

ABSTRACT
Kindlins are essential FERM-domain-containing focal adhesion (FA) proteins required for proper integrin activation and signaling. Despite the widely accepted importance of each of the three mammalian kindlins in cell adhesion, the molecular basis for their function has yet to be fully elucidated, and the functional differences between isoforms have generally not been examined. Here, we report functional differences between kindlin-2 and -3 (also known as FERMT2 and FERMT3, respectively); GFP-tagged kindlin-2 localizes to FAs whereas kindlin-3 does not, and kindlin-2, but not kindlin-3, can rescue α5β1 integrin activation defects in kindlin-2-knockdown fibroblasts. Using chimeric kindlins, we show that the relatively uncharacterized kindlin-2 F2 subdomain drives FA targeting and integrin activation. We find that the integrin-linked kinase (ILK)–PINCH–parvin complex binds strongly to the kindlin-2 F2 subdomain but poorly to that of kindlin-3. Using a point-mutated kindlin-2, we establish that efficient kindlin-2-mediated integrin activation and FA targeting require binding to the ILK complex. Thus, ILK-complex binding is crucial for normal kindlin-2 function and differential ILK binding contributes to kindlin isoform specificity.
KEY WORDS: Focal adhesion, Integrin, Integrin activation, Integrin-linked kinase, Kindlin
INTRODUCTION
Integrins are essential heterodimeric transmembrane adhesion receptors that mediate bi-directional signaling between cells and the extracellular matrix (ECM) (Harburger and Calderwood, 2009). As such, integrins govern a range of physiological processes including cell migration, tissue organization, angiogenesis, hemostasis and the immune response. Integrins can be found in an ‘inactive’ state with relatively low affinity for ECM ligands or in an ‘active’ conformation with high ligand-binding affinity (Calderwood, 2004a). Integrin activation (the conversion from inactive to active states) can be triggered by binding to ECM or by intracellular signaling events that culminate in the binding of the cytoskeletal adaptor protein talin to the cytoplasmic tail of the integrin β subunit (Calderwood et al., 2013; Shattil et al., 2010; Ye et al., 2012). Once activated or engaged by ECM molecules, integrin receptors can cluster to form focal adhesions (FAs) (Harburger and Calderwood, 2009; Wehrle-Haller, 2012). These complex multi-protein assemblies contain a large array of cytoskeletal, scaffolding and signaling proteins (Geiger and Zaidel-Bar, 2012; Harburger and Calderwood, 2009) and serve as anchorage points for the cytoskeleton and sites at which the cell senses and applies mechanical force.
Kindlins are a family of proteins that bind to the integrin cytoplasmic tail and that are crucial for normal integrin regulation and signaling (Calderwood et al., 2013; Karaköse et al., 2010; Larjava et al., 2008). Loss of the mainly epithelial kindlin-1 (also known as FERMT1) leads to Kindler syndrome, manifesting in skin blistering and ulcerative colitis (Siegel et al., 2003; Ussar et al., 2008). Knockout of the ubiquitously expressed kindlin-2 (also known as FERMT2) halts murine development at the peri-implantation stage due to cell detachment from the basement membrane (Montanez et al., 2008). Finally, loss of the hematopoietic-specific kindlin-3 (also known as FERMT3) causes leukocyte adhesion deficiency type III (LADIII) (Kuijpers et al., 2009; Malinin et al., 2009; Moser et al., 2009; Svensson et al., 2009), which is characterized by immune defects and severe bleeding.
