

ABSTRACT
The interferon system provides the first line of host defense against virus infection. Mouse pathogenesis studies have revealed the importance of specific interferon-induced proteins in providing protection against specific viruses. We have previously reported that one such protein, Ifit2, protects neurons of the central nervous system from intranasal infection by the neurotropic rhabdovirus, vesicular stomatitis virus (VSV). Here, we demonstrate that Ifit2 protects the peripheral nervous system from VSV infection as well. In Ifit2−/− mice, VSV, injected subcutaneously into the footpad, entered the proximal lymph node, where it replicated and infected the nodal nerve endings. The infection spread to the sciatic nerve, the spinal cord, and the brain, causing paralysis. In contrast, in the wild-type mice, although VSV replicated equally well in the lymph node, infection of the sciatic nerve and the rest of the nervous system was impaired, thus preventing paralysis. Ifit2 protected only the nervous system from VSV infection; other tissues were well protected even in Ifit2−/− mice. These results indicate that Ifit2 is the interferon-induced protein that prevents VSV infection of neurons of both the peripheral and the central nervous systems, thus inhibiting the consequent neuropathy, but it is dispensable for protecting the cells of other tissues from VSV infection.
IMPORTANCE Although viral infection is quite common, the immune system effectively protects us from viral diseases. A major part of this protection is mediated by interferon, the antiviral cytokine secreted by virus-infected cells. To empower the neighboring uninfected cells in combating the oncoming infection, interferon induces the synthesis of more than 200 new proteins, many of which have antiviral activities. The virus studied here, vesicular stomatitis virus (VSV), like its relative, rabies virus, can cause neuropathy in mice if it enters the peripheral nervous system through skin lesions; however, interferon can protect neurons from VSV infection. We have identified a specific interferon-induced protein, Ifit2, as the protein that protects neurons from VSV infection. Surprisingly, Ifit2 was not needed to protect other cell types from VSV. Our results indicate that the effector antiviral proteins of the interferon system have highly specialized functions.
INTRODUCTION
The type I interferon (IFN) system provides the first line of host defense against virus infection. Diverse cellular pattern recognition receptors (PRR) recognize components of invading viruses and trigger IFN synthesis (1). The newly synthesized IFN is secreted and acts upon not only the infected cells but on neighboring uninfected cells as well. All type I IFNs use the same cell surface receptor, IFNAR (IFN-α/β receptor), to activate the JAK-STAT signaling pathway, which causes the transcriptional activation of more than 200 IFN-stimulated genes (ISGs) (2, 3). The proteins encoded by some of the ISGs provide antiviral innate immunity to the infected cells. In vivo, type I IFNs also augment the functions of cells of the adaptive immune system to facilitate elimination of the infected cells (4, 5). As expected from the above description, mice lacking the IFNAR gene (IFNAR−/−) are highly susceptible to pathogenesis caused by many viruses (6). On the other hand, many viruses have evolved various mechanisms to evade the IFN system by interfering with one or more steps of IFN synthesis and IFN actions (7). Thus, a dynamic equilibrium between virus replication and the antiviral action of the immune system is established in an infected animal.
The outcome of acute viral infection, which may cause pathogenesis and death in animals, is determined shortly after infection by the IFN system and its innate antiviral functions as the major immune defense of the host (4). For this reason, an understanding of the specific antiviral functions of different ISGs is critically important. Most of the ISGs are induced not only by IFN-elicited signaling but also by cellular PRR signaling pathways activated by viruses themselves (8), presumably to protect the initially infected cells before IFN has been induced and secreted. A few ISGs' biochemical and antiviral functions have been studied in depth, whereas those of others remain undefined. More recently, systematic investigations of the antiviral functions of the entire class of ISGs against multiple viruses have produced interesting results (9, 10). A few general principles of the antiviral actions of ISGs have emerged from these studies: different ISGs target different families of viruses, and several ISGs block different steps of replication of the same virus, thus providing cumulative antiviral effects. Recent pathogenesis studies using genetically altered mice, devoid of the gene of a given ISG, have revealed another unexpected feature of this system, namely, the tissue specificity of their actions (11–13).
We have been studying the biochemical and the antiviral properties of the Ifit family of ISGs (8). They are very strongly induced by type I IFNs and by many signaling pathways that activate IRF-3 or IRF-7. Such PRR pathways can be triggered by several membrane-bound Toll-like receptors or cytoplasmic receptors recognizing viral RNA or DNA (1, 8, 14). There are three members of the Ifit family of genes in mice: Ifit1 or ISG56, Ifit2 or ISG54, and Ifit3 or ISG49. They all encode proteins that are distinct, but related in structure, comprised of multiple tetratricopeptide repeats. The Ifit proteins have no recognizable enzyme activity, but they interact with specific cellular proteins, as well as cellular and viral RNAs. They can also form heteromeric complexes, indicating the possibility of imparting a variety of cellular functions through different complexes (8, 14, 15).
Recently, the generation of Ifit1 and Ifit2 gene knockout lines of mice has opened the way to investigate their roles in pathogenesis by different viruses. Such studies have shown the critical protective action of Ifit1 in West Nile virus pathogenesis and that of Ifit2 in neuropathogenesis caused by vesicular stomatitis virus (VSV) and mouse hepatitis virus but not encephalomyocarditis virus (11, 16–18). Using the model of intranasal infection of mice by VSV, we have shown that Ifit2−/− mice are highly susceptible to neuropathy caused by this virus (11). In this model, neurons of the central nervous system (CNS) present in the olfactory bulb of the brain are directly infected in the nasal cavities of mice (19). In Ifit2−/− mice, VSV efficiently spread to neurons in other parts of the brain, causing death within 6 to 7 days after infection. The virus replicated equally well in the olfactory bulbs of wild-type (wt) and Ifit2−/− mice, but IFN produced by the infected cells could induce Ifit2 in uninfected neurons of other parts of the brains of wt mice and thus protect them from oncoming VSV infection (11, 20). Surprisingly, the absence of Ifit2 had no effect on protecting other organs from VSV; because they were well protected in wt mice and Ifit2−/− mice but not in IFNAR−/− mice; hence, other ISGs must be responsible for guarding other organs.
