

HIV protease converts procaspase 8 into Casp8p41, which binds and activates Bak to induce cell death in infected CD4 T cells.
Abstract
Previous studies have shown that human immunodeficiency virus (HIV) protease cleaves procaspase 8 to a fragment, termed Casp8p41, that lacks caspase activity but nonetheless contributes to T cell apoptosis. Herein, we show that Casp8p41 contains a domain that interacts with the BH3-binding groove of pro-apoptotic Bak to cause Bak oligomerization, Bak-mediated membrane permeabilization, and cell death. Levels of active Bak are higher in HIV-infected T cells that express Casp8p41. Conversely, targeted mutations in the Bak-interacting domain diminish Bak binding and Casp8p41-mediated cell death. Similar mutations in procaspase 8 impair the ability of HIV to kill infected T cells. These observations support a novel paradigm in which HIV converts a normal cellular constituent into a direct activator that functions like a BH3-only protein.
Introduction
Apoptosis reflects the interplay between three groups of proteins: caspases, inhibitor of apoptosis (IAP) proteins, and Bcl-2 family members (Taylor et al., 2008; Strasser et al., 2011). Caspases play two previously recognized roles in this process (Earnshaw et al., 1999; Taylor et al., 2008), transducing signals such as death receptor ligation or mitochondrial cytochrome c release into protease activity (initiator caspases) and cleaving a wide variety of cellular constituents to yield the apoptotic phenotype (effector caspases). Some of these cysteine proteases are inhibited by X chromosome–linked IAP (Taylor et al., 2008; Fulda and Vucic, 2012). Moreover, Bcl-2 family members regulate the mitochondrial cytochrome c release that initiates caspase 9 activation. In particular, the proapoptotic Bcl-2 family members Bax and Bak, which are responsible for mitochondrial outer membrane (MOM) permeabilization (MOMP), are inhibited by binding to antiapoptotic family members such as Bcl-2 and Mcl-1, and are activated by BH3-only proteins through either direct interactions (Kim et al., 2009; Gavathiotis et al., 2010; Dai et al., 2011; Czabotar et al., 2013) or neutralization of antiapoptotic family members (Llambi et al., 2011; Strasser et al., 2011).
Viruses have evolved several strategies for impacting cellular apoptotic pathways. These include the expression of IAP proteins such as Op-IAP and antiapoptotic Bcl-2–like proteins such as Epstein-Barr virus BHLF1 (Miller, 1999; Galluzzi et al., 2008). Indeed, study of these proteins has informed current understanding of viral pathogenicity as well as apoptotic pathway regulation. HIV, however, has not previously been shown to impact the core apoptotic machinery directly.
HIV causes death of infected CD4 T cells in three different ways: by triggering pyroptosis during abortive infection (Doitsh et al., 2010), activating an integrase-initiated DNA damage response in cells that integrate the virus (Cooper et al., 2013), or inducing apoptosis in cells that are productively infected, i.e., that produce progeny virions. During HIV production, HIV protease is active in the cytoplasm and at the cell membrane (Kaplan et al., 1994), where it cleaves both viral and host substrates (Ventoso et al., 2001). Moreover, expression of HIV protease is known to induce apoptosis (Baum et al., 1990; Blanco et al., 2003), although the process also requires procaspase 8 (Nie et al., 2002; Nie et al., 2008). Curiously, procaspase 8 is directly cleaved by HIV-1 protease between F355 and F356, generating a 41-kD N-terminal fragment, termed Casp8p41, that lacks the catalytic cysteine of an active caspase (Nie et al., 2002; Nie et al., 2008). Nonetheless, protease inhibitor–resistant HIV isolates that replicate but do not cause CD4 T cell decline in patients (discordant responses) contain HIV protease variants that are impaired in their ability to generate Casp8p41 (Natesampillai et al., 2010), which indicates the importance of Casp8p41 in T cell demise during productive HIV infection. Further studies have shown that Casp8p41 traffics to mitochondria (Algeciras-Schimnich et al., 2007; Sainski et al., 2011) and requires procaspase 9 to induce apoptosis (Sainski et al., 2011). How Casp8p41, as a catalytically inactive caspase fragment, activates caspase 9 to induce apoptosis has remained unclear.
In the present study, we show that Casp8p41 binds the Bak BH3-binding groove, leading to Bak oligomerization, Bak-mediated MOMP, and cell death. These observations not only provide new insight into the mechanism of T cell death during productive HIV infection, but also suggest a new paradigm in which a protein that ordinarily does not bind Bcl-2 family members can be converted into a direct activator to affect apoptosis.
Results and discussion
Casp8p41-induced death requires Bak
To gain mechanistic insight into Casp8p41-mediated killing, we screened for lentiviral shRNAs that diminish Casp8p41-induced cell death (Fig. S1 A). Because EGFP-Casp8p41 induces death within 24 h (Sainski et al., 2011), cells expressing both EGFP-Casp8p41 and RFP from the shRNA constructs at 48 h must have been altered in a way that renders Casp8p41 less toxic. Of 27,000 shRNAs tested, 1,558 were at least threefold higher in RFP/EGFP double-positive cells compared with background. Of these overrepresented shRNAs, only three (FADD, procaspase 9, and Bak) target proapoptotic components of the core apoptotic machinery. Because procaspase 9, which is involved in HIV-induced T cell death (Sainski et al., 2011), is downstream of mitochondria (Earnshaw et al., 1999; Taylor et al., 2008), we focused on FADD and Bak. Casp8p41 induced similar levels of apoptosis in FADD-deficient and parental Jurkat cells (unpublished data). In contrast, siRNA-mediated Bak knockdown diminished Casp8p41 killing, which indicates an important role for Bak in Casp8p41-induced death (Fig. S1, B and C).
