

Abstract
Wound healing is a complex multi-step process that requires spatial and temporal orchestration of cellular and non-cellular components. Hypoxia is one of the prominent microenvironmental factors in tissue injury and wound healing. Hypoxic responses, mainly mediated by a master transcription factor of oxygen homeostasis, hypoxia-inducible factor-1 (HIF-1), have been shown to be critically involved in virtually all processes of wound healing and remodeling. Yet, mechanisms underlying hypoxic regulation of wound healing are still poorly understood. Better understanding of how the wound healing process is regulated by the hypoxic microenvironment and HIF-1 signaling pathway will provide insight into the development of a novel therapeutic strategy for impaired wound healing conditions such as diabetic wound and fibrosis. In this review, we will discuss recent studies illuminating the roles of HIF-1 in physiologic and pathologic wound repair and further, the therapeutic potentials of HIF-1 stabilization or inhibition.
Keywords: fibrosis, hypoxia, hypoxia-inducible factor-1, oxygen, wound healing
INTRODUCTION
Oxygen (O2) is essential for all aerobic organisms to produce energy via mitochondrial oxidative respiration as well as to perform other critical biological processes (Koch and Britton, 2008; Taylor and McElwain, 2010). Hypoxia is a condition in which tissues are inadequately oxygenated, and various physiological conditions, such as strenuous exercise or high altitudes, can result in inadequate O2 supply to support metabolic and energy requirements. Since depriving tissues of oxygen can adversely affect the function of these tissues, and consequently the cells of these tissues are significantly stressed and are unable to perform important biological processes, evolutionarily conserved adaptive processes (e.g. hypoxia-inducible factors, HIFs) are activated in order to match O2 supply with the metabolic and energy needs of the cells (Semenza, 2012; Weidemann and Johnson, 2008). Insufficient O2 supply in cells or tissues is also a prominent feature of a number of pathological conditions including cancer, obesity and ischemic diseases. Recently, it has been shown that wounds and tissue injuries cause the injured area to become hypoxic, presumably due to the disruption of vasculature and increased O2 consumption by cells within and bordering the injured tissue (Lokmic et al., 2012). Moreover, recent preclinical studies suggest that adaptation to hypoxic stress via HIF signaling plays a critical role in promoting wound healing processes (Andrikopoulou et al., 2011; Hong et al., 2014). Despite that molecular and cellular mechanisms underlying hypoxic regulation of wound healing are still poorly understood, HIF activation via the inhibition of oxygen sensor prolyl hydroxylase enzyme (PHD) has been intensively investigated with the aim to develop novel treatment regimens for wound healing and ischemic diseases (Myllyharju, 2009; 2013). Thus, it is of critical importance to improve our understanding of how hypoxic responses influence wound healing and tissue injury at the molecular level. In this review, we will focus on the mechanisms and processes regulated by hypoxia and HIF signaling in wound healing and tissue injury. We will further discuss how hypoxic responses impact pathologic wound healing (e.g. diabetic wounds) and fibrotic diseases in which wound healing processes become aberrant and fail. Finally, we will conclude with an overview of an exciting new treatment strategy that may prove effective for wound healing, tissue injury, and fibrosis.
HYPOXIA-INDUCIBLE FACTOR-1 (HIF-1)
In response to the low oxygen-tension in the tissue, cells attempt to restore oxygen homeostasis within the hypoxic microenvironment. A master regulator in the cellular hypoxic response is HIF-1 (Fig. 1). HIF-1 is a heterodimeric protein complex that is composed of an α and a β subunit, both of which belong to the basic helix-loop-helix Per/ARNT/Sim (bHLH-PAS) family of transcription factors and are constitutively expressed in cells (Wang et al., 1995). In contrast to HIF-1β proteins that are not regulated by the presence or absence of oxygen (Hoffman et al., 1991; Labrecque et al., 2013), HIF-1α proteins are regulated at the posttranscriptional level by cellular oxygen tension. In non-hypoxic conditions, HIF-1α proteins are rapidly degraded resulting in a half-life of less than 10 min (Berra et al., 2001). Proline residues of the oxygen-dependent degradation (ODD) domain of HIF-1α are hydroxylated in non-hypoxic conditions by PHDs (Ivan et al., 2001; Jaakkola et al., 2001). The hydroxylation of HIF-1α allows the von Hippel-Lindau protein (pVHL), an E3 ubiquitin ligase, to bind, polyubiquitinate, and target it for proteasomal degradation (Maxwell et al., 1999; Ohh et al., 2000). The lack of available oxygen for hydroxylation by PHDs in hypoxic conditions stabilizes HIF-1α, increasing cellular levels of the protein, and facilitating its translocation into the nucleus where it heterodimerizes with HIF-1β. The HIF-1 complex then binds to a 50-base pair cis-acting hypoxia response element (HRE) in the enhancer and promoter regions of genomic DNA, thereby activating HIF-1 target genes (Semenza and Wang, 1992; Wang and Semenza, 1993), which are involved in hypoxic adaptation and survival processes including anaerobic glycolysis, growth factor signaling, angiogenesis, cellular mobility, and erythropoiesis. When tissue is injured the regulation of key processes by HIF-1 enables the cell to efficiently adapt to the changing oxygen-tensions of the microenvironment and enables the dynamic capacity for the injured tissue to repair, remodel, and heal.
