in NK-92 cells is mediated through the NF-κB signaling pathway.
Serum from HBeAg-positive individuals downregulated IL-18-mediated IFN-γ production by NK cells.
Since the HBeAg protein downregulated IFN-γ expression in NK cells, we next were interested in determining whether endogenous HBeAg in serum from HBV-infected individuals similarly modulated IFN-γ expression in blood NK cells. We stimulated whole blood with exogenous IL-18 and IL-12 and measured NK cell IFN-γ expression in the presence of plasma treated with serum from HBeAg-positive and HBeAg-negative individuals. To ensure that the reduction in NK cell IFN-γ expression was due specifically to HBV-related factors and not to other factors present in serum resulting from chronic viral infection of the liver, we included serum from HCV-infected individuals. IFN-γ production by NK cells was significantly suppressed following treatment of whole blood with serum from chronic HBV HBeAg-positive individuals (range, 0 to 15%; median, 2.24%) (Fig. 3A [P = 0.004] and B [P = 0.001]) relative to uninfected controls. Further, there was a significantly lower level of production of IFN-γ by NK cells following treatment of whole blood with serum from HBeAg-positive individuals compared to HBeAg-negative individuals (P = 0.02). Although we observed a much greater range of IFN-γ responses following treatment of whole blood with serum from HBeAg-negative individuals (0 to 45%; median, 9.5%), overall IFN-γ expression in NK cells treated with this serum did not differ significantly from that of uninfected controls (Fig. 3A and B). This effect was observed in whole blood from multiple control donors (P = 0.0016) (Fig. 3C). There was no association between suppression of IFN-γ expression and HBsAg levels or alanine aminotransferase (ALT) (Table 1). Results were similar irrespective of whether we measured the percentage of NK cells expressing IFN-γ (Fig. 3A) or the mean fluorescence intensity of IFN-γ expression (Fig. 3B). Treatment with serum from HCV-infected individuals had no effect on IFN-γ expression by NK cells (Fig. 3A and B). This indicates that HBeAg protein in serum from infected individuals suppresses IL-18-stimulated IFN-γ production by NK cells.
IL-10 cytokine levels did not differ in serum derived from HBeAg-positive or HBeAg-negative individuals.
One mechanism by which HBeAg-positive HBV downregulates IFN-γ expression is via upregulation of immunosuppressive cytokines, such as IL-10, which have been shown to suppress NK cell IFN-γ expression (18). To determine if endogenous cytokines differed in patient serum used to stimulate NK cells in the whole blood assay, we measured cytokines using a highly sensitive flow cytometry assay. IL-10 levels were significantly higher in HBV-infected patients than in HCV-infected individuals (P = 0.02), which were predominantly below the limit of detection of the assay. No difference was observed in IL-10 levels in serum from HBeAg-negative and HBeAg-positive patients (Fig. 4 and Table 1). IL-10 levels in serum of uninfected control patients were highly variable, and overall they did not differ from levels in HBV-infected individuals, most likely due to the high standard deviations for this small sample size. We observed no difference in endogenous serum IL-12p70 or IFN-γ levels between groups (not shown). Together, these findings suggest that NK cell-mediated IFN-γ levels measured using our whole-blood assay were not influenced by endogenous serum IL-10, IL-12p70, or IFN-γ.
In vitro hepatoma cell line model to study IL-18 signaling.
We were interested next to determine whether the HBeAg protein also can downregulate IL-18-mediated NF-κB signaling in hepatocytes. We have previously used a luciferase reporter hepatocyte cell culture model to interrogate NF-κB signaling pathways stimulated by the related cytokine IL-1β (7). These cells support productive HBV replication, enabling interrogation of the effect of whole virus or viral proteins on cell signaling, which is not possible in NK cells. Indeed, NK-92 cells were recalcitrant to transfection with all constructs tested (not shown).
Previous studies using gel shift assays have shown that the transcription factor NF-κB is upregulated following stimulation with exogenous IL-18 (27). This is important, as although IL-18 signaling does not induce IFN-γ expression in hepatocytes, the transcription factor NF-κB promotes expression of numerous antiviral cytokines, including IFN-α, IFN-β, and tumor necrosis factor (TNF). We have previously shown that the HBeAg precursor proteins P25 and P22 suppress NF-κB signaling in hepatocyte cell lines following stimulation with the related cytokine IL-1β. We now show that the HBeAg precursor proteins similarly downregulate IL-18-mediated NF-κB signaling in Huh7 cells (Fig. 5A). We also showed that wild-type HBV expressing the HBeAg inhibited IL-18-stimulated, NF-κB-driven luciferase expression, whereas the G1896A mutant HBV that does not express HBeAg precursor proteins or HBeAg did not (Fig. 5A). In contrast, the HBV p21 core protein had no effect on NF-κB-driven luciferase expression despite high levels of protein expression (Fig. 5B).