Impaired integrin activation is a common feature of kindlin disease or knockout phenotypes, strongly implicating all three kindlins in integrin activation and integrin-mediated processes. This is supported by studies of knockout and knockdown cells and by overexpression experiments (Dowling et al., 2008; Harburger et al., 2009; Ma et al., 2008; Malinin et al., 2009; Moser et al., 2009; Moser et al., 2008; Shi et al., 2007; Ussar et al., 2008). For example, when kindlin-2 is depleted, cells exhibit defects in β1 and β3 integrin activation, FA formation and cell spreading (Montanez et al., 2008; Pluskota et al., 2011; Shi et al., 2007). Similar defects are reported following knockout or knockdown of kindlin-1 (Ussar et al., 2008), and loss of kindlin-3 results in impaired activation of β1, β2 and β3 integrins (Moser et al., 2009; Moser et al., 2008). However, although all three kindlin isoforms are key regulators of cell adhesion, they also appear to have isoform-specific functions. For instance, endogenous kindlin-2 is unable to fully compensate for loss of kindlin-1 in the skin or for non-functional kindlin-3 in hematopoietic cells (Bialkowska et al., 2010; Lai-Cheong et al., 2008; Malinin et al., 2009; Moser et al., 2009; Ussar et al., 2008). Furthermore, although all three isoforms bind to the integrin β subunit cytoplasmic tails, differences in subcellular and tissue localization have been observed. For example, whereas kindlin-1 and kindlin-2 both target to FAs in adherent cells, kindlin-1 targets preferentially to αvβ6-rich adhesions in β1-knockout cells (Bandyopadhyay et al., 2012), and kindlin-3 does not localize to FAs (Bialkowska et al., 2010; Ussar et al., 2006). The molecular basis for the differences in isoform localization and function is generally not understood.
Despite recent progress (Ye et al., 2013), the mechanisms by which kindlins affect integrin activation and signaling remain to be fully determined. Kindlins are made up of an atypical FERM (band 4.1, ezrin, radixin, moesin) domain composed of four subdomains (F0, F1, F2 and F3) with a pleckstrin homology (PH) domain nested within the F2 subdomain (Calderwood et al., 2013; Goult et al., 2009). The lack of recognizable enzymatic domains suggests that kindlins act as scaffold or adaptor proteins. Consistent with this, the F3 subdomain contains a binding site for integrin β-tails, and this binding is required for kindlins to enhance talin-mediated integrin activation (Harburger et al., 2009; Ma et al., 2008; Moser et al., 2008; Ussar et al., 2008). However, subdomain deletion and mutagenesis studies show that kindlin function, and localization to adhesions, also requires the F0, F1 and PH domains, at least in part because of membrane-binding sites in these domains (Bouaouina et al., 2012a; Harburger et al., 2009; Liu et al., 2011; Perera et al., 2011; Xu et al., 2013; Yates et al., 2012). The inability of isolated kindlin subdomains to support integrin activation or signaling has made characterization of the kindlin mechanism of action difficult.
Besides integrin, integrin-linked kinase (ILK) is one of the few known kindlin-binding proteins (Calderwood et al., 2013). ILK is a FA protein essential for development and normal integrin signaling (Friedrich et al., 2004; Sakai et al., 2003). ILK was originally identified as a binding partner for integrin β-tails in a yeast two-hybrid screen (Hannigan et al., 1996), and is composed of an N-terminal ankyrin repeat domain (ILK-ARD) (Chiswell et al., 2008) followed by a C-terminal kinase domain (ILK-KD) (Fukuda et al., 2009). The kinase domain lacks key catalytic residues, and ILK is generally thought to be a pseudokinase (Fukuda et al., 2009) functioning primarily as an adaptor protein. ILK partners with the LIM domain protein PINCH (through its ARD) and the calponin homology (CH) domain protein parvin (through its kinase domain) (Chiswell et al., 2010; Chiswell et al., 2008; Fukuda et al., 2009; Stiegler et al., 2013) and this tri-protein IPP complex is central to ILK function (Fukuda et al., 2003; Gkretsi et al., 2007; Stanchi et al., 2009; Wang et al., 2008). In addition to PINCH and parvin, ILK interacts directly or indirectly with a range of other FA proteins and is required to maintain FA architecture and undertake integrin-mediated signaling (Elad et al., 2013; Kogata et al., 2009; Sakai et al., 2003; Wickström et al., 2010). Notably, loss of ILK has been reported to inhibit integrin activation in vitro and in vivo as assessed by the binding of soluble ligands, ligand-mimetic antibodies and conformation-specific reporter antibodies (Friedrich et al., 2004; Honda et al., 2009; Tucker et al., 2008). However, little is known about how ILK exerts effects on integrin activation and signaling.