In the present study, we investigated whether Ifit2 can also offer protection against neuropathy caused by VSV when the virus initially enters through the neurons of the peripheral nervous system (PNS) instead of those of the CNS. The experimental model used here requires subcutaneous infection of the footpad of mice, mimicking the natural route of infection by this virus. VSV is quickly drained to the nearest lymph node (LN), the popliteal LN, where it replicates primarily in the macrophages of the subcapsular sinus (SCS) (21). These macrophages, along with attracted plasmacytoid dendritic cells (pDCs), produce IFN which in turn protects the local nervous system from infection; if the LNs are depleted of macrophages, 60% of the infected mice develop ascending paralysis. Using bone marrow-chimeric mice, it was shown that the IFNAR must be present for IFN to act not only on the macrophages but also on surrounding cell types, including the intranodal neurons (21).
Using the above model, our study identified Ifit2 as the ISG that protects PNS neurons from VSV infection. Similar to the LN macrophage-depleted mice, 65% of the infected Ifit2−/− mice developed paralysis, whereas the wt mice were protected because Ifit2 blocked efficient infection of the sciatic nerve (SN) that innervates the LN.
MATERIALS AND METHODS
Mice.
All mice had a C57BL/6 background, were 8 to 16 weeks old, and included both sexes; Ifit2−/− mice were previously described (11). Ifit1−/− mice were generated in collaboration with Herbert W. Virgin and Larissa Thackray at Washington University School of Medicine, St. Louis, MO. The exon 2 (i.e., the entire coding region of the gene) of Ifit1 was loxP flanked in 129 strain embryonic stem cells via homologous recombination with an electroporated linear targeting DNA, and then the entire exon 2 was deleted by Cre recombinase. The resulting mouse was backcrossed 12 times to a C57BL/6 background. Neither Ifit1−/− nor Ifit2−/− mice carry any residual transgenes such as neomycin resistance or lacZ. IFNAR−/− mice (lacking Ifnar1) were a gift from Kaja Murali-Krishna (Emory University, Atlanta, GA) (22). C57BL/6 wild-type mice were obtained from Taconic Farms. All animal procedures were approved by the Cleveland Clinic Institutional Animal Care and Use Committee.
Viruses and infections.
VSV Indiana was a gift from Amiya K. Banerjee, Lerner Research Institute, Cleveland, OH. VSV/green fluorescent protein (GFP) Indiana, which expresses free GFP as a nonstructural protein, was provided by Curt Horvath, Northwestern University, Chicago, IL. All infections were subcutaneous injections into the left hind-leg footpad of ketamine/xylazine-anesthetized mice, using a 31-gauge insulin syringe (Becton Dickinson) to inject 20 μl of endotoxin-free phosphate-buffered saline (PBS) containing, unless otherwise indicated, 106 PFU of VSV or VSV/GFP, or PBS only as a control. Acetaminophen was provided with drinking water. Mice were monitored daily for weight loss and symptoms of disease, which manifested as ascending paralysis of one and both hind legs.
Sciatic nerve resection.
Resections were performed as described previously (23). Briefly, mice were anesthetized with ketamine/xylazine, and the sciatic nerve of the shaven hind left limb was lifted through a 1-cm incision into the lateral midthigh; a 1.5-cm segment of the nerve was excised, and the wound was closed with 5-0 vicryl suture (Ethicon) and tissue glue. Buprenorphine was given for analgesia subcutaneously immediately after and every 12 h for 48 h; acetaminophen was provided with drinking water and with Hydrogel cups (ClearH2O, Portland, ME). At 24 h after resection, 106 PFU of VSV were injected into resected or the opposite leg's footpad.
Quantitative RT-PCR.
Mice were anesthetized with pentobarbital; blood was removed from organs by cardiac perfusion with 10 ml of PBS, and tissues or organs were harvested and snap-frozen in liquid nitrogen. Total RNA was extracted using TRIzol reagent (Invitrogen). DNase I treatment (DNAfree; Applied Biosystems/Ambion) and reverse transcription (RT) with random hexamers (ImProm-II; Promega) were performed according to manufacturer's instructions. Then, 0.5 ng of cRNA was used in 384-well-format real-time PCRs in a Roche LightCycler 480 II using PowerSYBR Green PCR 2× mix (Applied Biosystems). The PCR primers for VSV-N RNA (24), murine Ifit2, Ifit1, and IFN-β genes, as well as 18S rRNA genes, have been published previously (11). The relative expression levels, expressed as the ratios of target RNA over 18S rRNA, were graphed by using Prism 5.02 software (GraphPad).
Virus quantification by plaque assay.
For quantification of infectious VSV in organs, mice were anesthetized with pentobarbital; blood was removed from organs by cardiac perfusion with 10 ml of PBS. Organs were snap-frozen in liquid nitrogen, weighed, and pestle/tube homogenized (Kimble/Kontes) in PBS, and virus titers were determined in 10-fold serial dilutions on Vero cells by plaque assay. The results are expressed as PFU per gram of tissue or PFU per ml of serum.
Immunohistofluorescence and microscopy.