Fig. 1.
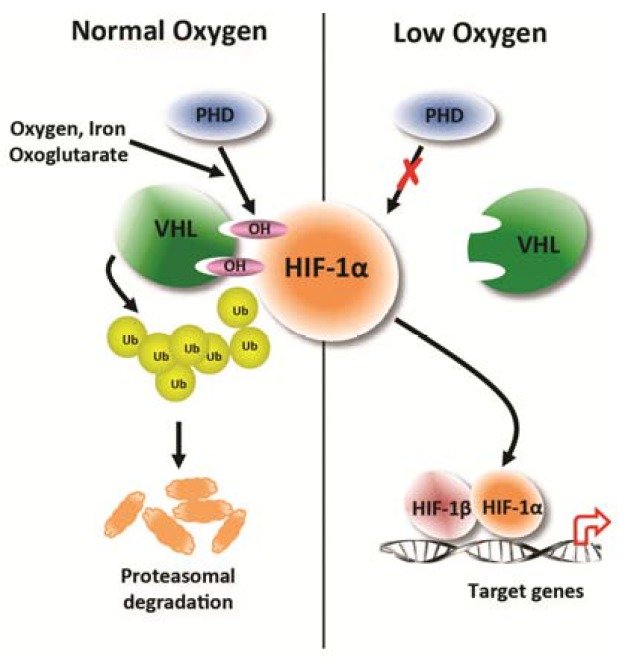
Post-transcriptional regulation of HIF-1α by cellular O2 level. In normal oxygen tension level conditions, proline residues in the oxygen-dependent domain (ODD) of the HIF-1α subunit are hydroxylated by prolyl hydroxylases (PHDs) that require oxygen, iron, and 2-oxoglutarate as substrates. Von Hippel-Lindau tumor suppressor protein (VHL) recognizes and binds the hydroxylated HIF-1α subunit targeting it for polyubiquitination and proteasomal degradation. In low oxygen tension level conditions, hydroxylation of HIF-1α subunits by PHDs is inhibited, stabilizing HIF-1α and allowing translocation to the nucleus where with binding partner HIF-1β, the HIF-1 complex binds to hypoxia-response elements (HREs) in the promoter of target genes that are involved in hypoxic adaptation and survival processes.
HYPOXIA, HIF-1, AND THE WOUND HEALING PROCESS
The process of tissue repair and wound healing is comprised of three continuous and overlapping phases: the inflammatory phase, the proliferation phase, and the remodeling phase (Hong et al., 2014). This process requires highly orchestrated temporal and spatial regulation of various cell types and mediators in the injured tissue area. One key microenvironmental condition that contributes to this complex process is hypoxic oxygen tension. The inflammatory phase is marked by vascular responses and inflammatory cell recruitment. Initial tissue injury causes blood vessel damage that leads to acute tissue hypoxia. Moreover, elevated oxygen consumption by infiltrated inflammatory and stromal cells further lowers the tissue oxygen tension leading to prolonged chronic hypoxia. An early study by Remensnyder and Majno made a seminal observation that wounded tissue displays a steep hypoxic gradient in the cremaster muscle of rats that is strongly correlated with the level of revascularization (Remensnyder and Majno, 1968). Later, Elson et al. demonstrated a coordinated transient induction of HIF-1α and its target genes, GLUT-1, PGK-1, and VEGF, in epidermal wound healing (Elson et al., 2000). These initial studies highlight the temporal and spatial activation and regulation of HIF-1 signaling in wound healing processes. Further, one can reason that adaption to the hypoxic microenvironment by hypoxic cells within the injured area, including inflammatory cells, keratinocytes, endothelial cells, and mesenchymal stromal cells, is essential for the cells to exert their wound healing activities.
During wound healing or tissue injury repair, after platelets begin the coagulation process, monocytes infiltrate the injury or wound and start to differentiate into macrophages. At this point, macrophages become the predominant cell population in the inflammatory phase of wound healing. Biological significance of hypoxic adaptation of macrophages has been investigated utilizing a macrophage-specific HIF-1α knockout animal model (Cramer et al., 2003). HIF-1α-null macrophages displayed impaired recruitment into inflamed areas, metabolic dysfunction characterized by ATP depletion, and decreased bactericidal capacity indicating that HIF-1α activation is an essential determinant for macrophage survival and function in hypoxic injury microenvironments.