Casp8p41 binds Bak
Based on the observation that Casp8p41, like a BH3-only protein, translocates to mitochondria while inducing apoptosis (Algeciras-Schimnich et al., 2007; Sainski et al., 2011), we assessed the possibility that Casp8p41 and Bak might directly interact. For these studies we initially immobilized recombinant His-tagged Bak lacking the transmembrane domain (His6 BakΔTM) on a surface plasmon resonance (SPR) chip and assessed binding of GST-Casp8p41. Binding occurred in a dose-dependent manner, with an equilibrium dissociation constant (Kd) of 16 ± 5 nM (Fig. 1, A and C; and Fig. S2 A), which is tighter than BakΔTM binding to Bim under comparable conditions (Dai et al., 2011). In contrast, full-length procaspase 8 fused to GST-bound BakΔTM 100-fold less tightly (Fig. 1, B and C; and Fig. S2 B), potentially explaining why full-length procaspase 8 does not independently induce apoptosis.
Figure 1.
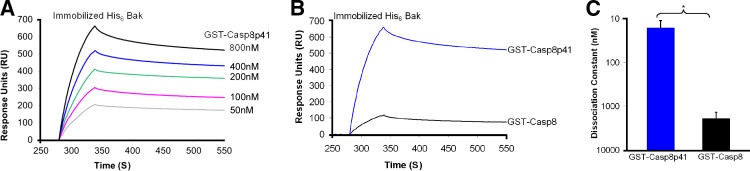
Casp8p41 binds Bak. (A) Binding of 50–800 nM GST-Casp8p41 to immobilized His6-BakΔTM as assessed by SPR. Representative of n = 3. (B) His6-BakΔTM immobilized on an SPR chip was exposed to 400 nM GST-Casp8p41 or GST–procaspase 8. Representative of n = 3. (C) Summarized results from three independent experiments (*, P < 0.05). Error bards indicate SD.
Casp8p41 contains a latent Bak activator domain
Using protein threading (Wu and Zhang, 2008) and multiple molecular dynamic simulations (57 independent 10-ns simulations with a time step of 1 fs) performed according to published protocols (Dai et al., 2011; Pang et al., 2012), we generated a three-dimensional model of Casp8p41 in order to gain insight into residues of Casp8p41 involved in the interaction with Bak. This model suggested that amino acids 141–160 of Casp8p41 adopt a loop conformation in their free state and an α-helical conformation when bound to the Bak BH3-binding groove (Fig. 2, A and B). Even though this Casp8p41 region exhibits only 35% sequence similarity to classical BH3 domains, the spatial positions of residues critical for Bak binding resemble those of a BH3 domain (Fig. 2 A, bottom). In contrast, in full-length procaspase 8, amino acids 141–160 are buried beneath the C-terminal domain, which folds back on the rest of the protein (Yu et al., 2009). Thus, the ability of HIV-1 protease to proteolytically remove the procaspase 8 C terminus, exposing a loop we term the “latent Bak activator domain” in Casp8p41, accounts for the preferential binding of Casp8p41 but not procaspase 8 to Bak.
Figure 2.

The latent Bak activator domain in Casp8p41 interacts with the BH3-binding groove in Bak. (A) Sequence alignment of Bim BH3, Puma BH3, Noxa BH3, and Casp8p41. Multiple molecular dynamics simulations revealed that the three BH3 domains form an α-helix in the Bak BH3-binding groove. Key interactions of the three BH3 domains with the Bak groove involving two hydrophobic residues are highlighted in green (e.g., L141Puma and L148Puma) and one anionic residue in red (e.g., D146Puma). Residues of the latent Bak activator domain spatially approximate these critical BH3 domain residues. (B) Close-up view of the multiple molecular dynamics simulation-refined model of the activator domain of Casp8p41 (green) binding the Bak BH3-binding groove. (C) Immobilized GST-Casp8p41 was exposed to 125 nM His6-BakΔTM or His6-BakΔTM R127E. Representative of n = 3. (D) SPR analysis of GST fused to Casp8p41, Casp8p41 V150A/L157A, or Casp8p41 V150E/L157K (400 nM) binding to immobilized His6-BakΔTM. Representative of n = 3. (E) Summarized results from three independent experiments (**, P < 0.01). (F) Peptides used in G and H. (G) Immobilized His6-BakΔTM was exposed to 400 nM Casp8p41 latent Bak activator peptide, Bim BH3 peptide, or Casp8p41 N-terminal peptide. Representative of n = 3. (H) Summarized results from three independent experiments (***, P < 0.001). (I) Pull-downs from 293T cells transfected with empty vector, HA-Casp8p41, HA-Casp8p41 V150A/L157A, or HA-Casp8p41 V150E/L157K along with S peptide–Bak (left) or S peptide–empty vector (right). Representative of n = 4. Error bars indicate SD.
Further simulations predicted that Val150 and Leu157 in the latent Bak activator domain of Casp8p41 (Fig. 2, A and B) dock in two previously described hydrophobic holes in the Bak BH3 binding groove (Dai et al., 2011; Pang et al., 2012). To test this model, we mutated residues in Bak or Casp8p41 predicted to be critical for the interaction. When Bak Arg127, a conserved residue in the BH3-binding grooves of all Bcl-2 family members (Dai et al., 2011), was mutated to Glu, Casp8p41 binding diminished markedly (Fig. 2 C), which implicates the Bak BH3-binding groove in Casp8p41 binding. Likewise, Casp8p41 V150E/L157K bound Bak much less tightly than wild type (wt) Casp8p41 (Kd 400 ± 80 nM vs. 19 ± 3 nM; Fig. 2, D and E; and Fig. S2 C), highlighting the importance of the latent Bak activator domain in this binding. In contrast, a more conservative Casp8p41 V150A/L157A mutant, which was predicted by the simulations to maintain affinity for Bak, bound indistinguishably from the wt construct (Kd 22 ± 5 nM vs. 19 ± 3 nM; Fig. 2, D and E; and Fig. S2 D).
To further confirm that the latent Bak activator domain is responsible for Bak binding, a synthetic peptide consisting of amino acids 130–160 of procaspase 8 (Bak activator peptide, Fig. 2 F) was also examined. Binding of the synthetic Bak activator peptide to immobilized Bak (Fig. 2, G and H) was similar to that of full-length Casp8p41 (Fig. 1, B and C) and tighter than binding of synthetic Bim BH3 peptide (Fig. 2, G and H). In contrast, a control peptide from the Casp8p41 N-terminal domain (Fig. 2 F) failed to bind Bak at all (Fig. 2, G and H).