Anesthetized mice were intracardially perfused with 10 ml of PBS, followed by perfusion with 10 ml of 4% paraformaldehyde–PBS for fixation. Popliteal lymph nodes were placed in 4% paraformaldehyde overnight for complete fixation, submerged in 30% sucrose–PBS overnight for cryoprotection, and frozen in OCT compound (Sakura Finetek USA, Torrance, CA). Next, 10-μm sections were cut at −18°C in a Leica CM1900 cryostat, mounted on coated slides (Superfrost Plus, Fisherbrand; Fisher Scientific). The cellular membranes were permeabilized by 0.2% Triton X-100–PBS treatment for 15 min. For immunolabeling, rabbit anti-mouse Ifit2/P54 (25) or rabbit anti-VSV-P protein (a gift from Amiya K. Banerjee, Lerner Research Institute, Cleveland, OH) in 5% normal goat serum were used overnight at 4°C as primary antibodies; labeled sections were then stained with a 1:1,000 dilution of anti-mouse–Alexa Fluor 488 or anti-rabbit–Alexa Fluor 594 secondary antibodies (Invitrogen/Molecular Probes, Carlsbad, CA). All objects were mounted with VectaShield with DAPI (Vector Laboratories, Burlingame, CA), examined with a Leica DRM fluorescence microscope at ×50 magnification in the Lerner Research Institute Imaging Core, and processed with Adobe Photoshop CS4 software.
Flow cytometry of lymph node cell suspensions.
Eight popliteal wt or Ifit2−/− lymph nodes, 14 h after VSV/GFP footpad injection, were pooled to make single cell suspensions. After mincing with a scalpel, the lymph nodes were digested in 2.5 ml of Dulbecco modified Eagle medium with Liberase TL (Roche) and DNase I (Roche) for 30 min at 37°C, during which the cells were dissociated once by aspiration with a P1000 pipette. The cells were then strained into single-cell suspensions with 100-, 70-, and 40-μm-pore-size sieves, counted, and spun for 5 min at 400 × g, and then 2 × 106 cells/labeling sample were resuspended in 200 μl of fluorescence-activated cell sorting (FACS) buffer (PBS with 1 mM EDTA, 25 mM HEPES [pH 7.0], and 1% fetal bovine serum). Nonspecific IgG binding was blocked with FcγR block (anti-mouse CD16/CD32; BD Pharmingen) for 30 min at 4°C, and cell-type surface markers were labeled by adding either anti-B220-PE (RA3-6B2; BD), anti-CD3-PerCP (145-2C11; BD), anti-CD11b-PerCP-Cy5.5 (M1/70; BD), or anti-CD11c-PE (N418; eBioscience), all at 1:100 in FACS buffer for 45 min at 4°C. After three washes with FACS buffer, the cells were fixed for 20 min with 2% paraformaldehyde–PBS to fix VSV, washed twice with FACS buffer, and subjected to flow cytometry using an LSRFortessa (Becton Dickinson) in the Lerner Research Institute Flow Cytometry Core. FlowJo v9 (Tree Star, Inc.) software was used for analysis: VSV/GFP-positive cells (consistently ∼1.5% of lymph node cells) were gated and analyzed for cell marker expression, comparing unlabeled samples (as controls) with marker-labeled samples. The results are presented as “percent cell type marker-positive cells, out of all GFP-positive cells.”
IFN-β enzyme-linked immunosorbent assay (ELISA).
At indicated times after VSV footpad infection, popliteal lymph nodes were frozen in liquid nitrogen until processing by the addition of 200 μl of PBS with 0.2% Triton X-100 and Complete protease inhibitor (Roche), followed by homogenization using a pestle/tube (Kimble/Kontes) and centrifugation at 10,000 × g for 10 min. Cleared homogenates were diluted 1:2 with dilution buffer, and samples, together with murine IFN-β standards, were subjected to VeriKine murine IFN-β ELISA (PBL Assay Science, Piscataway, NJ).
Statistical analysis.
Where appropriate, t tests were conducted to confirm significance of difference between two groups, using an unpaired two-tailed t test with a 95% confidence interval, employing Prism 5.02 software (GraphPad). P values are given in the figure legends; for P values larger than 0.05, the differences were considered not significant (n.s.).
RESULTS
Ifit2 protects mice from neuropathy caused by peripheral VSV infection.
To evaluate the potential role of Ifit2 in preventing neuropathogenesis in mice caused by peripheral infection with VSV, we used a previously characterized model of subcutaneous infection with the virus into the footpad of the left hind leg (21). In this model, VSV is captured by the macrophages present in SCS of the popliteal lymph node near the site of infection. These infected macrophages recruit pDCs to jointly produce type I IFN. IFN, in turn, acts upon the intranodal neurons to prevent VSV infection of the PNS and the CNS neurons. We challenged different genetically modified mouse lines with VSV, using the footpad route, and scored for hind-limb paralysis as a measure of the development of neuropathy. Over a 2-week period, very few wt mice were paralyzed at this dose of infection (106 PFU). However, 65% of the Ifit2−/− mice developed paralysis within 8 days postinfection (p.i.), indicating a protective role of Ifit2 in this pathogenesis model. Ifit2+/− mice, carrying only one allele of Ifit2, were half as sensitive as the complete knockout mice. In contrast, Ifit1−/− mice were completely resistant, as the wt mice (Fig. 1). These results demonstrated that Ifit2 protects mice from neuropathy caused by peripheral infection of VSV and that the effect is Ifit2 gene dosage dependent. Moreover, a related gene, Ifit1, cannot substitute for this function of Ifit2.
FIG 1.
