

ABSTRACT
Mutations in the STAG2 gene are present in ∼20% of tumors from different tissues of origin. STAG2 encodes a subunit of the cohesin complex, and tumors with loss-of-function mutations are usually aneuploid and display elevated frequencies of lagging chromosomes during anaphase. Lagging chromosomes are a hallmark of chromosomal instability (CIN) arising from persistent errors in kinetochore–microtubule (kMT) attachment. To determine whether the loss of STAG2 increases the rate of formation of kMT attachment errors or decreases the rate of their correction, we examined mitosis in STAG2-deficient cells. STAG2 depletion does not impair bipolar spindle formation or delay mitotic progression. Instead, loss of STAG2 permits excessive centromere stretch along with hyperstabilization of kMT attachments. STAG2-deficient cells display mislocalization of Bub1 kinase, Bub3 and the chromosome passenger complex. Importantly, strategically destabilizing kMT attachments in tumor cells harboring STAG2 mutations by overexpression of the microtubule-destabilizing enzymes MCAK (also known as KIF2C) and Kif2B decreased the rate of lagging chromosomes and reduced the rate of chromosome missegregation. These data demonstrate that STAG2 promotes the correction of kMT attachment errors to ensure faithful chromosome segregation during mitosis.
KEY WORDS: STAG2, Aneuploidy, Kinetochore, Merotely, Mitosis, Chromosomal instability
INTRODUCTION
Chromosomes must be faithfully segregated during mitosis to allow for normal cellular growth and development of organisms. Chromosome missegregation leads to aneuploidy, a common state of cells in solid tumors (Fang and Zhang, 2011; Holland and Cleveland, 2009). Evidence suggests that an important distinction between normal diploid cell populations and aneuploid cancer cells is the tolerance for aneuploid genomes. Diploid cells are intolerant of chromosome missegregation and either arrest in the cell cycle through a p53-dependent mechanism (Thompson and Compton, 2010) or display growth retardation (Williams et al., 2008). In addition to a state of aneuploidy, many solid tumors continuously missegregate chromosomes at high rates in a phenomenon called chromosomal instability (CIN) (Lengauer et al., 1998; Holland and Cleveland, 2009; Thompson et al., 2010). This high rate of missegregation has been clinically correlated with metastasis, drug resistance and poor patient prognosis (Walther et al., 2008; Choi et al., 2009; Heilig et al., 2010; Lee et al., 2011; Bakhoum et al., 2011; Bakhoum and Compton, 2012). Direct analysis of CIN cancer cells has shown that the most common cause of whole chromosome missegregation is the persistence of errors in the attachment of chromosomes to spindle microtubules (Thompson and Compton, 2008). These errors involve the attachment of single kinetochores to microtubules oriented towards both spindle poles (i.e. merotely) leading to lagging chromosomes in anaphase and chromosome non-disjunction (Salmon et al., 2005; Cimini, 2007).
Dysfunction of multiple mitotic processes can give rise to persistent merotelic kinetochore–microtubule (kMT) attachments (Thompson et al., 2010; Bakhoum and Compton, 2012). For example, defects that increase the rate of formation of merotelic kMT attachments cause CIN. Extra centrosomes cause multipolar spindles prior to bipolarization, and this transient defect in spindle geometry substantially elevates the frequency of merotelic attachments (Loncarek et al., 2007; Ganem et al., 2009; Silkworth and Cimini, 2012). Alterations in centromere geometry also increase the propensity for kMT attachment errors (Manning et al., 2010). By contrast, defects that decrease the rate of error correction also contribute to CIN. kMT attachment errors are corrected by the release of microtubules from kinetochores, making error correction dependent on the dynamic association and dissociation of microtubules from kinetochores. Studies have shown that CIN cancer cells have hyperstable kMT attachments, which erodes their ability to correct attachment errors (Bakhoum et al., 2009a; Bakhoum et al., 2009b). Importantly, strategic destabilization of kMT attachments can restore faithful chromosome segregation to otherwise CIN cancer cells (Bakhoum et al., 2009a), indicating a causal relationship between kMT attachment errors and CIN.
Genome sequencing of human cancers has revealed few recurrent mutations in genes responsible for mitosis, with the exception of members of the cohesin complex (Barbero, 2011; Kon et al., 2013; Mannini and Musio, 2011; Thol et al., 2013; Bailey et al., 2013; The Cancer Genome Atlas Research Network, 2014). The vertebrate cohesin core complex is composed of two structural maintenance of chromosome proteins (SMC1 and SMC3), the kleisin subunit Scc1/RAD21 and a stromal antigen protein (STAG). This complex is responsible for sister chromatid cohesion and participates in important cellular functions, including transcription regulation, DNA repair and chromosome segregation during mitosis (Peters et al., 2008; Barbero, 2009; Mehta et al., 2013). Inherited mutations in genes encoding cohesin subunits or their regulators cause cohesinopathies, such as Cornelia de Lange Syndrome and Roberts Syndrome. Somatically acquired mutations in CIN cancers have been identified in genes encoding SMC1, SMC3 and other cohesin-related proteins (Barber et al., 2008; Barbero, 2011; Yan et al., 2012). Recently, mutations causing loss of expression of the cohesin subunit STAG2 (also known as SA2) have been identified in numerous cancers (Solomon et al., 2011; Kim et al., 2012; Bernardes et al., 2013; Bailey et al., 2013). Microarray analysis has shown that STAG2 deficiency does not significantly alter gene expression (Solomon et al., 2011). Instead, fixed-cell imaging has revealed that STAG2-deficient cells show an increase in the frequency of chromosome bridges and lagging chromosomes at anaphase (Solomon et al., 2011). This suggests that loss of STAG2 generates CIN by either increasing the formation rate or decreasing the correction rate of kMT attachment errors in mitosis. Here, we use cell-based assays to show that STAG2 plays a role in regulating kMT attachment stability to promote efficient error correction, thereby ensuring faithful chromosome segregation.