To assess the ability of Casp8p41 and Bak to interact in intact cells, Bak and wt or mutant Casp8p41 constructs were expressed in HEK 293T cells. Despite the transient nature of BH3 peptide–Bak interactions (Dai et al., 2011), small amounts of Bak could be pulled down with Casp8p41 and the V150A/L157A mutant but not the Casp8p41 V150E/L157K mutant (Fig. 2 I).
Collectively, the results in Fig. 2 identify Casp8p41 residues 130–160 as a peptide that binds the BH3-binding groove of Bak in vitro and in intact cells.
Casp8p41 binding to Bak causes Bak activation
Based on recent studies showing that binding to activator BH3-only proteins such as Bim and truncated Bid induces Bak oligomerization and membrane permeabilization (Dai et al., 2011; Moldoveanu et al., 2013), we assessed whether Casp8p41 activates Bak. When His6-Bak was incubated with liposomes composed of MOM lipids, cross-linked with bismaleimidohexane, subjected to SDS-PAGE, and blotted for Bak, Casp8p41 induced Bak oligomerization, as indicated by readily detectable Bak dimers, whereas Casp8p41 V150E/L157K induced less dimerization (Fig. 3 A). Size exclusion chromatography demonstrated that Casp8p41 also induced higher-order Bak oligomers (Fig. 3 B).
Figure 3.

Casp8p41 facilitates Bak oligomerization and Bak-mediated MOMP. (A) Dextran-FITC–loaded liposomes composed of MOM lipids were incubated with 50 nM His6-BakΔTM in the absence or presence of 200 nM GST, GST-Casp8p41, or GST-Casp8p41 V150E/L157K at 37°C for 30 min. Bim BH3 peptide served as a positive control. After bismaleimidohexane cross-linking, samples were subjected to SDS-PAGE and blotted for Bak. (B) After His6-BakΔTM was incubated with GST or GST-Casp8p41 in buffer containing 1% (wt/vol) CHAPS, fractions from size exclusion chromatography were blotted for Bak and GST. Representative of n = 3. (C) FITC-dextran release over time from FITC-dextran–loaded liposomes treated as in A. Representative of n = 3. (D) Summarized results from three independent experiments at 300 s of FITC-dextran release from liposomes. (E) FITC-dextran release from liposomes treated with 50 nM His6-BakΔTM in the presence of 200 nM GST-Casp8p41, GST-Casp8p41 V150E/L157K (EK), or GST. 100% release in D and E was determined by treating liposomes with Triton X-100. Error bars indicate ±1 SD from three independent experiments (*, P < 0.05; ***, P < 0.001). (F–J) After mitochondria (F) from wt (G), Bax−/−Bak−/− double knockout (H), Bax−/− (I), or Bak−/− (J) MEFs were incubated with 200 (1), 500 (2), or 1,000 nM (3) of purified protein for 1 h, sedimented, and washed, the supernatants and pellets were blotted for cytochrome c (Cyto C) and, as a control, the mitochondrial matrix protein Hsp60. Bim BH3 (200 nM), which induces CytoC release in the presence of either Bak or Bax, served as a positive control. Representative of n = 3.
When release of FITC-labeled dextran from liposomes composed of MOM lipids was assessed (Kuwana et al., 2005; Dai et al., 2011), Casp8p41 had no effect on these liposomes by itself but nonetheless increased Bak-mediated release (Fig. 3, C and D). In contrast, Casp8p41 V150E/L157K, which exhibits diminished Bak binding (Fig. 2, D and E) and oligomerization (Fig. 3 A), induced no additional FITC-dextran release above that seen with GST alone (Fig. 3 E). Likewise, Casp8p41 increased the ability of Bak to release cytochrome c from mitochondria, whereas Casp8p41 V150E/L157K did not (Fig. 3, G and I). Importantly, this MOMP depended on the presence of Bak in mitochondria and was not observed in mitochondria from Bak−/− or Bax−/−Bak−/− cells (Fig. 3, H and J).
In additional experiments, we tracked Bak activation using a well-described conformation-sensitive antibody. Like Jurkat cells treated with agonistic anti-Fas antibody (Fig. 4 A), cells transfected with EGFP-Casp8p41 exhibited increased binding of this antibody (Fig. 4 B). Casp8p41-positive peripheral blood mononuclear cells from viremic HIV-infected patients also displayed increased binding of the activation-sensitive anti-Bak antibody relative to Casp8p41-negative cells from the same patients (Fig. 4, C and D).
Figure 4.

Casp8p41 expression is accompanied by Bak activation in vitro and in HIV-infected patients. (A) After Jurkat T cells were treated for 6 h with agonistic CH.11 anti-Fas antibody (solid line) or IgM control (broken line), the binding of an antibody that recognizes an active conformation of Bak was assessed by flow cytometry. The shaded area represents isotype control. Representative of n = 5. (B) 6 h after Jurkat cells transfected with EGFP (broken line) or EGFP-Casp8p41 (solid line), the binding of an antibody that recognizes an active conformation of Bak was assessed in GFP-positive cells by flow cytometry. The shaded area represents isotype control. Representative of n = 3. (C) Peripheral blood mononuclear cells from viremic, HIV-infected patients were fixed, permeabilized, and stained simultaneously with murine monoclonal antibody that specifically recognizes Casp8p41 and murine monoclonal antibody that recognizes active Bak. Results using isotype control (left) and anti-Casp8p41 (middle) are shown. Bak expression was compared between Casp8p41− and Casp8p41+ cells (right). Representative of n = 5. (D) Mean fluorescence intensity of Bak in Casp8p41− and Casp8p41+ PBMCs from five HIV-infected patients was compared.
Collectively, these results indicate that Casp8p41 induces Bak oligomerization and enhances Bak-mediated MOMP, leading to cytochrome c release in vitro, whereas Casp8p41 V150E/L157K is impaired in these functions. Moreover, the presence of Casp8p41 tracks with the activated Bak conformation in vitro and in vivo.