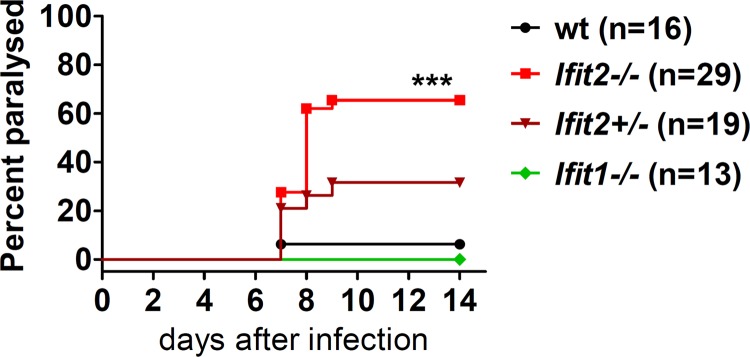
Ifit2 protects mice from paralytic VSV neuropathogenesis after footpad infection. Hind limb paralysis of wt, Ifit2−/−, Ifit2+/−, and Ifit1−/− mice after subcutaneous injection of 106 PFU of VSV was examined (n = number of animals). The asterisks (***) indicate statistical significance (wt versus Ifit2−/−; P < 0.0005).
Ifit2 has no role in VSV replication in the lymph nodes.
To investigate at what stage of viral spread Ifit2 intervenes, we monitored viral replication at several points along the route of infection, starting from the nearest lymph node (LN) up to the brain. VSV-infected cells in the popliteal LNs were detected microscopically by immunostaining of the viral P protein of wt-VSV (red in the upper panel of Fig. 2A) or by fluorescence of GFP produced by a recombinant VSV/GFP as a nonstructural protein (green in the lower panel of Fig. 2A). By both methods, we observed that the infected cells were all localized to the SCS at the outer rim of the LN; more importantly, there was no visible difference in this respect between wt and Ifit2−/− mice. In contrast, in IFNAR−/− mice, many cells in other parts of the LN were infected as well (upper panel, Fig. 2A). Shortly after infection, the total viral loads in LNs of wt and Ifit2−/− mice were very similar (Fig. 2B). This viral load decreased gradually over a 6-day period after infection, and the rates of virus clearance were very similar in the two lines of mice (Fig. 2C). Finally, to determine whether Ifit2 caused any distortions in the type of cells that were infected in the LN, we infected mice with VSV/GFP for 14 h and subjected suspended LN cells to flow cytometric analysis. Using different surface markers for different immune cell populations, we observed that among the GFP-positive infected cells, the distributions of cell types were very similar between wt and Ifit2−/− mice (Fig. 2D).
FIG 2.

Ifit2 does not restrict VSV replication in lymph nodes. (A) The top panels show the localization of VSV P protein in wt, Ifit2−/−, and IFNAR−/− popliteal lymph node sections 14 h after VSV footpad injection by immunohistofluorescence microscopy. The bottom panels show the localization of active viral gene expression in wt and Ifit2−/− popliteal lymph node sections by fluorescence microscopy of VSV-encoded GFP (arrowheads) 14 h after VSV/GFP footpad injection. All photos were taken at ×50 magnification. (B) Infectious VSV titers in wt and Ifit2−/− popliteal lymph nodes at 14 h after infection, plotted on a log scale. (C) VSV-N RNA levels in wt and Ifit2−/− popliteal lymph nodes, measured by real-time RT-PCR, plotted on a log scale. (D) Expression of cell type markers on VSV/GFP-infected wt and Ifit2−/− lymph node cells at 14 h after infection, measured by flow cytometry. Only GFP-positive cells from eight pooled lymph nodes per experiment were subjected to analysis. Dashed lines indicate the threshold of detection. Abbreviations: LN, lymph nodes; pDC, plasmacytoid dendritic cell; mDC, myeloid dendritic cell; n.s., not significant.
Next, we examined the levels of induction of IFN and IFN-induced genes in the infected LNs. IFN-β mRNA was strongly induced in wt mice and even more so in Ifit2−/− mice (Fig. 3A), and the same was true for Ifit1 mRNA. As expected, Ifit2 mRNA was induced in wt mice only. Upon immunostaining for Ifit2-encoded P54 protein expression in LN sections of infected wt mice, we observed that Ifit2 was induced in almost all cells (Fig. 3B), although VSV was mostly restricted to the SCS (Fig. 2A), indicating that IFN secreted by the infected cells induce Ifit2 expression in other cells as well. This conclusion was further supported by the staining results of LN from IFNAR−/− mice: although most cells were infected by VSV (see Fig. 2A), there was little induction of Ifit2 (Fig. 3B), most probably because IFN could not act on the cells to induce it. As expected, no Ifit2 was detected in the LN of Ifit2−/− mice. IFN-β production in the LN, measured by ELISA, was transient, diminishing by 48 p.i. (Fig. 3C), and again there was little difference between the two lines of mice. The above results demonstrated that there were no differences between wt and Ifit2−/− mice in the level of VSV replication or the IFN response in LNs.
FIG 3.

Induction of IFN-β and IFN-stimulated genes in lymph nodes after VSV footpad infection does not require Ifit2. (A) mRNA levels of IFN-β and the IFN-stimulated genes Ifit2 and Ifit1 in the popliteal lymph nodes of wt and Ifit2−/− mice, 14 h after footpad infection, as measured by real-time RT-PCR (three to four infected mice per group). (B) Ifit2 (= P54) protein expression in popliteal lymph nodes of wt, Ifit2−/−, and IFNAR−/− mice at 14 h after footpad infection, detected by immunohistofluorescence microscopy on tissue sections at ×50 magnification. Sections shown are adjacent sections from lymph nodes in Fig. 2A. (C) IFN-β protein production in popliteal lymph nodes after VSV infection of wt and Ifit2−/− mice, as measured by ELISA (three infected mice per group). Asterisks indicate statistical significance (**, P < 0.005). Dashed lines indicate the threshold of detection.