In this study, we investigated the isoform specificity of kindlin functions as a means to identify key kindlin subdomains and their functionally relevant binding partners. We initially observed striking differences in the localization of overexpressed kindlin-3 and kindlin-1 and -2. Most noticeably, kindlin-3 did not localize to FAs. In addition, kindlin-3 expression was unable to reverse the suppression of α5β1 integrin activation triggered by kindlin-2 knockdown in fibroblasts. We found that binding to the IPP complex differs between these two kindlin isoforms. We localized the kindlin-binding site to the ILK-KD–parvin-CH2 domain complex, mapped the ILK-binding site to the kindlin F2 domain, and identified a point mutation that strongly inhibited ILK binding. By using kindlin-2–kindlin-3 chimeras in which the overall kindlin domain structure was preserved, and kindlin mutants defective in binding the ILK complex, we showed that strong ILK-complex binding supports FA targeting and β1 integrin activation. Overall, our work shows that ILK drives kindlin subcellular localization and kindlin-mediated integrin activation, and provides insight into how kindlin and ILK contribute to integrin signaling.
RESULTS
GFP-tagged kindlin-1 and -2, but not kindlin-3, target to β1-integrin-rich FAs in CHO cells
To compare the ability of the three mammalian kindlins to accumulate in FAs, N-terminally GFP-tagged kindlin-1, -2 and -3 were each overexpressed in CHO cells plated on fibronectin and their localization was assessed by fluorescence microscopy. As previously reported (Bouaouina et al., 2012a; Goult et al., 2009; Harburger et al., 2009), GFP–kindlin-1 and -2 were readily detected in FAs at 24 h after replating (Fig. 1A). In clear contrast, GFP–kindlin-3 was always cytoplasmic and did not target to FAs, regardless of expression level (Fig. 1A). We assessed the extent of FA targeting by quantifying blindly the percentage of cells in which the GFP-tagged protein clearly colocalizes with vinculin-rich FAs (Fig. 1B). GFP–kindlin-1 and -2 targeted to FAs in most expressing cells, and kindlin-2 consistently targeted better than kindlin-1 (99% and 86%, respectively). However, we could not observe GFP–kindlin-3 targeting to FAs. As expected (Harburger et al., 2009; Montanez et al., 2008; Ussar et al., 2008), the integrin-binding defective mutants GFP–kindlin-1-W612A and GFP–kindlin-2-W615A failed to accumulate in FAs (Fig. 1B), confirming the specificity of our assays and demonstrating that integrin-binding is required for GFP–kindlin-1 and -2 accumulation at FAs in CHO cells.
Fig. 1.
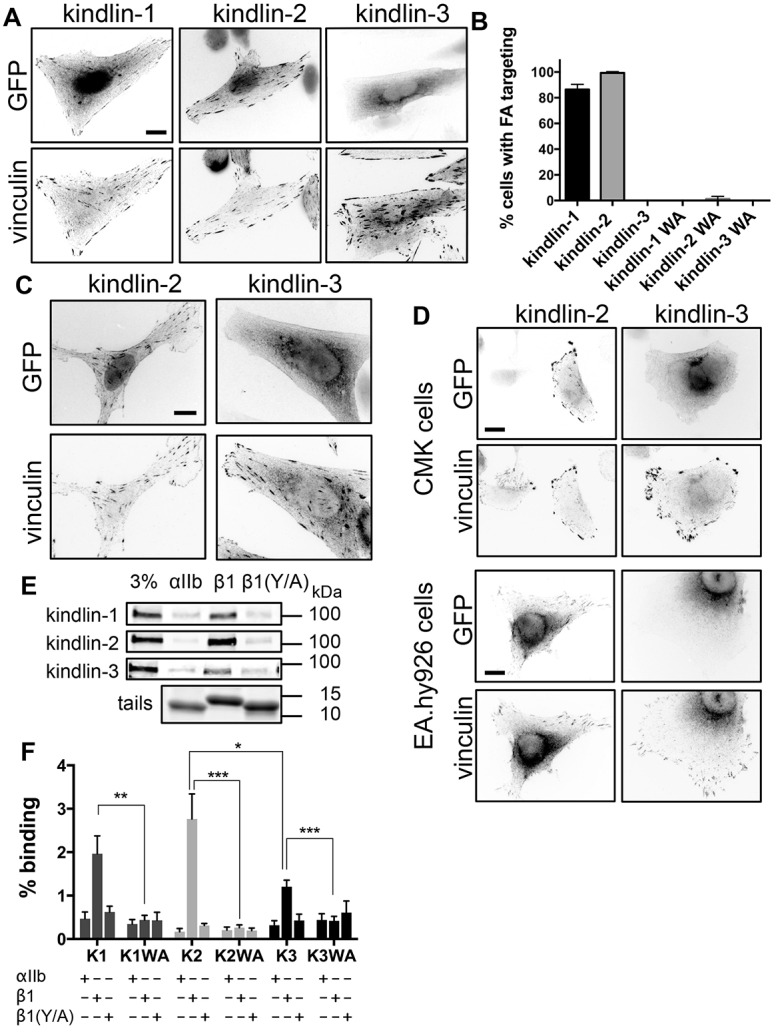