The inflammatory phase of wound healing is also governed by macrophage-derived reactive oxygen species (ROS). Although the rise in ROS production is the main defense against bacterial infection by acting as intracellular signaling mediators which activate cytokine activity and angiogenesis, a balance within the injured tissue must be maintained because high levels of the ROS can further damage the area due to the high reactivity of the molecules. Indeed, chronic non-healing wounds have demonstrated over production of ROS resulting in oxidative stress within the tissue (Schafer and Werner, 2008). In this regard, HIF-1-mediated switching to glycolytic metabolism from mitochondrial oxidative respiration in the hypoxic macrophages may be important for preventing the excess generation of ROS and impaired wound resolution (Kim et al., 2006). In addition, in response to hypoxia, macrophages release chemotactic factors and secrete growth factors important for cell migration and proliferation leading to tissue repair. Macrophage-secreted growth factors such as VEGF, platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), transforming growth factor-α (TGF-α), and TGF-β have been reported to be induced by hypoxia (Murdoch et al., 2005). In addition to being a chemoattractant for monocytes, macrophages, and neutrophils, PDGF has been shown to induce mitosis in fibroblasts and smooth muscle cells in vitro. Endothelial cell migration as well as fibroblast migration and deposition of extracellular matrix proteins (e.g. collagen) are induced by TGF-β secreted by macrophages and these events lead to the formation of granulation tissue (Li et al., 2007). These studies highlight the prominent implications of macrophage hypoxic responses in the wound healing process.
The proliferative phase of wound healing consists of several processes: angiogenesis and vasculogenesis to reestablish blood supply to the injured tissue; fibroplasia, the formation of fibrous tissue, which contributes to granulation tissue development; re-epithelialization driven by keratinocyte proliferation and migration; and wound contraction mediated by myofibroblasts. Healing tissue requires new blood vessel growth, via angiogenesis and vasculogenesis, to re-establish the vasculature necessary to deliver oxygen and nutrients to the injured tissue. Hypoxia, through the accumulation of HIF-1α and formation of the HIF complex, activates several angiogenic growth factor genes including VEGF, angiopoietin 2 (ANGPT2), and stromal cell-derived factor-1 (SDF-1) (Ceradini et al., 2004; Forsythe et al., 1996; Pichiule et al., 2004; Yamakawa et al., 2003). Multiple cell types, including fibroblasts and endothelial cells, additionally produce VEGF, the key mediator of angiogenesis. Work by Ahluwalia and Tarnawski provides evidence that HIF-1α increases in endothelial cells that line newly formed capillaries and that it co-localizes with VEGF in these cells (Ahluwalia and Tarnawski, 2012). Angiogenesis is also stimulated by the secretion of cytokines by macrophages (Murdoch et al., 2005), the low-oxygen tension resulting from the hypoxic microenvironment, and the increased lactic acid within the tissue resulting from the high metabolic demands of the cellular influx into the damaged tissue area (Remensnyder and Majno, 1968; Taraboletti et al., 2006). The activation of angiogenesis allows for the re-establishment of vasculature within the injured tissue site enhancing its capacity to heal.
In addition to angiogenesis, the re-establishment of vasculature within injured tissue can occur by the process called vasculogenesis where bone marrow-derived circulating endothelial progenitor cells (EPCs) have the ability to form new blood vessels (Ceradini and Gurtner, 2005). These EPCs are selectively recruited to injured tissue areas or wound regions exhibiting ischemia. Hypoxia, among other environmental stimuli, promotes recruiting these progenitor cells to the ischemic tissue via a HIF-1α direct target, stromal cell-derived factor-1 (SDF-1) (Ceradini et al., 2004). SDF-1, also known as C-X-C-motif chemokine 12 (CXCL12), is a chemokine that attracts hematopoietic and endothelial progenitor cells expressing its receptor CXCR4. Using a mouse model with graded soft tissue ischemia, Ceradini et al. demonstrated that reduction in the oxygen tension of tissue was directly proportionate to the expression of SDF-1 (Ceradini et al., 2004). Additionally, in vitro assay by Tepper et al. showed that EPCs preferentially proliferate in hypoxic conditions (Tepper et al., 2005). Taken together, these studies suggest that HIF-1α plays an important role in the localization or “homing” of circulating EPCs to injured tissues exhibiting low oxygen tension to promote the restoration of damaged blood vessels.
Another process occurring during the proliferation phase of wound healing and one that is essentially important for wound contraction is fibroplasia, the formation of fibrous tissue. Fibroplasia is initiated by fibroblast migration into the damaged tissue or wound region. The fibroblasts begin to proliferate and differentiate into myofibroblasts resulting in the generation of excess extracellular matrix (ECM) proteins aiding in granulation tissue development. Proliferation and differentiation of fibroblasts is induced by various growth factors such as FGF and TGF-β, whose expression and activity can be further enhanced by hypoxic microenvironment (Li et al., 2007). Intriguingly, recent studies have shown that increased lactic acid within tissue, due to high demand of HIF-1-mediated glycolytic metabolism of cells in the damaged hypoxic tissue area, is important in the activation of TGF-β signaling and myofibroblastic differentiation (Kottmann et al., 2012; Tuder et al., 2012). This study revealed a previously unknown pH-dependent mechanism of TGF-β activation in which hypoxia may provide fibroblasts an acidic microenvironment that enhances their differentiation to myofibroblasts.