Virus infection spreads through a neuronal route in Ifit2−/− mice.
When IFNAR−/− mice were footpad infected with VSV, viremia ensued, and the virus spread into many organs, including the brain, by 1 to 2 days p.i. (Fig. 4B), and the infected mice died within 3 to 4 days (data not shown). In contrast, we could not detect any virus between 6 to 8 days p.i. in the sera, spleens, and lungs of Ifit2−/− mice (Fig. 4A), demonstrating that the virus did not cause a systemic infection. These results also indicated that other ISGs, not Ifit2, protect the majority of cell types in the body. These observations led us to investigate a neuronal route of VSV transmission through the nervous system. Because the neuronal axon endings that innervate the popliteal LNs are connected to the sciatic nerve (SN), we focused our attention on the SN of the infected leg. At 1 day p.i., Ifit2 mRNA was induced in the SNs, as well as in the LNs, of wt and Ifit2+/− mice but not in those of Ifit2−/− and IFNAR−/− mice (Fig. 5A). In contrast, although VSV was present in the LNs of all lines of mice, the virus could be detected only in the SNs of IFNAR−/− mice at that time (Fig. 5B). Taken together, these data indicate that in wt mice, IFN produced by cells in the LN acts upon cells in the SN to induce Ifit2 at 1 day p.i. Although there was no detectable virus in the SNs of Ifit2−/− mice by 1 or 2 days p.i., there were abundant quantities of virus later after infection (Fig. 5C). In contrast, in wt mice, there was little virus in the SN, with the exception of a few escapees. Thus, the first measurable difference between wt and Ifit2−/− mice in the virus route toward the CNS was at the stage of LN-to-SN transmission. Once in the SN, the virus moved on efficiently to the spinal cord (SC); very high levels of viral RNA were detected in SCs of Ifit2−/− mice at 8 days p.i. (Fig. 6A). To definitively demonstrate that VSV was entering the SC via the SN, we resected the SNs of the left hind legs of the Ifit2−/− mice. When these mice were infected via the right footpad, high levels of VSV RNA in the SC were found at 8 days p.i., but when the mice had received ipsilateral infections, no viral RNA and paralysis were detected (Fig. 6B).
FIG 4.

Interferon protects non-neuronal tissues from peripheral VSV infection even in the absence of Ifit2. (A and B) Infectious VSV titers in tissues at different times after infection of Ifit2−/− (A) and IFNAR−/− (B) mice, plotted on log scale. Dashed lines indicate the threshold of detection.
FIG 5.

Interferon-induced Ifit2 suppresses VSV replication in sciatic nerves after footpad infection. (A and B) Ifit2 mRNA (A) and VSV-N RNA (B) levels in popliteal lymph nodes and sciatic nerves of wt, Ifit2−/−, and IFNAR−/− mice, 1 day after infection, as measured by real-time RT-PCR (four to six infected mice per group). (C) VSV-N RNA levels in wt and Ifit2−/− sciatic nerves, measured by real-time RT-PCR. All VSV RNA levels were plotted on log scale. Asterisks indicate statistical significance (***, P = 0.0007; **, P < 0.003). Dashed lines indicate the threshold of detection.
FIG 6.

The sciatic nerve is the exclusive entry route of VSV into the spinal cord after footpad infection. (A) VSV-N RNA levels in wt and Ifit2−/− spinal cords at different times after footpad infection, measured by real-time RT-PCR. (B) VSV-N RNA levels in Ifit2−/− spinal cords of mice, after surgical removal of the midthigh sciatic nerve segment (i.e., resection), followed by VSV infection of the same or the opposite leg's footpad. RNA was measured at 8 days after infection by real-time RT-PCR. All VSV RNA levels were plotted on a log scale. Asterisks indicate statistical significance (*, P = 0.02; ***, P = 0.0002). Dashed lines indicate the threshold of detection.
To determine the kinetics and the nature of VSV transmission from the PNS to CNS, we quantified VSV in the left SN, lower SC, upper SC, brain, and right SN. We also distinguished between Ifit2−/− mice which were paralyzed from those that were not (Fig. 7). By 6 days p.i., only the SN and lower SC showed VSV RNA, and mice were not paralyzed. In contrast, by 8 days p.i., the majority of the mice were paralyzed. These mice also had high levels of virus in the upper SC and the brain; the same was true for the appearance of the virus in the right SN (Fig. 7A). These observations were strengthened by determining infectious virus titers in the SCs and brains of Ifit2−/− and wt mice at 8 days p.i. There was no detectable virus in either tissue of wt mice, but there were high virus titers in SC and brain of Ifit2−/− mice that were paralyzed. These results demonstrate that in Ifit2−/− mice, VSV travels on a route from the left SN to the SC to the brain, as well as the right SN.
FIG 7.

VSV spread into the CNS precedes hind-limb paralysis of Ifit2−/− mice after footpad infection. (A) VSV-N RNA levels in left and right leg sciatic nerves, in the lower and upper segments of spinal cord, and in brains of Ifit2−/− mice at different times after footpad infection, as measured by real-time RT-PCR and plotted on a log scale. (B) Infectious VSV titers in tissues 8 days after footpad infection of wt and Ifit2−/− mice, plotted on a log scale. The tissues of mice showing leg paralysis at the time of harvest are labeled with empty squares. Asterisks indicate statistical significance (**, P = 0.004; *, P = 0.04). Dashed lines indicate the threshold of detection.