Kindlin isoforms have different subcellular localization and integrin binding. (A) CHO cells overexpressing GFP-tagged kindlins were plated on fibronectin, fixed 24 h later and stained for endogenous vinculin. (B) Percentage of transfected cells (±95% confidence interval) in which the GFP-tagged kindlins and mutants (kindlin-1-W612A, kindlin-2-W615A and kindlin-3-W597A, indicated by WA) target to FAs. n = 160–280 cells from three independent experiments. (C) αIIbβ3-integrin-expressing CHO cells overexpressing GFP-tagged kindlins were plated on fibrinogen, fixed and stained for endogenous vinculin. (D) CMK or EA.hy926 cells overexpressing GFP-tagged kindlins were plated on fibronectin and stained for endogenous vinculin. (E) Pulldown of GFP-tagged kindlins from CHO cell lysates with purified recombinant αIIb, β1 or β1Y795A (β1Y/A) integrin tails. Tail loading was assessed by Coomassie Blue staining. The 3% lane indicates 3% of input lysate. (F) GFP–kindlin-1 (K1), GFP–kindlin-1-W612A (K1WA), GFP–kindlin-2 (K2), GFP–kindlin-2-W615A (K2WA), GFP–kindlin-3 (K3) and GFP–kindlin-3-W597A (K3WA) binding to purified recombinant αIIb, β1 and β1Y/A tails was quantified and expressed as a percentage of input (mean±s.e.m.; n = 4). *P<0.05, **P<0.01, ***P<0.001 (Student's t-test). Scale bars: 10 µm.
The inability of kindlin-3 to accumulate in FAs is not specific to α5β1-mediated adhesions as, consistent with earlier reports (Moser et al., 2008), kindlin-3 also failed to accumulate in αIIbβ3-rich FAs in αIIbβ3-integrin-expressing CHO cells spread on fibrinogen, whereas GFP–kindlin-2 did target (Fig. 1C). Kindlin-3 expression is normally restricted to hematopoietic (Ussar et al., 2006) and endothelial (Bialkowska et al., 2010) cells, therefore a cell-specific cofactor could be needed for kindlin-3 targeting. However, both in megakaryocytic (CMK) and endothelial-like (EA.hy926) cells, GFP–kindlin-2 targeted to FAs, whereas GFP–kindlin-3 did not (Fig. 1D), indicating intrinsic differences between kindlin isoforms that might be due to differences in binding to one or several partners crucial for localization to FAs.
Kindlin-3 binds β1 integrin tails less well than other isoforms
Integrin binding is crucial for kindlin targeting to FAs (Fig. 1B). We therefore compared kindlin isoform binding to recombinant integrin cytoplasmic tails. Lysates from CHO cells expressing GFP-tagged kindlins were incubated with beads coated with purified His-tagged integrin cytoplasmic tails (Lad et al., 2007). All three kindlins bound wild-type β1-tails (Fig. 1E). Binding was specific as it was reduced to background (αIIb) levels by point mutating the membrane-distal NPxY motif in β1 integrin or the conserved tryptophan in the F3 subdomain of the kindlins (Fig. 1E,F). Notably, we observed that β1-tails pulled down significantly less kindlin-3 than kindlin-2 (Fig. 1F), suggesting that this could contribute to the inability of kindlin-3 to target to FAs.