RESULTS
To determine how loss-of-function mutations in the STAG2 gene cause lagging chromosomes in anaphase, we used siRNA to knock down STAG2 expression in immortalized, non-transformed and diploid RPE1 cells, transformed and near-diploid HCT116 cells, as well as aneuploid, chromosomally unstable and STAG2-deficient H4 cells (Fig. 1; supplementary material Fig. S1A,B). Immunoblot analysis for STAG2 showed that the efficiency of knockdown with this siRNA treatment varied from 40% to 70%, and that this effect was specific because no significant changes were observed in the levels of other mitotic proteins such as Bub1 or Aurora B kinase (supplementary material Fig. S1A). The fractional diminution of STAG2 levels observed by immunoblot under these conditions is reflected in the fraction of cells displaying loss of STAG2 as judged by fluorescence microscopy (supplementary material Fig. S1B), consistent with the effects of STAG2 siRNA published previously (Kong et al., 2014). RPE1 and HCT116 cells displayed a significant increase in the frequency of lagging chromosomes in anaphase, as judged by staining for chromosomes and centromeres, but H4 cells did not (Fig. 1A,B). H4 cells harbor a frameshift mutation in the STAG2 gene and do not express the STAG2 protein (Solomon et al., 2011). Thus, H4 cells are insensitive to siRNA targeting STAG2 (Fig. 1B), and this demonstrates that the lagging chromosomes induced by STAG2 siRNA are likely to be due to a specific effect of the siRNA on STAG2. Thus, depletion of STAG2 increases the frequency of lagging chromosomes during mitosis as reported previously (Solomon et al., 2011). The frequency of lagging chromosomes in anaphase under these conditions following STAG2 knockdown is an underestimate, because we did not pre-select STAG2-negative cells prior to quantifying lagging chromosomes.
Fig. 1.

STAG2 deficiency causes lagging chromosomes and increases centromere stretch. (A) Anaphase in HCT116 cells that were untreated (control) or transfected with siRNA specific to STAG2 (STAG2 KD) and stained for STAG2 (yellow), centromeres (red), tubulin (green) and DNA (blue) as indicated. Arrows indicates a lagging chromosome in a STAG2-deficient cell. KD, knockdown. Scale bar: 5 µm. (B) Percentage of anaphase cells displaying at least one lagging chromosome in anaphase in diploid RPE1 cells, near-diploid HCT116 cells and aneuploid H4 cells that were either untreated or transfected with siRNA specific for STAG2. n = 300 cells. (C) Percentage of untreated HCT116 cells and cells transfected with siRNA specific to STAG2 that were in different phases of mitosis. n = 1000 cells. (D) Centromere length in HCT116 cells that were untreated or transfected with siRNA specific to STAG2, measured between centroids of staining with the anti-centromere antibody; n = 100 centromeres. Data in B,D show the mean±s.e.m.; *P≤0.001.
An increase in the frequency of lagging chromosomes in anaphase can result from insults that either elevate the rate of formation of merotelic kMT attachments or decrease the efficiency of correction of merotelic kMT attachments (Thompson et al., 2010). To discriminate between these mechanisms, we examined different aspects of mitosis in STAG2-depleted cells. Depletion of STAG2 from RPE1 or HCT116 cells did not alter mitotic progression as measured by the fractions of cells in each phase of mitosis (Fig. 1C) or the mitotic index of the population (supplementary material Fig. S1C). Moreover, STAG2-depleted cells are checkpoint proficient, because disruption of spindles using nocodazole increases the mitotic index (supplementary material Fig. S1C). We observed an increase in cell death in STAG2-depleted cells during prolonged mitotic arrest, which explains the difference in mitotic index between control cells and STAG2-depleted cells after extended nocodazole treatment (e.g. 6 and 12 hours), although we currently do not know whether these cells die in mitosis or following mitotic exit. STAG2-depleted cells also did not show a significant difference in the frequency of multipolar spindles (supplementary material Fig. S1D). Thus, loss of STAG2 does not elevate the frequency of lagging chromosomes in anaphase by disruption of mitotic progression, the spindle assembly checkpoint or spindle bipolarity.
Because STAG2 functions in centromere cohesion (Canudas and Smith, 2009), we measured centromere stretch by staining with human centromere-specific antiserum or antibodies against the outer kinetochore component Hec1 (also known as NDC80). Both HCT116 (Fig. 1D) and RPE1 (supplementary material Fig. S2A) cells depleted of STAG2 displayed a significant increase in centromere and/or inter-kinetochore stretch relative to that of control cells in metaphase. Centromere distance was unchanged in STAG2-deficient cells relative to control cells when treated with nocodazole (supplementary material Fig. S2B), indicating that excessive centromere stretch in cells lacking STAG2 requires force from kMT attachments.
Next, we measured the stability of attachment of microtubules to kinetochores using fluorescence dissipation after photoactivation (Zhai et al., 1995) (Fig. 2). The turnover of fluorescently activated GFP–tubulin in spindle microtubules undergoes a double exponential decay, with the fast component representing the non-kinetochore microtubules (non-kMT) and the slow component representing the kMT (Fig. 2A,B). STAG2-depleted cells showed a significant increase in the stability of kMT attachments compared with that of control cells in both prometaphase and metaphase (Fig. 2C). There was no significant change in the stability of non-kMT dynamics between control cells and STAG2-depleted cells (Fig. 2D). These data demonstrate that loss of STAG2 results in hyperstable kMT attachments. This leads to improper regulation of kMT attachments, such that erroneous attachments persist into anaphase and thereby cause lagging chromosomes and CIN.
Fig. 2.

Measurement of kMT dynamics in cells depleted of STAG2. (A) DIC and time-lapse fluorescence images of mitotic RPE1 cells that were untreated (control) or transfected with siRNA specific to STAG2 (STAG2 KD) before (Pre-PA) and at indicated times after photoactivation (PA). KD, knockdown. Scale bar: 5 µm. (B) Normalized fluorescence intensity over time after photoactivation of spindles in untreated (white circles) and STAG2-deficient (filled circles) prometaphase and metaphase cells. (C) kMT half-life in untreated and STAG2-deficient RPE1 cells in prometaphase and metaphase. (D) Non-kMT microtubule half-life in RPE1 photoactivatable cells with or without STAG2 knockdown. Data represent the mean±s.e.m. (n≥10 cells); *P≤0.001.