VSV, after peripheral entry, infects neurons in all caudal regions of the brain.
Previously, we observed that when wt mice were infected with VSV intranasally, some regions of the brain, such as the cerebellum, were preferentially protected. In the current footpad infection model, all rear (caudal) brain regions of paralyzed Ifit2−/− mice, including the cerebellum, were strongly infected (Fig. 8A). To test whether Ifit2 affects the regional pattern of VSV infection in the brain after peripheral infection, we performed a similar experiment with wt mice. By increasing the dose of infection by 35-fold, we observed an increase to ca. 15% of wt mice displaying paralysis (Fig. 8B). When different caudal brain regions of paralyzed wt mice were analyzed for viral RNA, we observed a distribution pattern similar to Ifit2−/− brains, with no statistically significant differences between wt and Ifit2−/−. These results indicate that after peripheral infection, Ifit2 blocks the transmission of VSV through the PNS into the CNS, but it does not influence the infection patterns in different regions of the brain.
FIG 8.

VSV infection spreads to all regions of the brain. (A and C) VSV-N RNA levels in different regions of the brains of paralyzed mice. (A) Four Ifit2−/− mice, numbered 1 to 4, after infection with 106 PFU of VSV; (C) six wt mice, numbered 1 to 6, after infection with 3.5 × 107 PFU of VSV. (B) Hind-limb paralysis of wt mice after footpad injection of 106 PFU (from Fig. 1) or 3.5 × 107 PFU of VSV (n = number of animals). No significant difference in RNA levels was found between respective wt and Ifit2−/− brain regions in panels A and C. The dashed lines in panels A and B indicate the threshold of detection.
DISCUSSION
The pivotal role of Ifit2 in protecting CNS neurons from VSV infection was established in our previous study (11); in a new investigation, reported here, we demonstrate that the same is true for peripheral neurons. We used a well-characterized mouse model for studying the peripheral infection by VSV (21). The subcutaneous injection of the virus into the footpad keeps the infection restricted to the LNs, and it does not disseminate to the rest of the body, unless the type I IFN system is made dysfunctional. The presence of IFNAR in both hematopoietic and stromal compartments, including the local nerves, is necessary to prevent viral pathogenesis (21). In this route of infection, the virus is captured by macrophages in draining LNs, which prevents its systemic dissemination. A specific subpopulation of macrophages, the CD169+ CD11b+ MHC-II+ macrophages, present in the SCS of LNs, are the primary recipients of VSV (26). These macrophages not only remove the virus from the lymph but also present it to the adjacent LN B cells to initiate the humoral immune response. Importantly, they prevent the entry of the virus to the nervous system by blocking infection of peripheral nerve neurons in the LN. VSV replicates in the SCS macrophages, which attract pDCs, and both types of cells produce type I IFN. In this model, the IFN production, but not the B cell activation, turns out to be the critical function of the SCS macrophages in preventing neuronal infection by VSV (21, 27). Neuropathy is prevented by the action of IFN, produced by the infected LN macrophages and pDCs, on the local nerve. Our results demonstrate that this particular anti-VSV effect of IFN is mediated by Ifit2, one among the hundreds of ISGs. Because Ifit2 protects only the neurons, other cell types of the infected Ifit2−/− mice were still well protected. This is in contrast to the IFNAR−/− mice, which are susceptible to systemic infection; consequently, the threshold of virus inoculum to which they succumb is lower (21) than that required for pathogenesis in Ifit2−/− mice. The infected IFNAR−/− mice die much sooner after infection due to other organ failures before neuropathy sets in. It appears, however, that in the context of the neuronal infection, Ifit2 is the antiviral protein used by the IFN system. It remains to be seen whether Ifit2 is also the effector protein against other rhabdoviruses, such as rabies virus, and other unrelated neurotropic viruses that can enter the body through skin lesions or by animal and insect bites.
We have delineated the route of VSV spread within the host in Ifit2−/− mice from the initial site of entry to different regions of the brain (Fig. 9). Instead of following the migration of labeled particles from end to end, we measured virus replication at different steps on its route at different time points. This exercise also revealed the exact point where Ifit2 establishes its roadblock. VSV was detectable in the LN early after infection and replicated to high titers in LN before being cleared over the next few days (Fig. 2C). In the LN, the infection caused strong induction of IFN-β, which in turn induced Ifit2 (Fig. 3). Up to this point, there was no major difference between the responses elicited in wt and Ifit2−/− mice. In the SN, VSV was not detectable even at 2 days p.i., but thereafter the viral RNA levels increased over time in Ifit2−/− SN (Fig. 5C). This step in the route of spread within the mouse is where Ifit2 had an inhibitory effect. In the presence of Ifit2 in wt mice, VSV was detectable in the SN of only a very small percentage of mice, whereas 100% of the Ifit2−/− mice accumulated virus in their SNs. Ifit2 could have blocked virus entry via the neuronal endings innervating the LN, or blocked replication of VSV in the neurons of the SN; our experiments cannot distinguish between the two possibilities. The long delay between LN replication and the appearance of the virus in the SN could be due to its slow kinetics of transport or the limits of the sensitivity of our assay for viral RNA detection. From the SN, the virus entered the CNS through the lower SC, from where it proceeded to the upper SC and eventually to the brain. The virus also spread into the contralateral right SN, probably from within the SC. The infection of all parts of the PNS and CNS with VSV resulted from spread originating at its PNS entry point in the LN; since there was no viremia, it is unlikely that distant regions of the nervous system were de novo infected with virus received via body fluids. Strong support for the exclusive transmission of VSV through the nervous system in this experimental model came from our SN resection experiment (Fig. 6C). Our observations demonstrate that IFN produced by myeloid cells in the LN can act remotely on the SN neurons to induce Ifit2. Similar long-distance IFN signaling within the brain has recently been reported (20).