Differences in FA targeting of kindlin isoforms are only partially due to differences in the F3 subdomain
We and others have shown that deletion or mutation of the kindlin F0, F1, PH or F3 domain impairs FA targeting (Bouaouina et al., 2012a; Goult et al., 2009; Harburger et al., 2009; Liu et al., 2011; Montanez et al., 2008; Moser et al., 2008; Perera et al., 2011; Ussar et al., 2008; Yates et al., 2012). To map the regions responsible for isoform-specific differences in FA targeting, we therefore generated chimeras by swapping predicted FERM subdomains between kindlin isoforms (subdomain boundaries and kindlin conservation are indicated in supplementary material Fig. S1). We named chimeric kindlins using four digits, representing the kindlin F0, F1, F2 or F3 subdomain, with the number indicating the isoform from which the subdomain originates (Fig. 2A). To test whether the lack of FA localization of kindlin-3 could be explained by reduced integrin binding, we swapped the integrin-binding F3 subdomain between kindlin-2 and kindlin-3. The chimeric kindlins expressed well in CHO cells, were of the expected molecular masses and bound β1 integrin tail specifically (Fig. 2B). Notably, the chimera K3332 had enhanced integrin binding, comparable to that of kindlin-2, whereas K2223 had diminished integrin binding, like that of kindlin-3 (Fig. 2B,C). Hence, we conclude that the difference between kindlin-2 and kindlin-3 in binding integrin is due to properties of their F3 subdomains.
Fig. 2.

The kindlin F3 subdomain contributes to isoform-specific FA targeting through differences in integrin binding. (A) Schematic representation of chimeric kindlins and chimera nomenclature. (B,C) Binding of GFP-tagged kindlin chimeras from CHO cell lysates to αIIb, β1 and β1 Y795A (β1Y/A) was assessed by immunoblotting, quantified (C) and normalized to starting material loading (mean±s.e.m.; n = 5). The 3% lane indicates 3% of input lysate. *P<0.05, **P<0.01 (Student's t-test). (D) Localization of GFP-tagged kindlins, and endogenous vinculin, in CHO cells plated on fibronectin. Scale bars: 10 µm. (E) Percentage of transfected cells (±95% confidence interval) in which the GFP-tagged kindlins and chimeras target to FAs. n = 160 cells from three independent experiments.
When we examined subcellular localization of these GFP-tagged chimeras, we found that K2223 exhibited diminished FA targeting when compared to kindlin-2 (Fig. 2D,E). Conversely, K3332 had a modest gain-of-function when compared to kindlin-3 (Fig. 2D). In addition to scoring the percentage of cells exhibiting FA targeting, we used a blinded scoring system to categorize the intensity of GFP signal at adhesions. Briefly, a score of 0 indicated no targeting, 1 indicated low targeting, 2 indicated medium targeting (like that of GFP–kindlin-1), 3 indicated strong targeting (like that of GFP–kindlin-2), and 4 indicated very strong targeting. When assessed by this method, both GFP–K2223 and GFP–K3332 targeted significantly less well (P<0.05, paired Student's t-test) than wild-type GFP–kindlin-2 [FA targeting indices of 1.75±0.25 for K2223 and 1.25±0.25 for K3332 versus 3.0±0.00 for kindlin-2 (mean±s.e.m., n = 4)]. These results show that the F3 subdomain contributes to the varied abilities of kindlins to target to FAs. However, quantification of targeting (Fig. 2E) indicates that K2223 still targets in most cells (78%) suggesting that the F3 subdomain accounts for only part of the isoform differences. Likewise, despite binding integrin at comparable levels to kindlin-2 (Fig. 2C), K3332 targeted to FAs in only 37% of cells and its intensity in FAs was weak (Fig. 2D). Taken together, these data suggest that another kindlin subdomain makes important contributions to FA targeting.
Differences in the F2F3 subdomains drive differences in FA targeting between kindlin isoforms
Having shown that F3 swaps partially affect kindlin-2 and kindlin-3 FA targeting, we next tested whether swapping both the F2 and F3 subdomains (F2F3) (Fig. 3A) produced a more dramatic effect. All six chimeras resulting from swapping F2F3 between kindlin-1, -2 and -3 expressed well and were of the expected molecular mass (data not shown). Notably, chimeras bearing the kindlin-3 F2F3 (K1133 and K2233) failed to target to FAs, whereas those bearing a F2F3 from kindlin-1 (K3311 and K2211) or kindlin-2 (K3322 and K1122) targeted to FAs as well as kindlin-1 and -2, respectively (Fig. 3B,C). We therefore conclude that the differences within their F2F3 regions are the major drivers of differences in FA targeting between kindlin isoforms.
Fig. 3.