To determine how STAG2 affects kMT dynamics, we examined the distribution of proteins responsible for regulating kMT attachments (Figs 3, 4). Using fluorescence microscopy, we found that Aurora B kinase did not localize efficiently to the inner centromere in STAG2-depleted cells (Fig. 3A). STAG2 depletion did not alter the overall quantity of Aurora B kinase in cells (supplementary material Fig. S1A), but there was a 40% decrease in the amount of Aurora B kinase localized to centromeres. Instead, Aurora B kinase localized along the length of chromosome arms, similar to the localization observed when Bub1, Sgo1 or Wapl are disrupted (Ricke et al., 2012; Rivera et al., 2012; Haarhuis et al., 2013). This change in localization of Aurora B kinase was mimicked by the distribution of INCENP (Fig. 3B), demonstrating that the entire chromosome passenger complex (CPC) is improperly localized in STAG2-depleted cells. Moreover, the activity of Aurora B kinase was reduced without STAG2, as judged by the quantity of phosphorylated (phospho)-histone H3 serine 10 and phospho-Knl1 (Fig. 3C,D), two common Aurora B substrates (Crosio et al., 2002; Hirota et al., 2005; Welburn et al., 2010). In STAG2-depleted cells, the quantity of phospho-histone H3 declined by 20% in prometaphase cells and 37% in metaphase cells and phospho-Knl1 at kinetochores was decreased by ∼30% in prometaphase cells. These data support the role of STAG2 in the recruitment and activity of Aurora B kinase during mitosis.
Fig. 3.

STAG2 influences the centromeric localization and activity of Aurora B kinase in HCT116 cells. Fluorescence intensities of (A) Aurora B kinase, (B) INCENP, (C) phospho-histone 3 serine 10 (pH3 S10) and (D) phospho-Knl1 (pKnl1) in prometaphase and metaphase HCT116 cells that were untreated (control) or transfected with siRNA specific to STAG2 (STAG2 KD). KD, knockdown; A.U., arbitrary units. Scale bars: 5 µm. Insets in A show a single focal plane image of an enlarged (2.5×) chromosome arm with Aurora B localization. D shows de-convolved phospho-Knl1 staining for clarity; insets show an enlarged (3×) centromere pair (red) with phospho-Knl1 localization. Quantitative data show the mean±s.e.m. [n≥200 centromeres (A,B,D), n = 10 cells (C)]; *P≤0.001 (A,B,D); *P≤0.01 (C).
Fig. 4.

STAG2 influences the centromeric localization and activity of Bub1 kinase in HCT116 cells. Fluorescence intensities of (A) Bub1, (B) phospho-histone 2A threonine 120 (pH2A T120), (C) phospho-histone 3 threonine 3 (pH3 T3) and (D) Bub3 in prometaphase and metaphase in HCT116 cells that were untreated (control) or transfected with siRNA specific to STAG2 (STAG2 KD). KD, knockdown; A.U., arbitrary units. Scale bars: 5 µm. Quantitative data show the mean±s.e.m. [n = 25 centromeres (A), n = 200 centromeres (B), n≥25 prometaphase and n≥10 metaphase cells (C), n>200 centromeres (D)]; *P≤0.01 (A); *P≤0.001 (B,D).
Recently, it was demonstrated that Bub1 is required for efficient centromere localization of the CPC (Ricke et al., 2012). One function of Bub1 is to localize the CPC to the centromere by phosphorylating threonine 120 on histone H2A (phospho-H2A T120) (Kawashima et al., 2010; Watanabe, 2010). Therefore, we examined both Bub1 localization and phosphorylation activity in STAG2-depleted cells. Bub1 kinetochore localization was decreased in both prometaphase (∼50%) and metaphase (∼90%) relative to that of control cells (Fig. 4A), although Bub1 protein levels remained unchanged (supplementary material Fig. S1A) and there must be sufficient Bub1 activity to mount a checkpoint response (supplementary material Fig. S1C). Correspondingly, the levels of phospho-histone H2A decreased by 15% in prometaphase and 8% in metaphase in STAG2-depleted cells relative to those of control cells (Fig. 4B). Phosphorylation of histone H3 on threonine 3 (phospho-H3 T3) by haspin is also involved in CPC localization (Dai et al., 2005), but we found no change in phospho-H3 T3 levels in either prometaphase or metaphase (Fig. 4C). By contrast, the quantity of Bub3 at kinetochores was increased by nearly 50% during prometaphase in STAG2-depleted cells (Fig. 4D). Bub3 is a binding partner of Bub1, so it is curious that these proteins would respond differently to the loss of STAG2. Perhaps the change in abundance of Bub3 at kinetochores contributes to altering the stability of kMT attachments, because Bub3 overexpression has been shown to rescue some cold-sensitive microtubule mutants in budding yeast (Guénette et al., 1995).
These data indicate that STAG2 promotes the correction of errors in kMT attachment. This predicts that loss-of-function mutations in STAG2 could be rescued by destabilizing kMT attachments to enhance error correction. To test this prediction, we overexpressed GFP alone, GFP–STAG2, and the microtubule-destabilizing kinesins GFP–MCAK and GFP–Kif2b in H4 cells, which harbor a frameshift mutation in the STAG2 gene and do not produce functional STAG2 protein. Immunoblotting confirmed the expression of each transgene in these cells (supplementary material Fig. S3A). We then quantified the frequency of lagging chromosomes in anaphase (Fig. 5A,B), and, as expected, the expression of STAG2 significantly reduced the fraction of anaphase cells displaying lagging chromosomes relative to that of cells expressing GFP alone (Fig. 5B). Importantly, the expression of GFP–MCAK or GFP–Kif2b also significantly reduced the fraction of anaphase cells displaying lagging chromosomes with an efficiency that matched the expression of STAG2 (Fig. 5A,B). To ensure the specificity of this strategy, we overexpressed GFP alone and GFP–Kif2B in HCT116 cells that were depleted of STAG2 by transfection with STAG2-specific siRNA. GFP expression did not alter the fraction of anaphase cells with lagging chromosomes relative to control cells. However, GFP–Kif2b expression significantly reduced the fraction of anaphase cells displaying lagging chromosomes induced by loss of STAG2 expression (supplementary material Fig. S3B).
Fig. 5.

STAG2 promotes faithful chromosome segregation. (A) Anaphase in STAG2-deficient H4 cells expressing GFP only (GFP) or GFP-tagged Kif2b (GFP–Kif2b), showing GFP fluorescence (green), centromere staining (red) and DNA (blue). The arrow indicates a lagging chromosome. Scale bar: 5 µm. (B) Quantification of the percentage of anaphase cells with lagging chromosomes in H4 cells overexpressing GFP, GFP–Kif2b, GFP–MCAK or GFP–STAG2. Data show the mean±s.e.m. (n≥300 cells); *P≤0.01. (C) Model of STAG2 activity during mitosis. STAG2 promotes the localization and activity of Bub1 and Aurora B kinase to promote the destabilization of kMT attachments and enhance error correction. In the absence of STAG2, Bub1 and Aurora B kinase do not localize properly, causing hyperstabilization of kMT attachments and reduced error correction efficiency.