Mutations in the latent Casp8p41 Bak activator domain impair killing by HIV
If these observations are pertinent to Casp8p41-mediated killing, the V150E/L157K mutation should diminish apoptosis, whereas the V150A/L157A mutation with preserved Bak binding should not. To test these predictions, we initially transfected plasmids encoding wt EGFP-Casp8p41, the V150A/L157A mutant, or the V150E/L157K mutant into Jurkat T cells and assessed death by annexin V binding 6 h later. Wt Casp8p41 killed 36% of the cells in which it was expressed, and Casp8p41 V150E/L157K killed fewer cells (17%, P < 0.05 vs. wt), whereas Casp8p41 V150A/L157A induced killing similar to wt (28%, P > 0.05 vs. wt; Fig. 5, A and C). Similar results were observed at different time points (Fig. S3 A) and when death was assessed by TUNEL (Fig. 5, B and D). Despite these differences in killing, all Casp8p41 variants trafficked to mitochondria normally (Fig. S3, B and C).
Figure 5.

Mutations in the latent Bak activator domain impair killing by Casp8p41 or HIV. (A and B) Flow cytometry data from Jurkat T cells transfected with empty vector or EGFP fused to Casp8p41, Casp8p41 V150A/L157A, Casp8p41 V150E/L157K, or procaspase 8 (Casp8) and stained 6 h later with Annexin V (A) or TUNEL (B). Numbers on the top left indicate percentages of EGFP+ cells that are Annexin or TUNEL positive. Representative of n = 3. (C and D) Pooled data from three independent experiments measuring Annexin V (C) or TUNEL (D) positivity in EGFP+ cells. Error bars indicate ±1 SD. (E) I9.2 cells transfected with EGFP, EGFP–procaspase 8, or EGFP–procaspase 8 C360S were infected with VSV-G–HIV-1, and the EGFP-positive cells were monitored for viability over time. (F) I9.2 cells reconstituted with EGFP fused to procaspase 8, procaspase 8 V150E/L157K, or procaspase 8 V150A/L157A were infected with VSV-G–HIV-1, and the EGFP-positive cells were monitored for cell viability over time. *, P < 0.05; **, P < 0.01 by linear regression. Error bars indicate SD.
In further experiments, procaspase 8–deficient Jurkat I9.2 cells were transfected with empty vector or plasmids encoding EGFP fused to various procaspase 8 constructs and subsequently infected with vesicular stomatitis virus (VSV) pseudotyped HIV-1 reporter virus (HXB, Δvpu, Δvpr). Viral replication in the various transfected (EGFP+) cells was similar (Fig. S3, E–G). When viability of the transfected (EGFP+) cells was assessed on day 3 after HIV infection, procaspase 8–deficient cells sustained negligible death (82% viable), whereas cells expressing EGFP fused to wt procaspase 8 or the C360S (catalytically inactive) mutant were killed equally (35% and 38% viable, respectively; Fig. 5 E), which confirms that procaspase 8 is required for HIV-induced killing but that caspase 8 catalytic activity is not (Algeciras-Schimnich et al., 2007; Nie et al., 2008).
Consistent with the protein binding data, HIV-induced killing was diminished in cells expressing EGFP–procaspase 8 V150E/L157K compared with the wt construct (61% vs. 35% viable on day 3, P = 0.005; Fig. 5 F). Not surprisingly, HIV-induced killing was not completely eliminated by the V150E/L157K mutation, as other HIV-encoded proteins known to induce cell death such as Tat and nef are still expressed, and infected cell killing by activation of the DNA damage response or accumulation of reverse transcripts can still occur (Doitsh et al., 2010; Cooper et al., 2013). Nonetheless, the decrease in cell killing when HIV-producing cells express EGFP–procaspase 8 V150E/L157K at levels similar to wt EGFP–procaspase 8 (Fig. S3 D) demonstrates that HIV-induced death depends substantially upon the ability to generate Casp8p41 that can bind the Bak BH3-binding groove. Consistent with this conclusion, EGFP–procaspase 8 V150A/L157A, which generates Casp8p41 V150A/L157A that binds Bak almost as strongly as wt Casp8p41 (Fig. 2), supports death of infected cells at a rate that approaches that of cells expressing wt procaspase 8 (Fig. 5 F, 48% vs. 35% viable, P = 0.09).
Collectively, these observations suggest a novel and unexpected mechanism by which Casp8p41, a catalytically inactive caspase fragment, activates the mitochondrial apoptotic pathway. In particular, our results show for the first time that Casp8p41 interacts with Bak to induce Bak oligomerization, Bak-induced MOMP, and Bak-mediated T cell killing (Figs. 1, 3, and 5). Further experiments have traced the Casp8p41–Bak interaction to a domain in Casp8p41 that is unmasked when the C-terminal end of procaspase 8 is removed by HIV protease (Fig. 2). Importantly, a synthetic peptide containing this activator domain binds to Bak (Fig. 2, G and H) as tightly as Casp8p41 and >100-fold more tightly than full-length procaspase 8 (Fig. 1 B and Fig. S2). Moreover, the latent Bak activator domain in Casp8p41, like the BH3 domain of Bim, binds the BH3-binding groove of Bak (Fig. 2 G) to trigger subsequent events. Further, charge-altering mutation of two critical residues in the activator domain markedly diminishes binding of Casp8p41 to Bak ex vivo (Fig. 2, D and E) as well as T cell killing by Casp8p41 (Fig. 5, A–D) or HIV (Fig. 5 F), which confirms the importance of the Casp8p41-induced Bak activation mechanism described here.
These results have important implications for understanding HIV-induced T cell death. Although HIV-1 can undoubtedly kill both infected and bystander CD4 T cells via multiple mechanisms, our observation that killing of productively infected cells is diminished by critical mutations in the Casp8p41 activator domain (Fig. 5 F) suggests that Casp8p41 plays a prominent role in the death of cells productively infected with HIV. These results also provide an explanation for the clinical observation that impaired Casp8p41 production is associated with smaller declines in CD4+ cell counts (Natesampillai et al., 2010).
The present observation that a domain of a non–Bcl-2 family member generated or unmasked during cell signaling can serve as a direct activator to trigger mitochondrial apoptosis also provides a new paradigm in apoptotic regulation. Previous studies of Bax and Bak activation have focused exclusively on the role of BH3-only proteins (Cheng et al., 2001; Letai et al., 2002; Walensky et al., 2006; Dai et al., 2011; Llambi et al., 2011; Czabotar et al., 2013; Moldoveanu et al., 2013). Our observation that proapoptotic Bak can be directly activated by Casp8p41 raises a question of whether similar unmasking of latent activator domains in other proteins is used by additional viruses or other cell death stimuli as well.