The formation of granulation tissue is followed by the reepithelialization process, which is characterized by keratinocyte proliferation and migration into the wounded area to establish a protective barrier. Kalucka et al. (2013) have recently investigated the implication of hypoxic responses in this process in keratinocyte-specific PHD2 knockout mice that exhibit constitutively stable HIF-1α activity. The authors demonstrated that keratinocyte PHD2 ablation results in an elevated migratory capacity of keratinocytes in their skin wound healing model. This enhanced migration was mediated by HIF-1-dependent integrin-β3 that previously has been shown to play a critical role in wound healing (De Luca et al., 1994). Moreover, suppressing the growth inhibitory effects of TGF-β signaling significantly enhanced the proliferation of Phd2-null keratinocytes.
HIF-1 IN ABNORMAL WOUND HEALING
In order for wounds and injured tissues to be properly repaired, each step of the wound healing cascade essentially requires a fine orchestration among various cellular components with support from a physiological microenvironmental milieu. Defects in the normal wound repair process or lack of cellular or non-cellular components leads to the development of chronic or pathologic wounds. Among many impaired wound diseases, we will describe here diabetic wounds and fibrosis focusing on how hypoxia and HIF-1 signaling are implicated with the pathogenesis of these conditions. We will further discuss targeting hypoxic signaling as a potential therapeutic strategy for aberrant wound healing disorders.
DIABETIC WOUND HEALING
Impaired wound healing in diabetic patients (e.g. diabetic foot ulcers) is one of the major medical and public health issues worldwide. More than 50% of non-traumatic lower limb amputations are caused by diabetic wound ulceration. Moreover, diabetic patients with foot ulcers are expected to have a poorer prognosis and significantly higher mortality (Moulik et al., 2003). Currently, there is no effective treatment strategy available mainly because the molecular pathogenesis that leads to wound repair failure is poorly understood. Given that hypoxia plays a significant role in physiologic wound healing processes, several groups have sought to determine how hypoxic responses and HIF-1 signaling activity are affected by delayed wound healing in diabetic animal models. Initially, Catrina et al. (2004) and Gao et al. (2007) reported that the hyperglycemic condition was sufficient to repress HIF-1α induction in hypoxic conditions. This appeared to be a proteasome-dependent mechanism as proteasomal inhibitor, MG-132, but not PHD inhibitor abolished the HIF-1α suppressive effect of hyperglycemia. Consistent with in vitro observations, diabetic ulcer biopsy displayed a significantly lower expression of HIF-1α as compared to non-diabetic chronic ulcer tissues emphasizing the impaired HIF-1 signaling in hyperglycemic diabetic wounds. In subsequent studies, pharmacological stabilization or genetic activation of HIF-1α was able to improve wound healing in genetic diabetic db/db mice (Botusan et al., 2008; Zhang et al., 2013). HIF-1α activation significantly induced neovascularization that is associated with the local recruitment of bone marrow derived endothelial progenitor cells into the wound area. This highlights the clinical implication of HIF-1α stabilization for the management of diabetic wounds given that impaired vasculature and blood perfusion is considered a major pathogenic mechanism.
FIBROSIS
During the normal progression of tissue repair, myofibroblasts ultimately become the predominant cell population secreting collagen and other ECM proteins to restore tissue homeostasis and promote healing. Fibrosis is the aberrant wound healing process where the repair of injured tissue is deregulated causing myofibroblasts to deposit excessive amounts of ECM (e.g. collagen) (Zeisberg and Kalluri, 2013). The excess ECM results in the overgrowth of tissue, hardening, and scar formation. The tissue overgrowth and hardening can lead to organ dysfunction and ultimately organ failure can occur if fibrosis advances. One of the important mechanisms underlying the pathogenesis of tissue fibrosis is epithelial-mesenchymal transition (EMT) (Lee and Nelson, 2012). EMT is defined by the loss of epithelial cell polarity and cell-cell adhesion enabling these cells to migrate and acquire the invasive properties of mesenchymal cells. Mesenchymal cells facilitate tissue regeneration in wound and tissue healing and can promote the pathogenesis of chronic conditions, such as fibrosis. A characteristic of fibrotic tissue is reduced capillary density within the tissue or organ resulting in decreased oxygen delivery to cells. As a result, fibrotic tissue becomes hypoxic and stabilizes the transcription factor HIF-1α transactivating a wide variety of genes including profibrotic genes. In support of this, recent preclinical and clinical studies show that initiation and progression of various tissue fibroses are closely correlated with HIF-1α activated, or hypoxia-induced, profibrotic genes (Kaminski and Rosas, 2006; Tzouvelekis et al., 2007).