Lagging chromosomes are symptomatic of chromosomal instability, but they do not always result in nondisjunction causing aneuploidy (Thompson and Compton, 2011). To directly test whether altering kMT error correction efficiency alters CIN in STAG2-deficient cells, we isolated clones of H4 cells expressing GFP alone, GFP–STAG2, or the microtubule-destabilizing kinesins GFP–MCAK or GFP–Kif2b. Each clone was grown for ∼25 generations and then processed for fluorescence in situ hybridization (FISH) using centromere-specific probes. Using FISH probes specific for four different chromosomes, we identified the modal number for each of these chromosomes and quantified the fraction of cells in the population deviating from that mode (Table 1). As expected, H4 cells expressing GFP alone are aneuploid and chromosomally unstable, as judged by the high percentage of cells in the population that have a chromosome number that deviates from the modal number for each chromosome (ranging from 19–65%, depending on the chromosome). Expression of GFP–STAG2 decreased karyotypic deviance from the mode for all four chromosomes tested, with two of the chromosomes showing significant reductions relative to cells expressing GFP alone. Similarly, expression of GFP–MCAK decreased karyotypic deviance from the mode for all four chromosomes tested, again with two of the chromosomes showing significant reductions relative to cells expressing GFP alone. Finally, expression of GFP–Kif2b induced a significant decrease in karyotypic deviation from the mode for all four chromosomes tested relative to cells expressing GFP alone. These data demonstrate that strategically destabilizing kMT attachments to increase the efficiency of error correction in STAG2-deficient cells decreases not only the rate of lagging chromosomes in anaphase, but the frequency of whole chromosome missegregation.
Table 1. Percent deviation from the chromosomal mode in H4 cell clones.
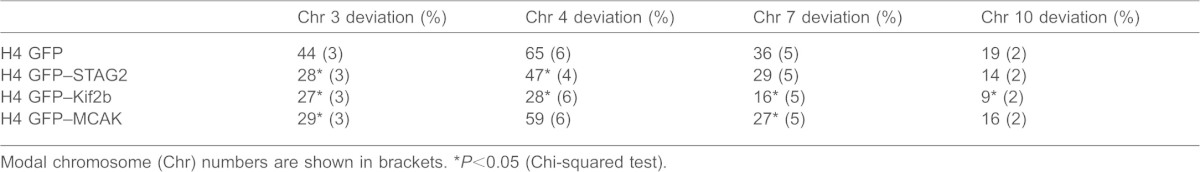
Modal chromosome (Chr) numbers are shown in brackets.
P<0.05 (Chi-squared test).
DISCUSSION
Loss of STAG2 has recently been detected in a variety of tumors and tumor-derived cell lines (Solomon et al., 2011; Kim et al., 2012; Bailey et al., 2013; Balbás-Martínez et al., 2013; Bernardes et al., 2013; Taylor et al., 2013; Lawrence et al., 2014; The Cancer Genome Atlas Research Network, 2014). In most of these cases, the loss of STAG2 correlates with aneuploidy and chromosomal instability. Moreover, STAG2 is one of only a handful of genes with a defined mitotic function that is frequently mutated in human tumors (Lawrence et al., 2014). These results suggest that the aneuploidy and chromosomal instability caused by the loss of STAG2 expression can act as a driver for cancer initiation. Moreover, because the tissue context determines whether aneuploidy promotes or suppresses cancer (Weaver et al., 2007), these results indicate that the tumors that most often harbor mutations in STAG2 might be derived from tissues that are most sensitive to transformation caused by karyotypic change.
In this work, we provide evidence that STAG2 ensures genomic integrity by promoting efficient correction of kMT attachment errors. Loss of STAG2 induces the hyperstabilization of kMT attachments, which impairs the error correction process and leads to elevated rates of chromosome missegregation (Fig. 5C). Several molecular models could explain how the loss of STAG2 stabilizes kMT attachments. One possibility is that STAG2 promotes the localization of Bub1 to kinetochores, allowing it to efficiently phosphorylate its substrates (e.g. histone H2A) and thereby encouraging proper CPC localization. Without STAG2, the CPC does not localize properly to the centromere, and Aurora B kinase fails to efficiently phosphorylate substrates involved in regulating kMT attachment stability (e.g. Knl1). This might precipitate a feedback network that is detrimental to kMT regulation, because Bub1 kinase has been shown to be a substrate of Aurora B kinase, especially for its checkpoint function. In this context, reductions in Aurora B activity might contribute to the mislocalization of Bub1 as was previously observed using small molecule inhibitors of Aurora B (Becker et al., 2010). Consistently, cells might have a more complicated response to STAG2 loss because we observed increases in Bub3 localization at kinetocores. Regardless of the specific molecular defects, we show that the strategic destabilization of kMT attachments in cells lacking STAG2 function is sufficient to increase the efficiency of correction of kMT attachment errors and restore faithful chromosome segregation in cells devoid of STAG2. This demonstrates that alterations in kMT attachment stability are the root cause of chromosomal instability caused by STAG2 loss of function.
STAG2 is not essential to the mechanical cohesion of sister chromatids during mitosis, because its loss does not induce premature sister separation as observed when other components of the cohesin ring are lost (Michaelis et al., 1997; Hoque and Ishikawa, 2002; Toyoda and Yanagida, 2006; Canudas and Smith, 2009; Solomon et al., 2011). This suggests that there might be partial compensation by other factors such as STAG1 or that the role played by STAG2 in cohesion is regulatory and not mechanical. Consistent with a regulatory role, the loss of STAG2 disrupts the localization and activity of multiple proteins and protein complexes at centromeres, including Aurora B kinase, Bub1 kinase and Bub3, and it might also disrupt the localization or activity of other centromere components to which it has been linked, including Plk1 and Sgo1 (Kettenbach et al., 2011; Liu et al., 2013; Shintomi and Hirano, 2009). However, these changes are selective, because not all kinases are affected by the loss of STAG2, as demonstrated by the quantity of phospho-H3 T3, which is a marker of haspin kinase activity.