FIG 9.

Route of spread of VSV infection from the footpad to the brain, blocked by Ifit2 at the sciatic nerve. Within minutes after subcutaneous deposition of VSV in the left footpad, virus drains to the local lymph node in the knee hole, replicates in its periphery, and induces IFN-β, which acts on sciatic nerve neurons innervating the lymph node to induce Ifit2 within less than 24 h after infection. Ifit2 blocks slow VSV spread via the sciatic nerve into spinal cord and brain, thereby preventing limb paralysis. Abbreviations: OB, olfactory bulbs; CX, cortex; MB, “midbrain”; CB, cerebellum; BS, brain stem.
Previously, using the intranasal model of VSV infection, we observed that in Ifit2−/− mice the virus replicated similarly well in all regions of the brain. In contrast, in wt mice, overall VSV replication was much lower and importantly, some regions, such as the cerebellum, were almost devoid of infection (11). This indicated that Ifit2 might be preferentially protecting the cerebellum. Such a notion is consistent with observations made by Cho et al. (28), according to which neurons from different regions of the brain display different levels of antiviral response to IFN. However, in the pathogenesis study reported here, using peripheral VSV infection, we did not observe such differential sensitivity. In paralyzed Ifit2−/− mice, all caudal regions of the brain, including the cerebellum, supported the replication of VSV. Importantly, the same was true for the few wt mice that had developed paralysis despite the presence of Ifit2. The fact that a much higher dose of VSV led to increased paralysis in wt mice also reinforced the concept of a dynamic equilibrium between the IFN system and the virus. Since the equilibrium can be shifted in favor of the virus by increasing the virus dose during infection, the restriction imparted by Ifit2 is not absolute.
How Ifit2 inhibits VSV infection of CNS and PNS is currently unknown. Whatever is the mechanism, there has to be a neuron-specific aspect to it. It is possible that Ifit2 functions in conjunction with a neuronal protein that is not expressed in other cell types. It is also possible that Ifit2 does not inhibit VSV replication per se but instead inhibits the transport of VSV particles along the long axons from their endings to the neuron cell bodies. The latter effect, if true, can readily explain why Ifit2's anti-VSV effects are restricted to the nervous system. More detailed information is available regarding the mechanism of the antiviral action of the related protein, Ifit1. Ifit1, but not Ifit2, recognizes mRNA 5′-caps that lack 2′-O-methylation of their first ribose and inhibits their translation (16, 29–31). Because many viral mRNAs do not contain 2′-O-methylated caps, they are subject to Ifit1-mediated restriction. Several Alphaviruses escape Ifit1-restriction in spite of the fact that their 5′-capped positive-strand genomic RNAs, which need to be translated, lack 2′-O-methylated caps. They achieve evasion by the presence of specific secondary structures adjacent to the 5′-cap in the untranslated region of their genome, which prevent Ifit1 binding (32). Neither of these mechanisms is relevant for explaining Ifit2's action on VSV, because Ifit1−/− mice were as protected as wt mice against VSV-induced neuropathy.
The present study demonstrates a neuron-specific antiviral action of Ifit2. Although abundant IFN and Ifit2 were induced in the infected LNs, they did not affect VSV replication in this tissue. Virus-specific antiviral properties of specific ISGs have been known for some time, but the realization of their tissue-specific action is rather recent. We observed the same tissue specificity of Ifit2 action against VSV in the intranasal infection model, and Zhao et al. observed a similar cell-type-specific action of the OAS/RNase L system on mouse hepatitis virus replication in the liver (11–13). The results presented here show that VSV infection spreads very efficiently to other organs in IFNAR−/− mice, but not in Ifit2−/− mice, except for the SN. This observation indicates that Ifit2 is not required to protect other tissues from VSV infection and that other ISGs perform this task instead. Challenging other ISG knockout mice using different routes of VSV infection will be required to further explore the basis of the tissue specificity of ISG actions.
ACKNOWLEDGMENTS
We thank Herbert W. Virgin and Larissa B. Thackray for collaborative contributions to the generation of Ifit1−/− mice, Stephen A. Stohlman for technical advice, Tomoaki Ogino for helpful discussion, and Liying Shi and Brandon Pool for technical assistance.
The study was supported in part by National Institutes of Health grants CA068782 and AI073303.