In studies using cell culture and genetic knockout mouse models, Higgins et al. (2007) showed that the expression of HIF-1α is associated with increased renal fibrosis by positive regulation of EMT. Loss of HIF-1α in primary renal epithelial cells resulted in a reduced capacity to undergo EMT as well as attenuated expression of profibrotic genes, including a collagen cross-linking enzyme lysyl oxidase (LOX). The authors further showed that targeted HIF-1α deletion in proximal tubular epithelial cells leads to reduced tubulointerstitial fibrosis in the model of unilateral urethral obstruction (UUO). Clinically, HIF-1α was shown to be highly expressed in renal biopsies from chronic kidney diseases. The transdifferentiation of the tubular epithelial cells into myofibroblasts is likely a result of HIF regulation of SDF-1 and its cytokine receptor, CXCR4 (Barriga et al., 2013). Additionally, HIF-1 has been shown to directly induce transcription of the pro-fibrotic factors tissue-inhibitor of metalloproteinases (TIMP)-1, plasminogen activator inhibitor (PAI)-1, and connective tissue growth factor (CTGF). Similar to VEGF transcription, a cooperation between hypoxia and TGF-β signaling has been suggested because the DNA binding sites for HIF-1 and Smads are near one another in target genes (e.g. type I collagen) of fibroblasts (Lee and Nelson, 2012). For example, Zhang et al. showed that the up-regulation of Smad3 by hypoxia leads to the activation of TGF-β signaling pathways (Zhang et al., 2003). Similarly, Basu et al. demonstrated that TGF-β directed expression of type I collagen could be decreased by inhibiting HIF-1 transcription (Basu et al., 2011).
While the pathological processes of renal fibrosis are extensively studied, the origin of kidney myofibroblasts is still a matter of debate. Recently, renal erythropoietin producing (REP) cells were identified (Pan et al., 2011), in which HIF-2 was abundantly expressed. Subsequent fate mapping analysis demonstrated that myelin protein zero-cre (P0-cre) lineage-labeled cells comprised the majority of REP cells. An animal model of renal fibrosis, unilateral ureteral obstruction, revealed that most of the kidney myofibroblasts were derived from P0-cre lineage-labeled cells, but not from injured tubular epithelial cells (Asada et al., 2011). More intriguingly, together with the differentiation into myofibroblasts, P0-cre lineage-labeled cells concomitantly lose erythropoietin production, which is one of the hallmarks of chronic kidney disease.
Cardiac fibrosis is closely related with dysfunctional cardiac remodeling after ischemic injury. Watson et al. (2014) reported a strong correlation between hypoxia and the expression of fibrotic gene products including collagens and α–smooth muscle actin (α-SMA), which are representative markers of myofibroblast differentiation, in human cardiac tissues. Under hypoxic conditions, human cardiac fibroblasts exhibited pro-fibrotic features including activated TGF-β signaling, which appears to be associated with global DNA methylation. Intriguingly, HIF-1α directly transactivated DNA methyltransferase 1 (DNMT1) and DNMT3B in cardiac fibroblasts in response to hypoxia. Pharmacological or genetic inhibition of DNMT abolished the hypoxic induction of pro-fibrotic genes suggesting that hypoxic induction of HIF-1α may contribute to ischemic fibrosis in cardiac tissue by epigenetic regulation of pro-fibrotic gene expression.
Another major target organ for fibrotic disorder is the lung (Loomis-King et al., 2013; Maher, 2012; Noble et al., 2012). Comparative expression profiling data of pulmonary fibrosis mouse models and idiopathic pulmonary fibrosis (IPF) patients demonstrated a significant increase of alveolar epithelial HIF-1α expression in an early stage of pulmonary fibrosis (Tzouvelekis et al., 2007). Immunohistochemical detection of HIF-1α in mice bearing bleomycin-induced pulmonary fibrosis and IPF patients further supports the potential implication of HIF-1α in fibrotic progression. A recent study by Kottman et al. (2012) showing the pH-dependent activation of TGF-β signaling and myofibroblastic differentiation by increased lactic acid within pulmonary fibrotic tissue provides an insight into the mechanisms underlying HIF-1-mediated fibrotic progression; aberrantly activated glycolytic metabolism may be implicated in fibrotic progression.
CONCLUSION
Wound healing involves a variety of processes including inflammation, angiogenesis, vasculogenesis, fibroplasia, and reepithelialization, all of which are critically regulated by hypoxic responses (Fig. 2). HIF-1α has been repeatedly shown to accelerate physiologic wound healing processes, as well as diabetic wound healing, providing a fundamental background to hypothesize that HIF-1 activation can be used to aid in the healing of normal and diabetic wounds. In light of this, inhibition of oxygen sensor proyly hydroxylase enzyme (PHD), which sensitizes HIF-1α for proteasomal degradation, has been intensively investigated and shown to be beneficial for diabetic wound healing (Kalucka et al., 2013; Myllyharju, 2009, 2013; Thangarajah et al., 2010; Zhang et al., 2013). Thus there is a potential for the use of PHD inhibitors to treat tissue injuries and wounds. For example, selective PHD inhibitors, such as FibroGen’s FG-4592 and FG-2216, are being developed and are currently undergoing FDA approved human clinical trials for use as oral drugs (Beuck et al., 2012). Although these PHD inhibitors are intended to treat anemia by acting upon HIF-2α-mediated erythropoietin (EPO) induction, they may potentially play a role in tissue injury and wound healing. While HIF-1α can accelerate normal and diabetic wound healing, it can also aggravate the pathogenic progression of fibrotic disorders such as fibrosis (Higgins et al., 2007; Kimura et al., 2008; Lokmic et al., 2012). Given that fibrotic progression is promoted by hypoxic response and its contribution to the differentiation into ECM-producing myofibroblasts, further study should be warranted to evaluate the inhibition of HIF-1α signaling as a potential therapeutic strategy for fibrotic diseases.