How STAG2 influences the localization and activity of some centromeric/kinetochore proteins remains an open question. One possibility is that it influences the activity or accessibility of the kinetochore protein Knl1, which, when phosphorylated by Mps1, has been shown to be a docking site for Bub3 and Bub1 kinase and a factor in Aurora B kinase localization and activity (Shepperd et al., 2012; Yamagishi et al., 2012; Caldas et al., 2013). Another possibility is that STAG2 stabilizes the positioning of cohesin rings on centromeric chromatin to create platforms for the appropriate phosphorylation of histone subunits and the recruitment of centromeric-binding proteins. These views are consistent with a recent report showing that the cohesin-interacting protein Pds5b also promotes Aurora B localization at centromeres (Carretero et al., 2013), although it affected haspin kinase activity, and not Bub1. This indicates that cohesin structure and function at centromeres is subject to regulation by multiple factors to ensure faithful chromosome segregation.
In conclusion, these data show that loss-of-function mutations in STAG2 directly undermine the fidelity of chromosome segregation, leading to whole-chromosome instability. This provides a straight-forward explanation for why tumors that have loss-of-function mutations in STAG2 are often aneuploid and display CIN. CIN generates intra-tumor genetic heterogeneity and correlates with tumor aggressiveness, drug-resistance and tumor recurrence (Choi et al., 2009; Heilig et al., 2010; Bakhoum et al., 2011; Bakhoum and Compton, 2012). Our data provide insight into strategies to suppress CIN caused by loss of STAG2 and, perhaps, alter the aggressiveness of tumors.
MATERIALS AND METHODS
Cell culture
RPE1, PA-RPE1 and H4 cells were grown in Dulbecco's Modification of Eagle's Medium (DMEM; Corning) and HCT116 cells were grown in McCoy's 5A (Iwakata and Grace Modification; Corning) at 37°C in a humidified atmosphere with 5% CO2. All medium was supplemented with 10% bovine growth serum (HyClone) and penicillin-streptomycin at 50 U penicillin and 50 µg streptomycin per ml (Lonza). Medium for cells stably expressing GFP constructs was supplemented with 1 mg/ml G418 Sulfate (InvivoGen) for clonal selection and 0.5 mg/ml for outgrowth. Nocodazole (EMD Millipore) for mitotic arrest was used at 300 ng/ml.
Cell transfection
Plasmids encoding GFP-tagged STAG2 (Addgene), MCAK, Kif2b and GFP vector control (Manning et al., 2007) were transfected into cells by using FuGENE 6 (Roche Diagnostics), and cells were analyzed at 24 hours post-transfection. For stable transgene expression, cells were placed under G418 selection. Clones were isolated after 14–18 days and screened for GFP expression. siRNA transfections were conducted using Oligofectamine (Invitrogen), and cells were analyzed 48 hours later. RNA duplexes for STAG2 (5′-CCGAAUGAAUGGUCAUCAC-3′) were purchased from Ambion. For STAG2 rescue experiments, cells were transfected with STAG2 siRNA using Oligofectamine (Invitrogen) and, 24 hours later, were transfected with GFP vector or GFP-Kif2B plasmids using FuGENE 6 (Roche Diagnostics); cells were analyzed 24 hours after GFP transfection (48 hours after RNAi).
Antibodies
Antibodies used for this study were against: ACA (CREST; Geisel School of Medicine, Dartmouth College, Hanover, NH), Hec1 (Novus Biologicals), actin (Seven Hills Bioreagents), tubulin (DM1α; Sigma-Aldrich), GFP (Invitrogen), STAG2 (Santa Cruz), Aurora B (Novus Biologicals), Bub1 (Abcam), Bub3 (Abcam), phospho-H3 S10 (Cell Signaling Technologies), phospho-Knl1 [a gift from Iain Cheeseman (Whitehead Institute for Biomedical Research, Cambridge, MA)], phospho-H3 T3 [a gift from Jonathan Higgins (Brigham and Women’s Hospital, Boston, MA)] and phospho-H2A T120 (Active Motif). Secondary antibodies were conjugated to fluorescein isothiocyanate (FITC) (Jackson ImmunoResearch), Texas Red (Jackson ImmunoResearch), Cy5 (Invitrogen) or HRP (Jackson ImmunoResearch). DNA was stained with Hoechst 33342 (VWR). Immunoblots were detected using Lumiglow (Kirkegaard & Perry Laboratories).
Fluorescence in situ hybridization
FISH analyses were performed as described previously (Thompson and Compton, 2008). Briefly, clones were isolated using glass cloning rings, grown in 100-mm dishes to >90% confluency and collected by trypsinization. Cells were washed with PBS and treated with 75 mM KCl for 15 min. Cells were then fixed in and washed twice with 75% methanol and 25% acetic acid fixation solution. FISH probes used in this study were Aquarius satellite enumeration probes (Cytocell, Rainbow Scientific) for chromosomes 3 (red, 3r), 4 (red, 4r), 7 (green, 7g) and 10 (green, 10g).
Photoactivation
Photoactivation was performed as described previously (Zhai et al., 1995; Kabeche and Compton, 2013). Fluorescence and differential interference contrast (DIC) microscopy images were acquired using a Quorum WaveFX-X1 spinning-disk confocal system (Quorum Technologies, Guelph, Canada) equipped with a Hamamatsu ImageEM camera (Bridgewater, NJ). Microtubules were locally activated on one half of the spindle using a Mosaic digital mirror (Andor Technology, South Windsor, CT). Fluorescence images were captured every 15 s for 4 min with a 100× oil-immersion 1.4 numerical aperture objective. DIC microscopy was used to select mitotic cells and to verify that a bipolar spindle was maintained throughout image acquisition and that cells had not entered anaphase.
To quantify fluorescence dissipation after photoactivation, pixel intensities were measured within a 1-µm rectangular area surrounding the region of highest fluorescence intensity, and background was subtracted using an equal area from the non-activated half spindle. The values were corrected for photobleaching by treating cells with 10 µM taxol and determining the percentage of fluorescence loss during 4 min of image acquisition after photoactivation. Fluorescence values were normalized to the first time-point after photoactivation for each cell and the average intensity at each time-point was fitted to a double exponential curve using MatLab (Mathworks): A1×exp(−k1t)+A2×exp(−k2t). Here, A1 represents the less stable non-kMT population and A2 is the more stable kMT population, with decay rates of k1 and k2, respectively. The turnover half-life for each process was calculated as ln2/k for each population of microtubules.