DISCUSSION
The ability of HBeAg-positive HBV to establish a chronic persistent infection suggests that HBeAg modulates immune responses against HBV. Indeed, it is known that the HBeAg modulates adaptive immune responses to the viral nucleocapsid (28) and downregulates TLR and IL-1 signaling pathways (6, 7, 25). It is becoming increasingly evident that NK cells play an important role in controlling HBV infection (16, 18), particularly early in the infection cycle (29). We now show that the HBeAg can specifically downregulate IL-18-stimulated IFN-γ expression by NK cells, providing a possible mechanism by which HBV suppresses NK cell-mediated antiviral activity. Studies using conditioned media showed that the HBeAg downregulated IFN-γ expression by NK cells compared to media expressing core protein or HBsAg, using either whole-blood assays or a clonal NK cell line. In addition, serum from subjects with HBeAg-positive CHB significantly downregulated IFN-γ expression in NK cells derived from uninfected control individuals compared to serum from HBeAg-negative patients or uninfected controls. Although we also observed an increase in IFN-γ expression following stimulation with LPS, which was also downregulated by the HBeAg, this increase most likely was due to indirect activation of IL-18 in other cells in the periphery (30) rather than direct activation of the LPS receptor TLR4. This was supported by our finding that LPS-stimulated IFN-γ expression was specifically blocked by antibodies to IL-18 receptors and the published observation that human NK cells do not express TLR4 (31), the major receptor for the LPS ligand.
It is possible that other factors in addition to the HBeAg in patient serum also contribute to our findings. It has been shown recently that the immunosuppressive cytokine IL-10 is upregulated during CHB and inhibits NK cell IFN-γ expression (18). Although we identified higher levels of IL-10 in the serum of HBV-infected patients than in HCV-infected patients, there was no significant difference in IL-10 levels in the serum of HBeAg-positive or HBeAg-negative patients used in our study. This suggests that IL-10 levels in patient serum did not affect IL-18-mediated NK cell IFN-γ expression in our whole-blood assay. We did not detect any difference in endogenous serum IL-12 levels, a key costimulatory molecule for IFN-γ expression, or IFN-γ itself (data not shown). We cannot rule out that other cytokines or other factors contribute to the inhibition of IFN-γ expression by NK cells in the presence of HBV sera. Despite this, our findings that HBeAg in conditioned medium suppressed IL-18-mediated IFN-γ expression and NF-κB signaling by NK cells suggests that the HBeAg in positive serum contributes to downregulation of IL-18-mediated NK cell IFN-γ expression in the periphery.
It has been proposed that regulation of NK cell-mediated IFN-γ expression plays an important role in the establishment of acute infection rather than the maintenance of persistent infection (29). This hypothesis is based largely on the finding that NK cell activity is lower in patients with chronic HBV, due in part to the upregulation of intracellular cytokines, including IL-10 (18). Others have concluded differently, suggesting that defects in the CD56dim subset in the periphery of patients with chronic HBV play a role in persistence (16). This is supported by the findings that activation of CD56dim NK cells was restored following antiviral treatment that decreased the level of serum HBV (16). Our findings that the HBeAg downregulates NK cell-mediated IFN-γ expression suggest that HBeAg contributes to the downregulation of NK cells during the establishment and/or maintenance of infection. However, a limitation of our study is that we are unable to draw conclusions regarding a role for HBeAg-mediated regulation of NK cell IFN-γ expression in the maintenance of persistent infection, as we did not compare NK cell-mediated IFN-γ expression in cells obtained from our HBeAg-positive or -negative CHB patients. If HBeAg-mediated downregulation of NK cells contributes to maintenance of persistence, we predict NK cell-mediated IFN-γ expression to have been lower in our HBeAg-positive patients. It is also possible that suppression of IFN-γ expression by the HBeAg plays a role in the establishment of persistent infection rather than the maintenance of the persistent state.