Fig. 2.
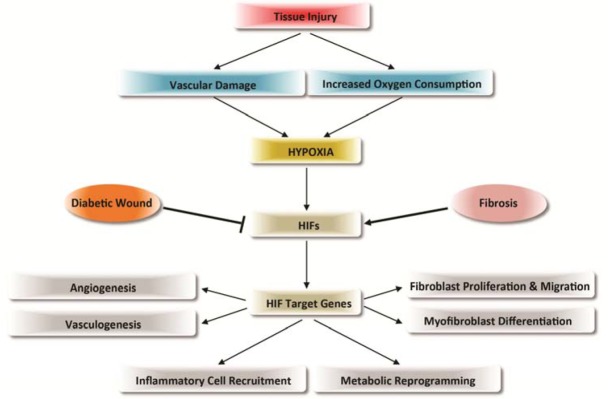
Regulation of wound healing and fibrosis by hypoxia and HIF-1. In tissue injury, vascular damage and increased oxygen consumption result in a hypoxic microenvironment that leads to the induction of HIFs. HIFs activate target genes that contribute to the processes of wound healing and remodeling; including angiogenesis, vasculogenesis, fibroblast proliferation and migration, myofibroblast differentiation, inflammatory cell recruitment, and metabolic reprogramming. The induction of HIF-1 is repressed in diabetic wounds resulting in delayed healing. In contrast, fibrosis is promoted by HIFs and hypoxia, aggravating the pathogenic progression of fibrotic disorders.
Acknowledgments
We thank the Kim Lab members for valuable discussions. This work was supported by the University of Texas at Dallas start-up grant. We sincerely apologize for inadvertent omission of any pertinent original references owing to space constraints.
REFERENCES
- Ahluwalia A, Tarnawski AS. Critical role of hypoxia sensor–HIF-1alpha in VEGF gene activation. Implications for angiogenesis and tissue injury healing. Curr Med Chem. 2012;19:90–97. doi: 10.2174/092986712803413944. [DOI] [PubMed] [Google Scholar]
- Andrikopoulou E, Zhang X, Sebastian R, Marti G, Liu L, Milner SM, Harmon JW. Current Insights into the role of HIF-1 in cutaneous wound healing. Curr Mol Med. 2011;11:218–235. doi: 10.2174/156652411795243414. [DOI] [PubMed] [Google Scholar]
- Asada N, Takase M, Nakamura J, Oguchi A, Asada M, Suzuki N, Yamamura K, Nagoshi N, Shibata S, Rao TN, et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest. 2011;121:3981–3990. doi: 10.1172/JCI57301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriga EH, Maxwell PH, Reyes AE, Mayor R. The hypoxia factor Hif-1alpha controls neural crest chemotaxis and epithelial to mesenchymal transition. J Cell Biol. 2013;201:759–776. doi: 10.1083/jcb.201212100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu RK, Hubchak S, Hayashida T, Runyan CE, Schumacker PT, Schnaper HW. Interdependence of HIF-1alpha and TGF-beta/Smad3 signaling in normoxic and hypoxic renal epithelial cell collagen expression. Am J Physiol Renal Physiol. 2011;300:F898–905. doi: 10.1152/ajprenal.00335.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berra E, Roux D, Richard DE, Pouyssegur J. Hypoxia-inducible factor-1 alpha (HIF-1 alpha) escapes O(2)-driven proteasomal degradation irrespective of its subcellular localization: nucleus or cytoplasm. EMBO Rep. 2001;2:615–620. doi: 10.1093/embo-reports/kve130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuck S, Schanzer W, Thevis M. Hypoxia-inducible factor stabilizers and other small-molecule erythropoiesis-stimulating agents in current and preventive doping analysis. Drug Test. Anal. 2012;4:830–845. doi: 10.1002/dta.390. [DOI] [PubMed] [Google Scholar]
- Botusan IR, Sunkari VG, Savu O, Catrina AI, Grunler J, Lindberg S, Pereira T, Yla-Herttuala S, Poellinger L, Brismar K, et al. Stabilization of HIF-1 alpha is critical to improve wound healing in diabetic mice. Proc Natl Acad Sci USA. 2008;105:19426–19431. doi: 10.1073/pnas.0805230105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catrina SB, Okamoto K, Pereira T, Brismar K, Poellinger L. Hyperglycemia regulates hypoxia-inducible factor-1alpha protein stability and function. Diabetes. 2004;53:3226–3232. doi: 10.2337/diabetes.53.12.3226. [DOI] [PubMed] [Google Scholar]
- Ceradini DJ, Gurtner GC. Homing to hypoxia: HIF-1 as a mediator of progenitor cell recruitment to injured tissue. Trends Cardiovasc. Med. 2005;15:57–63. doi: 10.1016/j.tcm.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. HIF-1 alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca M, Pellegrini G, Zambruno G, Marchisio PC. Role of integrins in cell adhesion and polarity in normal keratinocytes and human skin pathologies. J Dermatol. 1994;21:821–828. doi: 10.1111/j.1346-8138.1994.tb03296.x. [DOI] [PubMed] [Google Scholar]