The kindlin F2 subdomain drives isoform-specific FA targeting differences. (A) Schematic representation of the kindlin chimeras. (B) Subcellular localization of GFP-kindlins and endogenous vinculin in CHO cells plated on fibronectin. (C) Percentage of transfected cells (±95% confidence interval) in which the GFP-tagged kindlins and chimeras target to FAs. n = 123–162 from three independent experiments. (D) Subcellular localization of GFP-tagged kindlin chimeras and endogenous vinculin in CHO cells plated on fibronectin. (E) Percentage of transfected cells (±95% confidence interval) in which the GFP-tagged kindlins and chimeras target to FAs. n = 160–482 from three independent experiments. (F,G) Binding of chimeric kindlins to αIIb, β1 and β1 Y795A (β1Y/A) integrin tails was assessed by immunoblotting, quantified and expressed as a percentage of input (mean±s.e.m.; n = 5). The 3% lane indicates 3% of input lysate. **P<0.01 (Student's t-test). (H) Subcellular localization of GFP-tagged kindlin PH chimeras and endogenous vinculin in CHO cells plated on fibronectin. (I) Percentage of transfected cells (±95% confidence interval) in which the GFP-tagged kindlins and chimeras target to FAs. n = 120–400 from three independent experiments. Scale bars: 10 µm.
The F2PH subdomain dictates FA targeting, independently from the F3 subdomain
The preceding results suggest that a major FA-targeting motif resides within the relatively uncharacterized F2PH subdomain of kindlin-2. To test this, we generated chimeras by swapping only the F2 domain plus its inserted PH domain (F2PH) (Fig. 3A). Strikingly, introducing the kindlin-3 F2PH subdomain into kindlin-2 (K2232) was sufficient to abrogate FA targeting almost completely, and, more importantly, introducing the F2PH subdomain of kindlin-2 into kindlin-3 (K3323) generated FA targeting at almost wild-type kindlin-2 levels (Fig. 3D,E). Blind scoring of FA-targeting intensity showed that GFP–K3323 targeted only slightly less well than wild-type GFP–kindlin-2 [FA targeting index of 2.29±0.18 versus 2.86±0.14 (mean±s.e.m., n = 7; P<0.05, paired Student's t-test)]. As expected, both K1121 and K2212 targeted efficiently to FAs (Fig. 3E), but even here the small differences in the percentage of cells showing FA targeting between kindlin-1 and kindlin-2 tracked with the F2PH subdomain. Although the F2PH controlled FA targeting, it did not alter integrin binding, as K2232 and K3323 bound integrin tails similarly to kindlin-2 and kindlin-3, respectively (Fig. 3F,G). Thus K3323 targets to FAs almost as well as kindlin-2, even though it binds integrin tails less well, like kindlin-3. Therefore, our results show that the F2PH region of kindlin drives FA targeting in an isoform-specific fashion. However, consistent with published data describing the importance of F0, F1 and F3 subdomains for FA localization (Bouaouina et al., 2012a; Goult et al., 2009; Harburger et al., 2009; Ma et al., 2008), the isolated GFP-tagged F2PH domain of kindlin-2 was unable to target to FAs (data not shown).
The PH domain embedded in a predicted FERM F2 subdomain is a feature unique to kindlins, and it has been implicated in their FA targeting (Liu et al., 2011). To test whether the PH domain is responsible for isoform-specific FA targeting we swapped it between kindlin-2 and -3. As shown in Fig. 3H,I, this had little impact on targeting, as kindlin-2 containing the kindlin-3 PH domain (K2-K3PH) still efficiently targeted to FAs whereas kindlin-3 containing the kindlin-2 PH domain (K3-K2PH) still failed to target. Taken together, our work is the first to show that the F2PH subdomain bears a strong isoform-specific determinant of FA targeting and points to interactions outside of the PH domain in mediating this effect.
Kindlin-2 F2PH directly interacts with the kinase domain of ILK
To identify binding partners for the kindlin-2 F2PH that are potentially involved in FA targeting, we purified the recombinant GST-tagged F2PH domain of kindlin-2 (K2-F2PH) from E. coli and used it as bait in pulldown assays with CHO cell lysates. Testing candidate proteins, including known kindlin-binding proteins, revealed pulldown of endogenous ILK (Fig. 4A). This interaction is specific as vinculin, another FA protein, was not pulled down by GST–K2-F2PH (Fig. 4A). ILK is of particular interest as it is an essential FA protein previously shown to associate with kindlin in yeast two-hybrid and co-immunoprecipitation assays (Mackinnon et al., 2002; Montanez et al., 2008).