Materials and methods
Plasmids and peptides
Casp8p41 and procaspase 8 were cloned into pEGFP-C1, pcDNA3-HA, or pGEX (Nie et al., 2008). BakΔTM (aa 1–186) was cloned into pGEX-4T-1 or pSPN (Dai et al., 2011). Mutations introduced using site-directed mutagenesis (Agilent Technologies) were confirmed by sequencing. The Casp8p41 activator peptide (DMNLLDIFIEMEKRVILGEGKLDILKRVCAQ), Casp8p41 N-terminal control peptide (MDFSRNLYDIGEQLDSEDLASLK), and Bim BH3 peptide (RPEIWIAQELRRIGDEFNAYYARRVF; Dai et al., 2011) were synthesized in the Mayo Clinic Proteomics Research Center.
Cell culture
Jurkat cells, the Jurkat variant I9.2 lacking procaspase 8, and 293T cells were obtained from American Type Culture Collection and cultured as instructed by the supplier. Fibroblasts from wt, Bax−/−, Bak−/−, and Bax−/−/Bak−/− mouse embryos were obtained from Z. Dong (Mayo Clinic, Augusta, GA). The Bak−/− mice were generated by deletion of exons 3–6 of the murine Bak, and the Bax−/− mice were generated by deletion of exons 2–4 and part of 5 of the murine Bax gene (Wei et al., 2001). In both cases, neomycin cassettes were inserted by homologous recombination.
Transfections and infections
Jurkat and I9.2 cells were transfected with 2 µg of DNA per 105 cells using a square wave electroporator (BXT) at 320 V or 40 µg of DNA per 107 cells at 240 V for 10 ms. HIV-1 infections were performed with a single-cycle reporter virus, HIV-1, which is vpu, vpr negative. HIV-1 was prepared in 293T cells by cotransfection with a VSV glycoprotein G (VSV-G) expression plasmid (3 µg of HIV-1 plasmid and 1 µg of VSV-G plasmid). I9.2 cells were infected with 30,000 ng of p24 (measured by p24 ELISA) per 106 cells for 3 h. Cells were then washed three times and incubated in fresh medium.
Lentiviral shRNA library screening
293T cells (106) were infected with a library containing 27,000 shRNAs in a lentiviral backbone encoding RFP (Cellecta) at an MOI of 0.3–0.4. After 48 h, cells were selected with 1 µg/ml puromycin for 2 d. Surviving cells were transfected with EGFP-Casp8p41 using Lipofectamine 2000. Beginning 48 h later, RFP/EGFP double-positive cells were collected by cell sorting and analyzed by Cellecta for the shRNA insert barcodes to enumerate shRNAs present.
Flow cytometry
Cell death was measured using Annexin V (BD), LIVE/DEAD fixable aqua dead cell stain (Invitrogen), or TUNEL (Roche) and analyzed on a FACScan flow cytometer (BD). All gating was set first to control samples and then applied to experimental samples.
Microscopy
For fluorescence microscopy, 293T cells were transfected overnight with GFP, GFP-Casp8p41, GFP-Casp8p41 V150A/L157A, or GFP-Casp8p41 V150E/L157K. Cells were then fixed with 2% paraformaldehyde for 1 h at room temperature and then permeabilized with PBS containing 0.1% Nonidet P-40 and 5% bovine serum albumin. Cells were first stained with Bak (Clone TC-100; EMD Millipore) antibody or mouse IgG control (BD), washed three time with PBS, and stained with a goat anti–mouse PE secondary antibody (BD) before being washed three times with PBS. Slides were prepared with DAPI containing Vectashield (Vector Laboratories). Laser scanning confocal microscopy was performed using a confocal laser scanning microscope (LSM 510; Carl Zeiss). Images were taken at the Mayo Microscopy and Cell Analysis Core facility.
Images were analyzed with KS400 Image Analysis Software (Carl Zeiss). The threshold value was determined and separately set for green and red fluorescence channels. The same threshold values were used for all subsequent images. Areas of green, red, and colocalized pixels were measured and data were expressed as a total percent area of green that colocalized with red.
Immunoprecipitation and immunoblotting
293T cells transfected with HA-empty vector, HA-Casp8p41, HA-Casp8p41 V150E/L157K, or HA-Casp8p41 V150A/L157A and pSPN-empty vector or pSPN-Bak expression vector for 24 h were collected, washed, and lysed. Cells were lysed in 100 µl of lysis buffer (20 mM Tris/HCl, pH 7.5, 150 mM NaCl, 0.1% Triton X-100, 2 µg/ml aprotinin, 10 µg/ml leupeptin, 2 µg/ml pepstatin, and 1 mM PMSF) for 10 min on ice. Cells were then centrifuged at 400 g, for 5 min, at 4°C. For cell fractionation, the lysate was then further centrifuged at 15,000 g for 5 min at 4°C, resulting in a mitochondrial enriched pellet and cytosolic supernatant. Aliquots containing 900 µg of protein were incubated with 12 µl of anti-HA affinity matrix (Roche) or S-protein agarose (EMD Millipore) for 4 h at 4°C. After beads were washed twice with 150 µl TBST, bound protein was eluted. Samples were run on SDS-polyacrylamide gels and transferred to PVDF membranes for 2 h at 1,200 mAmps in transfer buffer (24 mM Tris and 192 mM glycine). The membranes were then blocked in TBST (20 mM Tris, 150 mM NaCl, and 0.05% Tween-20, pH 7.4) containing 2% bovine serum albumin (Sigma-Aldrich) for >1 h at 21°C or overnight at 4°C followed by immunoblotting, without stripping between blots. Primary antibodies used were: anti-HA peroxidase high-affinity 3F10 (rat; Roche) or anti–S-Peptide HRP (mouse; EMD Millipore).