- Elson DA, Ryan HE, Snow JW, Johnson R, Arbeit JM. Coordinate up-regulation of hypoxia inducible factor (HIF)-1alpha and HIF-1 target genes during multi-stage epidermal carcinogenesis and wound healing. Cancer Res. 2000;60:6189–6195. [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Ferguson G, Connell P, Walshe T, Murphy R, Birney YA, O’Brien C, Cahill PA. High glucose concentrations alter hypoxia-induced control of vascular smooth muscle cell growth via a HIF-1alpha-dependent pathway. J Mol Cell Cardiol. 2007;42:609–619. doi: 10.1016/j.yjmcc.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EC, Reyes H, Chu FF, Sander F, Conley LH, Brooks BA, Hankinson O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science. 1991;252:954–958. doi: 10.1126/science.1852076. [DOI] [PubMed] [Google Scholar]
- Hong WX, Hu MS, Esquivel M, Liang GY, Rennert RC, McArdle A, Paik KJ, Duscher D, Gurtner GC, Lorenz HP, et al. The Role of Hypoxia-Inducible Factor in Wound Healing. Adv Wound Care. 2014;3:390–399. doi: 10.1089/wound.2013.0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Kalucka J, Ettinger A, Franke K, Mamlouk S, Singh RP, Farhat K, Muschter A, Olbrich S, Breier G, Katschinski DM, et al. Loss of epithelial hypoxia-inducible factor prolyl hydroxylase 2 accelerates skin wound healing in mice. Mol Cell Biol. 2013;33:3426–3438. doi: 10.1128/MCB.00609-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski N, Rosas IO. Gene expression profiling as a window into idiopathic pulmonary fibrosis pathogenesis: can we identify the right target genes? Proc Am Thorac Soc. 2006;3:339–344. doi: 10.1513/pats.200601-011TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metabol. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kimura K, Iwano M, Higgins DF, Yamaguchi Y, Nakatani K, Harada K, Kubo A, Akai Y, Rankin EB, Neilson EG, et al. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol. 2008;295:F1023–1029. doi: 10.1152/ajprenal.90209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch LG, Britton SL. Aerobic metabolism underlies complexity and capacity. J Physiol. 2008;586:83–95. doi: 10.1113/jphysiol.2007.144709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, Honnons S, Jones C, Isern NG, Hu JZ, et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-beta. Am. J. Respir. Crit Care Med. 2012;186:740–751. doi: 10.1164/rccm.201201-0084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrecque MP, Prefontaine GG, Beischlag TV. The aryl hydrocarbon receptor nuclear translocator (ARNT) family of proteins: transcriptional modifiers with multi-functional protein interfaces. Curr Mol Med. 2013;13:1047–1065. doi: 10.2174/15665240113139990042. [DOI] [PubMed] [Google Scholar]
- Lee K, Nelson CM. New insights into the regulation of epithelial-mesenchymal transition and tissue fibrosis. Int Rev Cell Mol Biol. 2012;294:171–221. doi: 10.1016/B978-0-12-394305-7.00004-5. [DOI] [PubMed] [Google Scholar]
- Li J, Chen J, Kirsner R. Pathophysiology of acute wound healing. Clin Dermatol. 2007;25:9–18. doi: 10.1016/j.clindermatol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Lokmic Z, Musyoka J, Hewitson TD, Darby IA. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int Rev Cell Mol Biol. 2012;296:139–185. doi: 10.1016/B978-0-12-394307-1.00003-5. [DOI] [PubMed] [Google Scholar]
- Loomis-King H, Flaherty KR, Moore BB. Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis. Curr Opin Pharmacol. 2013;13:377–385. doi: 10.1016/j.coph.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher TM. Idiopathic pulmonary fibrosis: pathobiology of novel approaches to treatment. Clin Chest Med. 2012;33:69–83. doi: 10.1016/j.ccm.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Moulik PK, Mtonga R, Gill GV. Amputation and mortality in new-onset diabetic foot ulcers stratified by etiology. Diabetes Care. 2003;26:491–494. doi: 10.2337/diacare.26.2.491. [DOI] [PubMed] [Google Scholar]
- Murdoch C, Muthana M, Lewis CE. Hypoxia regulates macrophage functions in inflammation. J Immunol. 2005;175:6257–6263. doi: 10.4049/jimmunol.175.10.6257. [DOI] [PubMed] [Google Scholar]
- Myllyharju J. HIF prolyl 4-hydroxylases and their potential as drug targets. Curr Pharm Des. 2009;15:3878–3885. doi: 10.2174/138161209789649457. [DOI] [PubMed] [Google Scholar]