We have shown that the HBeAg downregulates IL-18-mediated signaling in NK cells and Huh7 hepatoma cells through modulation of NF-κB signaling. Although HBV does not infect NK cells, the HBeAg is secreted into the peripheral blood. It is possible that this effect is mediated by HBeAg interacting directly with IL-18 receptors (IL-18R1 and/or AcPL) on the NK cell membrane. Indeed, we showed that blocking these receptors abrogates IL-18-stimulated IFN-γ expression in NK cells. The AcPL is a membrane-spanning protein which binds to the alpha receptor IL-18R1 to initiate signaling (10). Direct interaction with the membrane-spanning AcPL provides one explanation for our findings that the secreted HBeAg downregulates IL-18-mediated IFN-γ expression in NK cells. It has recently been shown that the HBeAg directly interacts with the related IL-1 receptor AcP (32), and it remains to be determined if similar interactions are at play in IL-18 signaling. We also show that the intracellular precursor proteins p22 and p25 downregulate IL-18-mediated signaling in hepatocytes by downregulating NF-κB promoter activity in vitro. This is similar to our previous findings in hepatocytes treated with the related cytokine IL-1β (7) and shows that the HBeAg is a powerful suppressor of IL-18 signaling across numerous cell types.
Since IL-18-stimulated IFN-γ has direct antiviral effects against HBV and activation of IL-18 signaling decreases HBV replication in hepatocytes (14), it is not surprising that HBV evolved mechanisms to regulate IL-18 signaling and IFN-γ expression. HBV utilizes numerous mechanisms to regulate other innate immunity signaling pathways, including HBeAg and precore protein-mediated regulation of TLR2 (5, 6) and IL-1β (7), with the latter sharing a number of salient features with IL-18 signaling (33). The mechanisms by which HBV establishes and maintains chronic infection are still to be resolved. However, recent findings that NK cell-driven IFN-γ responses are lower in CHB patients with high HBV viral loads (16) and that upregulation of IL-1 or the related IL-18 signaling inhibits HBV replication in hepatocytes (4, 14) suggests that regulation of IL-18 is one mechanism by which HBV facilitates persistence. This is further supported by our findings that suppression of IFN-γ expression by NK cells is directly mediated both by factors present in serum from HBeAg-positive CHB patients and by the HBeAg protein itself. Modulation of immune cells by the secreted HBeAg may be contributing to the establishment of HBV infection and/or the maintenance of HBV persistence in the setting of HBeAg-positive disease.
ACKNOWLEDGMENTS
We thank Rachel Wilson for assistance with the luciferase assay and Andrew Bowie and Michael Gale for the pAcPL and NF-κB plasmids, respectively. We thank Elizabeth Vincan and Bang Tran for assistance with cell culture. We also thank the HBV- and HCV-infected subjects for donating serum and the many uninfected control volunteers who donated blood for the flow cytometry experiments.
S.J. was supported by an APA Ph.D. scholarship. S.H.A. was supported by the GlaxoSmithKline Research Fund of the Korean Association for the Study of the Liver and a grant from the Bilateral International Collaborative R and D Program from the Ministry of Knowledge Economy and the Brain Korea 21 Project of Medical Science, Republic of Korea. This study also was supported by NHMRC award 510448. A.J.T. was supported by an NHMRC Career Development Fellowship.