Immunofluorescence microscopy
Cells were grown on 18-mm coverslips in 12-well dishes (Nunc) and then fixed with 3.5% paraformaldehyde for 15 minutes, quenched twice with 500 mM ammonium chloride in PBS for 10 minutes each, washed with Tris-buffered saline containing 5% bovine serum albumin (BSA) (TBS-BSA) plus 0.5% Triton X-100 for 5 minutes, and washed with TBS-BSA for 5 minutes. For Bub3, phospho-Knl1 and phospho-H3 T3, cells were pre-treated with microtubule-stabilizing buffer [MTSB (pH 6.8); 4 M glycerol, 100 mM PIPES, 1 mM EGTA and 5 mM MgCl2]. They were then treated with MTSB plus 0.05% Triton X-100 and washed with MTSB again for 2 min each before fixing with paraformaldehyde. For Aurora B, phospho-H2A T120, Bub3, phospho-Knl1 and phospho-H3 T3 staining, cells were permeabilized with −20°C methanol for 5 minutes after paraformaldehyde fixation. Antibodies were diluted in TBS-BSA plus 0.1% Triton X-100, and coverslips were incubated for 1 hour at room temperature. After incubation, coverslips were washed with TBS-BSA for 5 minutes with shaking. Secondary antibodies were diluted in TBS-BSA plus 0.1% Triton X-100 with Hoechst 33342 and coverslips were incubated for 1 hour at room temperature.
Immunofluorescent staining for STAG2 was used to directly identify specific STAG2-deficient cells when scoring for Bub1 and phospho-histone H3 S10 localization. In other assays, STAG2 levels were not directly assessed, indicating that the reported values underestimate the effect of loss of STAG2. All experiments performed with GFP-tagged plasmids pre-selected individual cells based on GFP expression and localization of the transgene, if applicable. P-values for immunofluorescence quantification were calculated with the two-way ANOVA test using Graph Pad Prism 5 software.
Images for FISH, Bub1, phospho-H3 S10, Bub3, phospho-H3 T3, phospho-Knl1, mitotic analyses and lagging chromosomes were acquired with an Orca-ER Hamamatsu cooled CCD camera mounted on an Eclipse TE 2000-E Nikon microscope. Sister separation and Bub1, Bub3, phospho-H3 S10, phospho-Knl1 and phospho-H3 T3 staining images were taken with 0.2-µm optical sections in the z-axis and were collected with plan Apo 60× or 100× 1.4 NA oil-immersion objectives at room temperature. Iterative restoration was performed using Phylum Live software (Improvision) for DNA and centromere images; all but phospho-Knl1 panels are raw images to show pixel intensities. Images for Aurora B, INCENP and phospho-H2A T120 were acquired using a Quorum WaveFX-X1 spinning-disk confocal system (Quorum Technologies); 0.1-µm optical sections were taken in the z-axis at room temperature.
Anaphase chromatids were counted as lagging if they contained both Hoechst 33342 and centromere staining (using CREST antibody) and persisted in the spindle midzone, separate from centromeres segregating to the poles. For quantitative assessments, cells were fixed and stained for Aurora B, Bub1, phospho-H3 S10, phospho-H2A T120, Bub3, phospho-Knl1, phospho-H3 T3, CREST, STAG2 and DNA. Raw pixel intensities for Aurora B, Bub1, phospho-H2A T120, Bub3 and phospho-Knl1 staining were measured in 10–15 regions over the entire cell. Raw pixel intensities for phospho-H3 S10 and phospho-H3 T3 were measured at the chromosome region for each cell. Background fluorescence was subtracted, and the ratio of intensities was calculated and averaged over multiple kinetochores from multiple mitotic cells in three independent experiments (n≥10 cells).
Measurements of inter-centromere and inter-kinetochore distances were made with Phylum Live software (Improvision). Measurements were performed on ≥10 centromeres pairs per cell in n≥10 cells for three independent experiments. Error bars represent the standard error of the mean (s.e.m.). The Student's t-test was used to calculate the significance of the differences between samples.
Supplementary Material
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
M.K. and L.K. participated in experimental design and execution, data interpretation and manuscript preparation. D.A.C. participated in experimental design, data interpretation and manuscript preparation.
Funding
This work was supported by funding from the National Institutes of Health [grant numbers GM51542 to D.A.C. and GM008704 to L.K.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.151613/-/DC1
References
- Bailey M. L., O'Neil N. J., van Pel D. M., Solomon D. A., Waldman T., Hieter P. (2013). Glioblastoma cells containing mutations in the cohesin component, STAG2, are sensitive to PARP inhibition. Mol. Cancer Ther [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum S. F., Compton D. A. (2012). Chromosomal instability and cancer: a complex relationship with therapeutic potential. J. Clin. Invest. 122, 1138–1143 10.1172/JCI59954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum S. F., Thompson S. L., Manning A. L., Compton D. A. (2009a). Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat. Cell Biol. 11, 27–35 10.1038/ncb1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum S. F., Genovese G., Compton D. A. (2009b). Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr. Biol. 19, 1937–1942 10.1016/j.cub.2009.09.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum S. F., Danilova O. V., Kaur P., Levy N. B., Compton D. A. (2011). Chromosomal instability substantiates poor prognosis in patients with diffuse large B-cell lymphoma. Clin. Cancer Res. 17, 7704–7711 10.1158/1078-0432.CCR-11-2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbás-Martínez C., Sagrera A., Carrillo-de-Santa-Pau E., Earl J., Márquez M., Vazquez M., Lapi E., Castro-Giner F., Beltran S., Bayés M. et al. (2013). Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat. Genet. 45, 1464–1469 10.1038/ng.2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber T. D., McManus K., Yuen K. W. Y., Reis M., Parmigiani G., Shen D., Barrett I., Nouhi Y., Spencer F., Markowitz S. et al. (2008). Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. U.S.A. 105, 3443–3448 10.1073/pnas.0712384105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbero J. L. (2009). Cohesins: chromatin architects in chromosome segregation, control of gene expression and much more. Cell. Mol. Life Sci. 66, 2025–2035 10.1007/s00018-009-0004-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbero J. L. (2011). Sister chromatid cohesion control and aneuploidy. Cytogenet. Genome Res. 133, 223–233 10.1159/000323507 [DOI] [PubMed] [Google Scholar]