Protein expression and purification
Plasmids for GST- and His6-tagged proteins were transformed into Escherichia coli BL21 or DH5α by heat shock, grown to an optical density of 0.8, and induced with 1 mM IPTG for 24 h at 16°C or 3 h at 37°C. Bacteria were frozen and thawed on ice; suspended in calcium- and magnesium-free Dulbecco’s PBS containing 0.1% Triton X-100, 2 µg/ml aprotinin, 10 µg/ml leupeptin, 2 µg/ml pepstatin, and 1 mM PMSF (GST-tagged proteins) or 150 mM NaCl containing 10 mM Tris-HCl, pH 7.4, (TS buffer) and 1 mM PMSF (His6-tagged proteins); and sonicated three times for 15 s/min on ice. His6-tagged proteins were purified using Ni2+-NTA-agarose (EMD Millipore). Alternatively, GST-tagged proteins were purified with glutathione-agarose (Thermo Fisher Scientific).
SPR
All proteins for SPR were further purified by fast protein liquid chromatography (FPLC) on Superdex S200, concentrated in a centrifugal concentrator (Centricon; EMD Millipore), dialyzed against Biacore buffer (10 mM Hepes, pH 7.4, 150 mM NaCl, 0.05 mM EDTA, and 0.005% [wt/vol] Polysorbate 20), and stored at 4°C for <48 h before use. Binding assays were performed at 25°C on a Biacore 3000 biosensor using GST-Casp8p41 or His6-BakΔTM immobilized on a CM5 chip (GE Healthcare). Ligands were injected at 30 µl/min for 1 min in Biacore buffer. Bound protein was allowed to dissociate in Biacore buffer at 30 µl/min for 10 min and then desorbed with 2 M MgCl2. Binding kinetics were derived using BIA evaluation software (Biacore; GE Healthcare).
Preparation of FITC-Dextran lipid vesicles
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 1-plamitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine, l-α-phosphatidylinositol, cholesterol, cardiolipin, and 18:1 DGS-NTA(Ni) at a weight ratio of 36:22:9:8:20:5 were dried as thin films in glass test tubes under nitrogen and then under vacuum for 16 h. 50 mg of FITC-labeled dextran 10 (F-d10; Molecular Probes) was encapsulated in lipid in 1 ml of 20 mM Hepes, 150 KCl, pH 7.0, by sonication followed by extrusion 15 times through a 100-nm polycarbonate membrane. Gel filtration on Sephacryl S-300 HR (GE Healthcare) was used to remove untrapped F-d10. Phosphate was determined by colorimetric assay (Abcam).
Liposome release assay
Release of F-d10 from large unilamellar vesicles (LUVs) was monitored by fluorescence dequenching using a fluorimetric plate reader. Purified His6-Bak with or without other proteins was added to LUVs (final lipid concentration, 10 µg/ml) in 96 well plates, which were then incubated at 37°C and monitored (excitation, 485 nm; emission, 538 nm) every 10 s. The equation [(Fsample − Fblank)/(FTriton − Fblank) × 100] was used to calculate F-d10 release, where Fsample, Fblank, and FTriton are fluorescence of reagent-, buffer-, and 1% Triton X-100–treated LUVs.
Analytical gel filtration
Purified His6-Bak and Casp8p41 proteins were mixed in CHAPS buffer (1% CHAPS, 1% glycerol, 150 mM NaCl, 5 mM DTT, and 20 mM Hepes, pH 7.5) at 23°C for 1 h. 200-µl samples were subjected to FPLC at 4°C on a Superdex S200 (GE Healthcare). Fractions (500 µl) were analyzed by SDS-PAGE followed by immunoblotting. Molecular markers (Sigma-Aldrich) in CHAPS buffer were run through the same column.
Cytochrome c release
Purified GST, GST-Casp8p41, or GST-Casp8p41 EK were dialyzed against mitochondria buffer (150 mM KCl, 5 mM MgCl2, 1 mM EGTA, and 25 mM Hepes, pH 7.5). Mitochondria purified from wt, Bak−/−, Bax−/−, or Bak−/−Bax−/− MEFs were incubated with the indicated proteins at 23°C for 1 h. After centrifugation (10,000 g for 15 min), supernatants and pellets were analyzed by immunoblotting.
Clinical HIV samples
Blood was obtained from five HIV-infected, viremic patients according to a Mayo Clinic Institutional Review Board approved protocol. After patients signed informed consent, peripheral blood mononuclear cells were isolated by Ficoll gradient, fixed with 4% paraformaldehyde, permeabilized with PBS containing 0.1% Nonidet P-40 and 5% bovine serum albumin, and stained with a monoclonal antibody specific for Casp8p41 directly conjugated with Mix-n-Stain CF 640R (Sigma-Aldrich) overnight at 4°C. Cells were costained for the final 1 h with a monoclonal antibody specific for the active conformation of Bak (Clone TC-100; EMD Millipore) directly conjugated to Mix-n-Stain CF 532 (Sigma-Aldrich) at 4°C, then washed twice and fixed. Single color compensation controls and isotype controls were used for gating. Flow cytometry was performed on a flow cytometer (LSR II; BD) and data were analyzed with FlowJo Software (Tree Star, Inc.).
Active Bak expression was compared between Casp8p41-negative and Casp8p41-positive patient cells. Mean fluorescence intensity of Casp8p41-negative and Casp8p41-positive PBMCs were compared with a paired t test.
Statistics
One-way analysis of variance (ANOVA) followed by Tukey-Kramer multiple comparison tests was used for single time point samples in experiments containing more than two variables. For experiments in which aliquots were taken from the same sample over multiple days, linear regression models were used. P-values for linear regression used the F test to compare the elevations of the best-fit lines using the software GraphPad Prism. All but one of the experiments have been repeated at least three times independently.