Footnotes
Published ahead of print 28 May 2014
REFERENCES
- 1.Ait-Goughoulte M, Lucifora J, Zoulim F, Durantel D. 2010. Innate antiviral immune responses to hepatitis B virus. Viruses 2:1394–1410. 10.3390/v2071394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Revill P, Yuan Z. 2013. New insights into how hepatitis B virus manipulates the innate immune response to establish acute and persistent infection. Antiviral Ther. 18(1):1–15. 10.3851/IMP2542 [DOI] [PubMed] [Google Scholar]
- 3.Zeissig S, Murata K, Sweet L, Publicover J, Hu Z, Kaser A, Bosse E, Iqbal J, Hussain MM, Balschun K, Rocken C, Arlt A, Gunther R, Hampe J, Schreiber S, Baron JL, Moody DB, Liang TJ, Blumberg RS. 2012. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat. Med. 18:1060–1068. 10.1038/nm.2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson AJ, Colledge D, Rodgers S, Wilson R, Revill P, Desmond P, Mansell A, Visvanathan K, Locarnini S. 2009. Stimulation of the interleukin-1 receptor and Toll-like receptor 2 inhibits hepatitis B virus replication in hepatoma cell lines in vitro. Antivir. Ther. 14:797–808. 10.3851/IMP1294 [DOI] [PubMed] [Google Scholar]
- 5.Lang T, Lo C, Skinner N, Locarnini S, Visvanathan K, Mansell A. 2011. The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J. Hepatol. 55:762–769. 10.1016/j.jhep.2010.12.042 [DOI] [PubMed] [Google Scholar]
- 6.Visvanathan K, Skinner NA, Thompson AJ, Riordan SM, Sozzi V, Edwards R, Rodgers S, Kurtovic J, Chang J, Lewin S, Desmond P, Locarnini S. 2007. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 45:102–110. 10.1002/hep.21482 [DOI] [PubMed] [Google Scholar]
- 7.Wilson R, Warner N, Ryan K, Selleck L, Colledge D, Rodgers S, Li K, Revill P, Locarnini S. 2011. The hepatitis B e antigen suppresses IL-1beta-mediated NF-kappaB activation in hepatocytes. J. Viral Hepat. 18:e499–e507. 10.1111/j.1365-2893.2011.01484.x [DOI] [PubMed] [Google Scholar]
- 8.Messageot F, Salhi S, Eon P, Rossignol JM. 2003. Proteolytic processing of the hepatitis B virus e antigen precursor. Cleavage at two furin consensus sequences. J. Biol. Chem. 278:891–895. 10.1074/jbc.M207634200 [DOI] [PubMed] [Google Scholar]
- 9.Okamura H, Kashiwamura S, Tsutsui H, Yoshimoto T, Nakanishi K. 1998. Regulation of interferon-gamma production by IL-12 and IL-18. Curr. Opin. Immunol. 10:259–264. 10.1016/S0952-7915(98)80163-5 [DOI] [PubMed] [Google Scholar]
- 10.Cheung H, Chen NJ, Cao Z, Ono N, Ohashi PS, Yeh WC. 2005. Accessory protein-like is essential for IL-18-mediated signaling. J. Immunol. 174:5351–5357. 10.4049/jimmunol.174.9.5351 [DOI] [PubMed] [Google Scholar]
- 11.Kimura K, Kakimi K, Wieland S, Guidotti LG, Chisari FV. 2002. Interleukin-18 inhibits hepatitis B virus replication in the livers of transgenic mice. J. Virol. 76:10702–10707. 10.1128/JVI.76.21.10702-10707.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pasquetto V, Wieland SF, Uprichard SL, Tripodi M, Chisari FV. 2002. Cytokine-sensitive replication of hepatitis B virus in immortalized mouse hepatocyte cultures. J. Virol. 76:5646–5653. 10.1128/JVI.76.11.5646-5653.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wieland SF, Eustaquio A, Whitten-Bauer C, Boyd B, Chisari FV. 2005. Interferon prevents formation of replication-competent hepatitis B virus RNA-containing nucleocapsids. Proc. Natl. Acad. Sci. U. S. A. 102:9913–9917. 10.1073/pnas.0504273102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Li Y, Ma Y, Liu S, She Y, Zhao P, Jing M, Han T, Yan C, Wu Z, Gao J, Ye L. 2011. Dual effects of interleukin-18: inhibiting hepatitis B virus replication in HepG2.2.15 cells and promoting hepatoma cell metastasis. Am. J. Physiol. Gastrointest. Liver Physiol. 301:G565–G573. 10.1152/ajpgi.00058.2011 [DOI] [PubMed] [Google Scholar]
- 15.Guidotti LG, Chisari FV. 2001. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu. Rev. Immunol. 19:65–91. 10.1146/annurev.immunol.19.1.65 [DOI] [PubMed] [Google Scholar]
- 16.Tjwa ET, van Oord GW, Hegmans JP, Janssen HL, Woltman AM. 2011. Viral load reduction improves activation and function of natural killer cells in patients with chronic hepatitis B. J. Hepatol. 54:209–218. 10.1016/j.jhep.2010.07.009 [DOI] [PubMed] [Google Scholar]
- 17. Reference deleted.