- Becker M., Stolz A., Ertych N., Bastians H. (2010). Centromere localization of INCENP-Aurora B is sufficient to support spindle checkpoint function. Cell Cycle 9, 1360–1372 10.4161/cc.9.7.11177 [DOI] [PubMed] [Google Scholar]
- Bernardes V. F., Correa G. T. B., Loyola A. M., Cardoso S. V., de Paula A. M. B., Cabral M. M. D. Á., Gomez R. S., Gomes C. C. (2013). STAG2 expression in oral cancer and potentially malignant lesions. Tumour Biol. 35, 3641–3645 [DOI] [PubMed] [Google Scholar]
- Caldas G. V., DeLuca K. F., DeLuca J. G. (2013). KNL1 facilitates phosphorylation of outer kinetochore proteins by promoting Aurora B kinase activity. J. Cell Biol. 203, 957–969 10.1083/jcb.201306054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canudas S., Smith S. (2009). Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J. Cell Biol. 187, 165–173 10.1083/jcb.200903096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero M., Ruiz-Torres M., Rodríguez-Corsino M., Barthelemy I., Losada A. (2013). Pds5B is required for cohesion establishment and Aurora B accumulation at centromeres. EMBO J. 32, 2938–2949(advance online publication) 10.1038/emboj.2013.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C-M., Seo K. W., Jang S. J., Oh Y-M., Shim T-S., Kim W. S., Lee D-S., Lee S-D. (2009). Chromosomal instability is a risk factor for poor prognosis of adenocarcinoma of the lung: Fluorescence in situ hybridization analysis of paraffin-embedded tissue from Korean patients. Lung Cancer 64, 66–70 10.1016/j.lungcan.2008.07.016 [DOI] [PubMed] [Google Scholar]
- Cimini D. (2007). Detection and correction of merotelic kinetochore orientation by Aurora B and its partners. Cell Cycle 6, 1558–1564 10.4161/cc.6.13.4452 [DOI] [PubMed] [Google Scholar]
- Crosio C., Fimia G. M., Loury R., Kimura M., Okano Y., Zhou H., Sen S., Allis C. D., Sassone-Corsi P. (2002). Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol. Cell. Biol. 22, 874–885 10.1128/MCB.22.3.874-885.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J., Sultan S., Taylor S. S., Higgins J. M. G. (2005). The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev. 19, 472–488 10.1101/gad.1267105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X., Zhang P. (2011). Aneuploidy and tumorigenesis. Semin. Cell Dev. Biol. 22, 595–601 10.1016/j.semcdb.2011.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem N. J., Godinho S. A., Pellman D. (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guénette S., Magendantz M., Solomon F. (1995). Suppression of a conditional mutation in alpha-tubulin by overexpression of two checkpoint genes. J. Cell Sci. 108, 1195–1204 [DOI] [PubMed] [Google Scholar]
- Haarhuis J. H. I., Elbatsh A. M. O., van den Broek B., Camps D., Erkan H., Jalink K., Medema R. H., Rowland B. D. (2013). WAPL-mediated removal of cohesin protects against segregation errors and aneuploidy. Curr. Biol. 23, 2071–2077 10.1016/j.cub.2013.09.003 [DOI] [PubMed] [Google Scholar]
- Heilig C. E., Löffler H., Mahlknecht U., Janssen J. W. G., Ho A. D., Jauch A., Krämer A. (2010). Chromosomal instability correlates with poor outcome in patients with myelodysplastic syndromes irrespectively of the cytogenetic risk group. J. Cell. Mol. Med. 14, 895–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T., Lipp J. J., Toh B-H., Peters J-M. (2005). Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 438, 1176–1180 10.1038/nature04254 [DOI] [PubMed] [Google Scholar]
- Holland A. J., Cleveland D. W. (2009). Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 10, 478–487 10.1038/nrm2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M. T., Ishikawa F. (2002). Cohesin defects lead to premature sister chromatid separation, kinetochore dysfunction, and spindle-assembly checkpoint activation. J. Biol. Chem. 277, 42306–42314 10.1074/jbc.M206836200 [DOI] [PubMed] [Google Scholar]
- Kabeche L., Compton D. A. (2013). Cyclin A regulates kinetochore microtubules to promote faithful chromosome segregation. Nature 502, 110–113(advance online publication) 10.1038/nature12507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima S. A., Yamagishi Y., Honda T., Ishiguro K., Watanabe Y. (2010). Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science 327, 172–177 10.1126/science.1180189 [DOI] [PubMed] [Google Scholar]
- Kettenbach A. N., Schweppe D. K., Faherty B. K., Pechenick D., Pletnev A. A., Gerber S. A. (2011). Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal. 4, rs5 10.1126/scisignal.2001497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. S., Kim S. S., Je E. M., Yoo N. J., Lee S. H. (2012). Mutational and expressional analyses of STAG2 gene in solid cancers. Neoplasma 59, 524–529 10.4149/neo_2012_067 [DOI] [PubMed] [Google Scholar]
- Kon A., Shih L-Y., Minamino M., Sanada M., Shiraishi Y., Nagata Y., Yoshida K., Okuno Y., Bando M., Nakato R. et al. (2013). Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 45, 1232–1237 10.1038/ng.2731 [DOI] [PubMed] [Google Scholar]
- Kong X., Ball A. R., Pham H. X., Zeng W., Chen H. -Y., Schmiesing J. A., Kim J. -S., Berns M., Yokomori K. (2014). Distinct functions of human cohesin-SA1 and cohesin-SA2 in double-strand break repair. Mol. Cell. Biol. 34, 685–698 10.1128/MCB.01503-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M. S., Stojanov P., Mermel C. H., Robinson J. T., Garraway L. A., Golub T. R., Meyerson M., Gabriel S. B., Lander E. S., Getz G. (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 10.1038/nature12912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. J. X., Endesfelder D., Rowan A. J., Walther A., Birkbak N. J., Futreal P. A., Downward J., Szallasi Z., Tomlinson I. P. M., Howell M. et al. (2011). Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 71, 1858–1870 10.1158/0008-5472.CAN-10-3604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer C., Kinzler K. W., Vogelstein B. (1998). Genetic instabilities in human cancers. Nature 396, 643–649 10.1038/25292 [DOI] [PubMed] [Google Scholar]
- Liu H., Jia L., Yu H. (2013). Phospho-H2A and cohesin specify distinct tension-regulated sgo1 pools at kinetochores and inner centromeres. Curr. Biol. 23, 1927–1933 [DOI] [PubMed] [Google Scholar]