Computational model of Bak in complex with the Casp8p41 activator domain
The starting structure of the complex was generated by manually docking the activator domain (residues 142–162) in the α-helical conformation into the vacated BH3-binding groove of Bak (residues 21–183) that was taken from the reported Noxa–Bak complex (Dai et al., 2011; Pang et al., 2012). This manual docking placed V150Casp8p41 in the proximity of I114Bak and L118Bak, and L157Casp8p41 close to V129Bak and I85Bak. All His, Glu, Asp, Arg, and Lys residues were treated as HIP, GLU, ASP, ARG, and LYS, respectively. The topology and coordinate files of the docking-generated Casp8p41–Bak complex were generated by the PREP, LINK, EDIT, and PARM modules of the AMBER 5.0 program (Pearlman et al., 1995). The complex was refined by energy minimization using the SANDER module of the AMBER 5.0 program with a dielectric constant of 1.0 and 500 cycles of steepest-descent minimization followed by 10,000 cycles of conjugate-gradient minimization. The energy-minimized complex was solvated with 6,947 TIP3P water molecules (Jorgensen et al., 1983), leading to a system of 23,767 atoms. The water molecules were obtained from solvating the complex using a preequilibrated box of 216,000 TIP3P molecules, whose hydrogen atom charge was set to 0.4170, where any water molecule was removed if it had an oxygen atom closer than 2.2 Å to any solute atom or a hydrogen atom closer than 2.0 Å to any solute atom, or if it was located further than 10.0 Å along the x, y, or z axis from any solute atom. The solvated complex system was energy-minimized for 100 cycles of steepest-descent minimization followed by 100 cycles of conjugate-gradient minimization to remove close van der Waals contacts in the system, then heated from 0 to 300 K at a rate of 10 K/ps under constant temperature and volume, and finally simulated independently with a unique seed number for initial velocities at 300 K under constant temperature and pressure using the PMEMD module of the AMBER 8.0 program (Case et al., 2005) with the AMBER force field (ff99SB; Hornak et al., 2006; Wickstrom et al., 2009). All simulations used (1) a dielectric constant of 1.0, (2) the Berendsen coupling algorithm (Berendsen et al., 1984), (3) a periodic boundary condition at a constant temperature of 300 K and a constant pressure of 1 atm with isotropic molecule-based scaling, (4) the Particle Mesh Ewald method to calculate long-range electrostatic interactions (Darden et al., 1993), (5) a time step of 1.0 fs, (6) the SHAKE-bond-length constraints applied to all the bonds involving the H atom, (7) saving the image closest to the middle of the “primary box” to the restart and trajectory files, (8) a formatted restart file, and (9) default values of all other inputs of the PMEMD module. 57 different simulations (each lasting 10 ns) were performed on a cluster of Apple X servers with 590 G5 processors (2.2/2.3 GHz).
Online supplemental material
Fig. S1 shows results of the shRNA library screen. Fig. S2 shows binding isotherms of paired proteins analyzed sequentially on the same SPR chip. Fig. S3 shows equal infectivity of HIV in the various EGFP–procaspase 8 cell lines. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201405051/DC1.
Supplementary Material
Acknowledgments
We thank Greg Gores for helpful suggestions.
This work was supported by grants R56 AI102959 and RO1 AI110173 (to A.D. Badley), grant R01 CA166741 (to S.H. Kaufmann and Y.-P. Pang), and a predoctoral fellowship from the Mayo Foundation for Education and Research (to A.M. Sainski).
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- IAP
- inhibitor of apoptosis
- LUV
- large unilamellar vesicle
- MOM
- mitochondrial outer membrane
- MOMP
- MOM permeabilization
- SPR
- surface plasmon resonance
- VSV
- vesicular stomatitis virus
- VSV-G
- VSV glycoprotein G
- wt
- wild type
References
- Algeciras-Schimnich, A., Belzacq-Casagrande A.S., Bren G.D., Nie Z., Taylor J.A., Rizza S.A., Brenner C., and Badley A.D.. 2007. Analysis of HIV protease killing through caspase 8 reveals a novel interaction between caspase 8 and mitochondria. Open Virol. J. 1:39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, E.Z., Bebernitz G.A., and Gluzman Y.. 1990. Isolation of mutants of human immunodeficiency virus protease based on the toxicity of the enzyme in Escherichia coli. Proc. Natl. Acad. Sci. USA. 87:5573–5577 10.1073/pnas.87.14.5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen, H.J.C., Postma J.P.M., van Gunsteren W.F., Di Nola A., and Haak J.R.. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690 10.1063/1.448118 [DOI] [Google Scholar]
- Blanco, R., Carrasco L., and Ventoso I.. 2003. Cell killing by HIV-1 protease. J. Biol. Chem. 278:1086–1093 10.1074/jbc.M205636200 [DOI] [PubMed] [Google Scholar]
- Case, D.A., Cheatham T.E. III, Darden T., Gohlke H., Luo R., Merz K.M. Jr, Onufriev A., Simmerling C., Wang B., and Woods R.J.. 2005. The Amber biomolecular simulation programs. J. Comput. Chem. 26:1668–1688 10.1002/jcc.20290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, E.H., Wei M.C., Weiler S., Flavell R.A., Mak T.W., Lindsten T., and Korsmeyer S.J.. 2001. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell. 8:705–711 10.1016/S1097-2765(01)00320-3 [DOI] [PubMed] [Google Scholar]
- Cooper, A., García M., Petrovas C., Yamamoto T., Koup R.A., and Nabel G.J.. 2013. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature. 498:376–379 10.1038/nature12274 [DOI] [PubMed] [Google Scholar]
- Czabotar, P.E., Westphal D., Dewson G., Ma S., Hockings C., Fairlie W.D., Lee E.F., Yao S., Robin A.Y., Smith B.J., et al. 2013. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 152:519–531 10.1016/j.cell.2012.12.031 [DOI] [PubMed] [Google Scholar]
- Dai, H., Smith A., Meng X.W., Schneider P.A., Pang Y.-P., and Kaufmann S.H.. 2011. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J. Cell Biol. 194:39–48 10.1083/jcb.201102027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden, T., York D., and Pedersen L.. 1993. Particle mesh Ewald: an N log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092 10.1063/1.464397 [DOI] [Google Scholar]