- 18.Peppa D, Micco L, Javaid A, Kennedy PT, Schurich A, Dunn C, Pallant C, Ellis G, Khanna P, Dusheiko G, Gilson RJ, Maini MK. 2010. Blockade of immunosuppressive cytokines restores NK cell antiviral function in chronic hepatitis B virus infection. PLoS Pathog. 6:e1001227. 10.1371/journal.ppat.1001227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schlaak JF, Tully G, Lohr HF, Gerken G, Meyer zum Buschenfelde KH. 1999. HBV-specific immune defect in chronic hepatitis B (CHB) is correlated with a dysregulation of pro- and anti-inflammatory cytokines. Clin. Exp. Immunol. 115:508–514. 10.1046/j.1365-2249.1999.00812.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Locarnini S, Shaw T, Dean J, Colledge D, Thompson A, Li K, Lemon SM, Lau GG, Beard MR. 2005. Cellular response to conditional expression of the hepatitis B virus precore and core proteins in cultured hepatoma (Huh-7) cells. J. Clin. Virol. 32:113–121. 10.1016/j.jcv.2004.10.002 [DOI] [PubMed] [Google Scholar]
- 21.Thompson AJ, Nguyen T, Iser D, Ayres A, Jackson K, Littlejohn M, Slavin J, Bowden S, Gane EJ, Abbott W, Lau GK, Lewin SR, Visvanathan K, Desmond PV, Locarnini SA. 2010. Serum hepatitis B surface antigen and hepatitis B e antigen titers: disease phase influences correlation with viral load and intrahepatic hepatitis B virus markers. Hepatology 51:1933–1944. 10.1002/hep.23571 [DOI] [PubMed] [Google Scholar]
- 22.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 41:1715–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cabuang LM, Shaw T, Littlejohn M, Colledge D, Sozzi V, Soppe S, Warner N, Thompson A, Preiss S, Lam N, Walsh R, Lewin SR, Thio CL, Matthews G, Locarnini SA, Revill PA. 2012. In vitro replication phenotype of a novel (-1G) hepatitis B virus variant associated with HIV co-infection. J. Med. Virol. 84:1166–1176. 10.1002/jmv.23328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowie A, Kiss-Toth E, Symons JA, Smith GL, Dower SK, O'Neill LA. 2000. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl. Acad. Sci. U. S. A. 97:10162–10167. 10.1073/pnas.160027697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu S, Kanda T, Imazeki F, Arai M, Yonemitsu Y, Nakamoto S, Fujiwara K, Fukai K, Nomura F, Yokosuka O. 2010. Hepatitis B virus e antigen downregulates cytokine production in human hepatoma cell lines. Viral Immunol. 23:467–476. 10.1089/vim.2010.0042 [DOI] [PubMed] [Google Scholar]
- 26.Ganz M, Csak T, Nath B, Szabo G. 2011. Lipopolysaccharide induces and activates the Nalp3 inflammasome in the liver. World J. Gastroenterol. 17:4772–4778. 10.3748/wjg.v17.i43.4772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asakawa M, Kono H, Amemiya H, Matsuda M, Suzuki T, Maki A, Fujii H. 2006. Role of interleukin-18 and its receptor in hepatocellular carcinoma associated with hepatitis C virus infection. Int. J. Cancer 118:564–570. 10.1002/ijc.21367 [DOI] [PubMed] [Google Scholar]
- 28.Milich DR, Chen MK, Hughes JL, Jones JE. 1998. The secreted hepatitis B precore antigen can modulate the immune response to the nucleocapsid: a mechanism for persistence. J. Immunol. 160:2013–2021 [PubMed] [Google Scholar]
- 29.Maini MK, Peppa D. 2013. NK cells: a double-edged sword in chronic hepatitis B virus infection. Front. Immunol. 4:57. 10.3389/fimmu.2013.00057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, Quintal L, Sekut L, Talanian R, Paskind M, Wong W, Kamen R, Tracey D, Allen H. 1997. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 386:619–623. 10.1038/386619a0 [DOI] [PubMed] [Google Scholar]
- 31.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. 2002. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 168:4531–4537. 10.4049/jimmunol.168.9.4531 [DOI] [PubMed] [Google Scholar]
- 32.Yang CY, Kuo TH, Ting LP. 2006. Human hepatitis B viral e antigen interacts with cellular interleukin-1 receptor accessory protein and triggers interleukin-1 response. J. Biol. Chem. 281:34525–34536. 10.1074/jbc.M510981200 [DOI] [PubMed] [Google Scholar]
- 33.Dinarello CA. 2009. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27:519–550. 10.1146/annurev.immunol.021908.132612 [DOI] [PubMed] [Google Scholar]