- Loncarek J., Kisurina-Evgenieva O., Vinogradova T., Hergert P., La Terra S., Kapoor T. M., Khodjakov A. (2007). The centromere geometry essential for error-free mitosis is controlled by spindle forces. Nature 450, 745–749 10.1038/nature06344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning A. L., Ganem N. J., Bakhoum S. F., Wagenbach M., Wordeman L., Compton D. A. (2007). The kinesin-13 proteins Kif2a, Kif2b, and Kif2c/MCAK have distinct roles during mitosis in human cells. Mol. Biol. Cell 18, 2970–2979 10.1091/mbc.E07-02-0110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning A. L., Longworth M. S., Dyson N. J. (2010). Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 24, 1364–1376 10.1101/gad.1917310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannini L., Musio A. (2011). The dark side of cohesin: the carcinogenic point of view. Mutat. Res. 728, 81–87 10.1016/j.mrrev.2011.07.004 [DOI] [PubMed] [Google Scholar]
- Mehta G. D., Kumar R., Srivastava S., Ghosh S. K. (2013). Cohesin: functions beyond sister chromatid cohesion. FEBS Lett. 587, 2299–2312 10.1016/j.febslet.2013.06.035 [DOI] [PubMed] [Google Scholar]
- Michaelis C., Ciosk R., Nasmyth K. (1997). Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91, 35–45 10.1016/S0092-8674(01)80007-6 [DOI] [PubMed] [Google Scholar]
- Peters J-M., Tedeschi A., Schmitz J. (2008). The cohesin complex and its roles in chromosome biology. Genes Dev. 22, 3089–3114 10.1101/gad.1724308 [DOI] [PubMed] [Google Scholar]
- Ricke R. M., Jeganathan K. B., Malureanu L., Harrison A. M., van Deursen J. M. (2012). Bub1 kinase activity drives error correction and mitotic checkpoint control but not tumor suppression. J. Cell Biol. 199, 931–949 10.1083/jcb.201205115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera T., Ghenoiu C., Rodríguez-Corsino M., Mochida S., Funabiki H., Losada A. (2012). Xenopus Shugoshin 2 regulates the spindle assembly pathway mediated by the chromosomal passenger complex. EMBO J. 31, 1467–1479 10.1038/emboj.2012.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon E. D., Cimini D., Cameron L. A., DeLuca J. G. (2005). Merotelic kinetochores in mammalian tissue cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepperd L. A., Meadows J. C., Sochaj A. M., Lancaster T. C., Zou J., Buttrick G. J., Rappsilber J., Hardwick K. G., Millar J. B. A. (2012). Phosphodependent recruitment of Bub1 and Bub3 to Spc7/KNL1 by Mph1 kinase maintains the spindle checkpoint. Curr. Biol. 22, 891–899 10.1016/j.cub.2012.03.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintomi K., Hirano T. (2009). Releasing cohesin from chromosome arms in early mitosis: opposing actions of Wapl-Pds5 and Sgo1. Genes Dev. 23, 2224–2236 10.1101/gad.1844309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkworth W. T., Cimini D. (2012). Transient defects of mitotic spindle geometry and chromosome segregation errors. Cell Div. 7, 19 10.1186/1747-1028-7-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon D. A., Kim T., Diaz-Martinez L. A., Fair J., Elkahloun A. G., Harris B. T., Toretsky J. A., Rosenberg S. A., Shukla N., Ladanyi M. et al. (2011). Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 333, 1039–1043 10.1126/science.1203619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor C. F., Platt F. M., Hurst C. D., Thygesen H. H., Knowles M. A. (2013). Frequent inactivating mutations of STAG2 in bladder cancer are associated with low tumor grade and stage and inversely related to chromosomal copy number changes. Hum. Mol. Genet. 23, 1964–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network(2014). Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507, 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thol F., Bollin R., Gehlhaar M., Walter C., Dugas M., Suchanek K. J., Kirchner A., Huang L., Chaturvedi A., Wichmann M. et al. (2013). Mutations in the cohesin complex in acute myeloid leukemia: clinical and prognostic implications. Blood 123, 914–920 [DOI] [PubMed] [Google Scholar]
- Thompson S. L., Compton D. A. (2008). Examining the link between chromosomal instability and aneuploidy in human cells. J. Cell Biol. 180, 665–672 10.1083/jcb.200712029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S. L., Compton D. A. (2010). Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 188, 369–381 10.1083/jcb.200905057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S. L., Compton D. A. (2011). Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc. Natl. Acad. Sci. USA 108, 17974–17978 10.1073/pnas.1109720108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S. L., Bakhoum S. F., Compton D. A. (2010). Mechanisms of chromosomal instability. Curr. Biol. 20, R285–R295 10.1016/j.cub.2010.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda Y., Yanagida M. (2006). Coordinated requirements of human topo II and cohesin for metaphase centromere alignment under Mad2-dependent spindle checkpoint surveillance. Mol. Biol. Cell 17, 2287–2302 10.1091/mbc.E05-11-1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther A., Houlston R., Tomlinson I. (2008). Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut 57, 941–950 10.1136/gut.2007.135004 [DOI] [PubMed] [Google Scholar]
- Watanabe Y. (2010). Temporal and spatial regulation of targeting aurora B to the inner centromere. Cold Spring Harb. Symp. Quant. Biol. 75, 419–423 10.1101/sqb.2010.75.035 [DOI] [PubMed] [Google Scholar]
- Weaver B. A. A., Silk A. D., Montagna C., Verdier-Pinard P., Cleveland D. W. (2007). Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11, 25–36 10.1016/j.ccr.2006.12.003 [DOI] [PubMed] [Google Scholar]
- Welburn J. P. I., Vleugel M., Liu D., Yates J. R., 3rd, Lampson M. A., Fukagawa T., Cheeseman I. M. (2010). Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol. Cell 38, 383–392 10.1016/j.molcel.2010.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams B. R., Prabhu V. R., Hunter K. E., Glazier C. M., Whittaker C. A., Housman D. E., Amon A. (2008). Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322, 703–709 10.1126/science.1160058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi Y., Yang C-H., Tanno Y., Watanabe Y. (2012). MPS1/Mph1 phosphorylates the kinetochore protein KNL1/Spc7 to recruit SAC components. Nat. Cell Biol. 14, 746–752 10.1038/ncb2515 [DOI] [PubMed] [Google Scholar]
- Yan M., Xu H., Waddell N., Shield-Artin K., Haviv I., McKay M. J., Fox S. B. kConFab authors(2012). Enhanced RAD21 cohesin expression confers poor prognosis in BRCA2 and BRCAX, but not BRCA1 familial breast cancers. Breast Cancer Res. 14, R69 10.1186/bcr3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y., Kronebusch P. J., Borisy G. G. (1995). Kinetochore microtubule dynamics and the metaphase-anaphase transition. J. Cell Biol. 131, 721–734 10.1083/jcb.131.3.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.