Footnotes
Published ahead of print 2 July 2014
REFERENCES
- 1.Broz P, Monack DM. 2013. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 13:551–565. 10.1038/nri3479 [DOI] [PubMed] [Google Scholar]
- 2.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. U. S. A. 95:15623–15628. 10.1073/pnas.95.26.15623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schoggins JW, Rice CM. 2011. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 1:519–525. 10.1016/j.coviro.2011.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ivashkiv LB, Donlin LT. 2014. Regulation of type I interferon responses. Nat. Rev. Immunol. 14:36–49. 10.1038/nri3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopez CB, Hermesh T. 2011. Systemic responses during local viral infections: type I IFNs sound the alarm. Curr. Opin. Immunol. 23:495–499. 10.1016/j.coi.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. 10.1126/science.8009221 [DOI] [PubMed] [Google Scholar]
- 7.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signaling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47. 10.1099/vir.0.83391-0 [DOI] [PubMed] [Google Scholar]
- 8.Fensterl V, Sen GC. 2011. The ISG56/IFIT1 gene family. J. Interferon Cytokine Res. 31:71–78. 10.1089/jir.2010.0101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Ding SC, Cho H, Chung BC, Gale M, Jr, Chanda SK, Diamond MS. 2013. A short hairpin RNA screen of interferon-stimulated genes identifies a novel negative regulator of the cellular antiviral response. mBio 4:e00385-13. 10.1128/mBio.00385-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. 10.1038/nature09907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, Diamond MS, Virgin HW, Sen GC. 2012. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 8:e1002712. 10.1371/journal.ppat.1002712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao L, Jha BK, Wu A, Elliott R, Ziebuhr J, Gorbalenya AE, Silverman RH, Weiss SR. 2012. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 11:607–616. 10.1016/j.chom.2012.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao L, Rose KM, Elliott R, Van Rooijen N, Weiss SR. 2011. Cell-type-specific type I interferon antagonism influences organ tropism of murine coronavirus. J. Virol. 85:10058–10068. 10.1128/JVI.05075-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diamond MS, Farzan M. 2013. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 13:46–57. 10.1038/nri3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vladimer GI, Gorna MW, Superti-Furga G. 2014. IFITs: emerging roles as key anti-viral proteins. Front. Immunol. 5:94. 10.3389/fimmu.2014.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M, Jr, Shi PY, Diamond MS. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456. 10.1038/nature09489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szretter KJ, Daniels BP, Cho H, Gainey MD, Yokoyama WM, Gale M, Jr, Virgin HW, Klein RS, Sen GC, Diamond MS. 2012. 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 8:e1002698. 10.1371/journal.ppat.1002698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butchi NB, Hinton DR, Stohlman SA, Kapil P, Fensterl V, Sen GC, Bergmann CC. 2014. Ifit2 deficiency results in uncontrolled neurotropic coronavirus replication and enhanced encephalitis via impaired alpha/beta interferon induction in macrophages. J. Virol. 88:1051–1064. 10.1128/JVI.02272-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plakhov IV, Arlund EE, Aoki C, Reiss CS. 1995. The earliest events in vesicular stomatitis virus infection of the murine olfactory neuroepithelium and entry of the central nervous system. Virology 209:257–262. 10.1006/viro.1995.1252 [DOI] [PubMed] [Google Scholar]
- 20.van den Pol AN, Ding S, Robek MD. 2014. Long-distance interferon signaling within the brain blocks virus spread. J. Virol. 88:3695–3704. 10.1128/JVI.03509-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, Whelan SP, Guidotti LG, von Andrian UH. 2010. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature 465:1079–1083. 10.1038/nature09118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. 2005. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202:637–650. 10.1084/jem.20050821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shao C, Liu M, Wu X, Ding F. 2007. Time-dependent expression of myostatin RNA transcript and protein in gastrocnemius muscle of mice after sciatic nerve resection. Microsurgery 27:487–493. 10.1002/micr.20392 [DOI] [PubMed] [Google Scholar]
- 24.Simon ID, Publicover J, Rose JK. 2007. Replication and propagation of attenuated vesicular stomatitis virus vectors in vivo: vector spread correlates with induction of immune responses and persistence of genomic RNA. J. Virol. 81:2078–2082. 10.1128/JVI.02525-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terenzi F, White C, Pal S, Williams BR, Sen GC. 2007. Tissue-specific and inducer-specific differential induction of ISG56 and ISG54 in mice. J. Virol. 81:8656–8665. 10.1128/JVI.00322-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, Fink K, Henrickson SE, Shayakhmetov DM, Di Paolo NC, van Rooijen N, Mempel TR, Whelan SP, von Andrian UH. 2007. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature 450:110–114. 10.1038/nature06287 [DOI] [PubMed] [Google Scholar]
- 27.Moseman EA, Iannacone M, Bosurgi L, Tonti E, Chevrier N, Tumanov A, Fu YX, Hacohen N, von Andrian UH. 2012. B cell maintenance of subcapsular sinus macrophages protects against a fatal viral infection independent of adaptive immunity. Immunity 36:415–426. 10.1016/j.immuni.2012.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho H, Proll SC, Szretter KJ, Katze MG, Gale M, Jr, Diamond MS. 2013. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 19:458–464. 10.1038/nm.3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar P, Sweeney TR, Skabkin MA, Skabkina OV, Hellen CU, Pestova TV. 2014. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1-, and 5′ppp-mRNAs. Nucleic Acids Res. 42:3228–3245. 10.1093/nar/gkt1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Habjan M, Hubel P, Lacerda L, Benda C, Holze C, Eberl CH, Mann A, Kindler E, Gil-Cruz C, Ziebuhr J, Thiel V, Pichlmair A. 2013. Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS Pathog. 9:e1003663. 10.1371/journal.ppat.1003663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kimura T, Katoh H, Kayama H, Saiga H, Okuyama M, Okamoto T, Umemoto E, Matsuura Y, Yamamoto M, Takeda K. 2013. Ifit1 inhibits Japanese encephalitis virus replication through binding to 5′ capped 2′-O unmethylated RNA. J. Virol. 87:9997–10003. 10.1128/JVI.00883-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hyde JL, Gardner CL, Kimura T, White JP, Liu G, Trobaugh DW, Huang C, Tonelli M, Paessler S, Takeda K, Klimstra WB, Amarasinghe GK, Diamond MS. 2014. A viral RNA structural element alters host recognition of nonself RNA. Science 343:783–787. 10.1126/science.1248465 [DOI] [PMC free article] [PubMed] [Google Scholar]