- Doitsh, G., Cavrois M., Lassen K.G., Zepeda O., Yang Z., Santiago M.L., Hebbeler A.M., and Greene W.C.. 2010. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell. 143:789–801 10.1016/j.cell.2010.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw, W.C., Martins L.M., and Kaufmann S.H.. 1999. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68:383–424 10.1146/annurev.biochem.68.1.383 [DOI] [PubMed] [Google Scholar]
- Fulda, S., and Vucic D.. 2012. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 11:109–124 10.1038/nrd3627 [DOI] [PubMed] [Google Scholar]
- Galluzzi, L., Brenner C., Morselli E., Touat Z., and Kroemer G.. 2008. Viral control of mitochondrial apoptosis. PLoS Pathog. 4:e1000018 10.1371/journal.ppat.1000018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis, E., Reyna D.E., Davis M.L., Bird G.H., and Walensky L.D.. 2010. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol. Cell. 40:481–492 10.1016/j.molcel.2010.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak, V., Abel R., Okur A., Strockbine B., Roitberg A., and Simmerling C.. 2006. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 65:712–725 10.1002/prot.21123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, W.L., Chandreskhar J., Madura J.D., Impey R.W., and Klein M.L.. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935 10.1063/1.445869 [DOI] [Google Scholar]
- Kaplan, A.H., Manchester M., and Swanstrom R.. 1994. The activity of the protease of human immunodeficiency virus type 1 is initiated at the membrane of infected cells before the release of viral proteins and is required for release to occur with maximum efficiency. J. Virol. 68:6782–6786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H., Tu H.C., Ren D., Takeuchi O., Jeffers J.R., Zambetti G.P., Hsieh J.J., and Cheng E.H.. 2009. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell. 36:487–499 10.1016/j.molcel.2009.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana, T., Bouchier-Hayes L., Chipuk J.E., Bonzon C., Sullivan B.A., Green D.R., and Newmeyer D.D.. 2005. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell. 17:525–535 10.1016/j.molcel.2005.02.003 [DOI] [PubMed] [Google Scholar]
- Letai, A., Bassik M.C., Walensky L.D., Sorcinelli M.D., Weiler S., and Korsmeyer S.J.. 2002. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2:183–192 10.1016/S1535-6108(02)00127-7 [DOI] [PubMed] [Google Scholar]
- Llambi, F., Moldoveanu T., Tait S.W., Bouchier-Hayes L., Temirov J., McCormick L.L., Dillon C.P., and Green D.R.. 2011. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell. 44:517–531 10.1016/j.molcel.2011.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, L.K.1999. An exegesis of IAPs: salvation and surprises from BIR motifs. Trends Cell Biol. 9:323–328 10.1016/S0962-8924(99)01609-8 [DOI] [PubMed] [Google Scholar]
- Moldoveanu, T., Grace C.R., Llambi F., Nourse A., Fitzgerald P., Gehring K., Kriwacki R.W., and Green D.R.. 2013. BID-induced structural changes in BAK promote apoptosis. Nat. Struct. Mol. Biol. 20:589–597 10.1038/nsmb.2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natesampillai, S., Nie Z., Cummins N.W., Jochmans D., Bren G.D., Angel J.B., and Badley A.D.. 2010. Patients with discordant responses to antiretroviral therapy have impaired killing of HIV-infected T cells. PLoS Pathog. 6:e1001213 10.1371/journal.ppat.1001213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie, Z., Phenix B.N., Lum J.J., Alam A., Lynch D.H., Beckett B., Krammer P.H., Sekaly R.P., and Badley A.D.. 2002. HIV-1 protease processes procaspase 8 to cause mitochondrial release of cytochrome c, caspase cleavage, and nuclear fragmentation. Cell Death Differ. 9:1172–1184 10.1038/sj.cdd.4401094 [DOI] [PubMed] [Google Scholar]
- Nie, Z., Bren G.D., Rizza S.A., and Badley A.D.. 2008. HIV protease cleavage of procaspase 8 is necessary or death of HIV-infected cells. Open Virol. J. 2:1–7 10.2174/1874357900802010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, Y.-P., Dai H., Smith A., Meng X.W., Schneider P.A., and Kaufmann S.H.. 2012. Bak conformational changes induced by ligand binding: Insight into BH3 domain binding and Bak homo-oligomerization. Sci. Rep. 2:257 10.1038/srep00257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman, D.A., Case D.A., Caldwell J.W., Ross W.S., Cheatham T.E. III, Debolt S., Ferguson D., Seibel G., and Kollman P.A.. 1995. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91:1–41 10.1016/0010-4655(95)00041-D [DOI] [Google Scholar]
- Sainski, A.M., Natesampillai S., Cummins N.W., Bren G.D., Taylor J., Saenz D.T., Poeschla E.M., and Badley A.D.. 2011. The HIV-1-specific protein Casp8p41 induces death of infected cells through Bax/Bak. J. Virol. 85:7965–7975 10.1128/JVI.02515-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser, A., Cory S., and Adams J.M.. 2011. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 30:3667–3683 10.1038/emboj.2011.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, R.C., Cullen S.P., and Martin S.J.. 2008. Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 9:231–241 10.1038/nrm2312 [DOI] [PubMed] [Google Scholar]
- Ventoso, I., Blanco R., Perales C., and Carrasco L.. 2001. HIV-1 protease cleaves eukaryotic initiation factor 4G and inhibits cap-dependent translation. Proc. Natl. Acad. Sci. USA. 98:12966–12971 10.1073/pnas.231343498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky, L.D., Pitter K., Morash J., Oh K.J., Barbuto S., Fisher J., Smith E., Verdine G.L., and Korsmeyer S.J.. 2006. A stapled BID BH3 helix directly binds and activates BAX. Mol. Cell. 24:199–210 10.1016/j.molcel.2006.08.020 [DOI] [PubMed] [Google Scholar]
- Wei, M.C., Zong W.X., Cheng E.H., Lindsten T., Panoutsakopoulou V., Ross A.J., Roth K.A., MacGregor G.R., Thompson C.B., and Korsmeyer S.J.. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 292:727–730 10.1126/science.1059108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstrom, L., Okur A., and Simmerling C.. 2009. Evaluating the performance of the ff99SB force field based on NMR scalar coupling data. Biophys. J. 97:853–856 10.1016/j.bpj.2009.04.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, S., and Zhang Y.. 2008. MUSTER: Improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins. 72:547–556 10.1002/prot.21945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J.W., Jeffrey P.D., and Shi Y.. 2009. Mechanism of procaspase-8 activation by c-FLIPL. Proc. Natl. Acad. Sci. USA. 106:8169–8174 10.1073/pnas.0812453106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.