- Myllyharju J. Prolyl 4-hydroxylases, master regulators of the hypoxia response. Acta Physiol. 2013;208:148–165. doi: 10.1111/apha.12096. [DOI] [PubMed] [Google Scholar]
- Noble PW, Barkauskas CE, Jiang DH. Pulmonary fibrosis: patterns and Perpetrators. J Clin Invest. 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- Pan X, Suzuki N, Hirano I, Yamazaki S, Minegishi N, Yamamoto M. Isolation and characterization of renal erythropoietin-producing cells from genetically produced anemia mice. PLoS One. 2011;6:e25839. doi: 10.1371/journal.pone.0025839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichiule P, Chavez JC, LaManna JC. Hypoxic regulation of angiopoietin-2 expression in endothelial cells. J Biol Chem. 2004;279:12171–12180. doi: 10.1074/jbc.M305146200. [DOI] [PubMed] [Google Scholar]
- Remensnyder JP, Majno G. Oxygen gradients in healing wounds. Am J Pathol. 1968;52:301–323. [PMC free article] [PubMed] [Google Scholar]
- Schafer M, Werner S. Oxidative stress in normal and impaired wound repair. Pharmacol Res. 2008;58:165–171. doi: 10.1016/j.phrs.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboletti G, D’Ascenzo S, Giusti I, Marchetti D, Borsotti P, Millimaggi D, Giavazzi R, Pavan A, Dolo V. Bioavailability of VEGF in tumor-shed vesicles depends on vesicle burst induced by acidic pH. Neoplasia. 2006;8:96–103. doi: 10.1593/neo.05583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CT, McElwain JC. Ancient atmospheres and the evolution of oxygen sensing via the hypoxia-inducible factor in metazoans. Physiology. 2010;25:272–279. doi: 10.1152/physiol.00029.2010. [DOI] [PubMed] [Google Scholar]
- Tepper OM, Capla JM, Galiano RD, Ceradini DJ, Callaghan MJ, Kleinman ME, Gurtner GC. Adult vasculogenesis occurs through in situ recruitment, proliferation, and tubulization of circulating bone marrow-derived cells. Blood. 2005;105:1068–1077. doi: 10.1182/blood-2004-03-1051. [DOI] [PubMed] [Google Scholar]
- Thangarajah H, Vial IN, Grogan RH, Yao DC, Shi YB, Januszyk M, Galiano RD, Chang EI, Galvez MG, Glotzbach JP, et al. HIF-1 alpha dysfunction in diabetes. Cell Cycle. 2010;9:75–79. doi: 10.4161/cc.9.1.10371. [DOI] [PubMed] [Google Scholar]
- Tuder RM, Lara AR, Thannickal VJ. Lactate, a novel trigger of transforming growth factor-beta activation in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:701–703. doi: 10.1164/rccm.201208-1491ED. [DOI] [PubMed] [Google Scholar]
- Tzouvelekis A, Harokopos V, Paparountas T, Oikonomou N, Chatziioannou A, Vilaras G, Tsiambas E, Karameris A, Bouros D, Aidinis V. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am J Respir Crit Care Med. 2007;176:1108–1119. doi: 10.1164/rccm.200705-683OC. [DOI] [PubMed] [Google Scholar]
- Wang GL, Semenza GL. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem. 1993;268:21513–21518. [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson CJ, Collier P, Tea I, Neary R, Watson JA, Robinson C, Phelan D, Ledwidge MT, McDonald KM, McCann A, et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum Mol Genet. 2014;23:2176–2188. doi: 10.1093/hmg/ddt614. [DOI] [PubMed] [Google Scholar]
- Weidemann A, Johnson RS. Biology of HIF-1 alpha. Cell Death Differ. 2008;15:621–627. doi: 10.1038/cdd.2008.12. [DOI] [PubMed] [Google Scholar]
- Yamakawa M, Liu LX, Date T, Belanger AJ, Vincent KA, Akita GY, Kuriyama T, Cheng SH, Gregory RJ, Jiang C. Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ Res. 2003;93:664–673. doi: 10.1161/01.RES.0000093984.48643.D7. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Kalluri R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol. 2013;304:C216–C225. doi: 10.1152/ajpcell.00328.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Akman HO, Smith EL, Zhao J, Murphy-Ullrich JE, Batuman OA. Cellular response to hypoxia involves signaling via Smad proteins. Blood. 2003;101:2253–2260. doi: 10.1182/blood-2002-02-0629. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yan X, Cheng L, Dai J, Wang C, Han P, Chai Y. Wound healing improvement with PHD-2 silenced fibroblasts in diabetic mice. PLoS One. 2013;8:e84548. doi: 10.1371/journal.pone.0084548. [DOI] [PMC free article] [PubMed] [Google Scholar]