Fig. 4.

Kindlin-2 F2PH binds directly to ILK kinase domain. (A) Binding of endogenous ILK and vinculin from CHO cell lysates to immobilized GST or GST–kindlin-2-F2PH was assessed by immunoblotting (IB). GST protein loading was assessed by Ponceau S staining. (B) Direct binding of purified soluble ILK-ARD + PINCH-LIM1 domain or ILK-KD + parvin-CH2 domain to immobilized GST or GST–kindlin-2-F2PH was assessed by Coomassie Blue staining. (C) Binding of increasing concentrations of the ILK-KD–parvin-CH2 complex, or of parvin-CH2 as a control, to BLItz GST Biosensors coated with GST–kindlin-2-F2PH was measured at room temperature. Binding to GST alone was subtracted and fitted to a curve by nonlinear regression in GraphPad Prism. (D) Binding of GFP–kindlin-2-F2PH from CHO cell lysates to immobilized GST, the GST-tagged parvin-CH2 domain, or GST–ILK-KD in complex with parvin CH2 was assessed by immunoblotting. Inputs (5% or 3%) are shown for A, B and D.
ILK is composed of an N-terminal ankyrin repeat domain (ILK-ARD) and a C-terminal kinase domain (ILK-KD). In vivo, the ARD associates with the N-terminal LIM domain of PINCH, whereas the kinase domain associates with the C-terminal CH2 domain of parvin (Ghatak et al., 2013; Qin and Wu, 2012). Production of ILK-ARD or ILK-KD in E. coli requires co-purification with their respective binding partners (Chiswell et al., 2010; Chiswell et al., 2008; Fukuda et al., 2009; Yang et al., 2009). To identify the kindlin-interacting domain in ILK and test whether the interaction is direct we used GST–K2-F2PH to pull down a purified ILK-KD–α-parvin-CH2 complex and an ILK-ARD–PINCH1-LIM1 complex. As shown in Fig. 4B, GST–K2-F2PH bound the ILK-KD complex but not the ILK-ARD complex. Neither complex bound GST. Using bio-layer interferometry (BLItz, Pall Life Sciences) we estimated an apparent dissociation constant of 4.4±0.8 µM between the kindlin-2 F2PH domain and the ILK-KD complex (Fig. 4C). Although we cannot produce soluble recombinant ILK-KD in the absence of parvin-CH2, we tested whether kindlin bound parvin-CH2 alone. GST, GST–parvin-CH2 or GST–ILK-KD plus His–parvin-CH2 proteins were immobilized on glutathione–Sepharose beads and incubated with lysates of cells expressing GFP–K2-F2PH. The K2-F2PH domain only pulled down with the ILK-KD complex and not with the parvin-CH2 domain alone (Fig. 4D). This is consistent with yeast two-hybrid data (Mackinnon et al., 2002; Qadota et al., 2012) and strongly suggests that the kindlin-2 F2PH region mediates a direct interaction with the ILK kinase domain.
Kindlin-2 and -3 differ in their ILK-binding properties
To evaluate whether differences in ILK binding could explain differences in FA targeting of kindlin isoforms, we compared the ILK-KD binding of kindlin-2 and -3. Unfortunately, we were unable to purify soluble recombinant kindlin-3-F2PH from E. coli. We therefore expressed GFP–kindlin-2-F2PH (K2-F2PH) and GFP–kindlin-3-F2PH (K3-F2PH) in CHO cells and compared their binding to the GST–ILK-KD complex in pulldown assays. Although both F2PH subdomains bound the ILK-KD complex (Fig. 5A), the ILK-KD complex pulled down significantly less GFP–K3-F2PH (Fig. 5B). Similar results were obtained with full-length GFP-tagged kindlins (Fig. 5C,D). Notably, when we examined the F2PH swap-chimeras, the ILK-KD complex pulled down the FA-targeting K3323 chimera more efficiently than the non-targeting K2232 chimera (Fig. 5D). Hence, ILK binding to kindlin differs between isoforms and is determined by the F2PH region. Similar results were obtained using a minimal IPP complex of GST-tagged full-length ILK, complexed with His–parvin-CH2 and His–PINCH1-LIM1 (Stiegler et al., 2013). As we saw with the ILK-KD, kindlin-2 and the FA-targeting K3323 chimera bound preferentially to the minimal IPP (Fig. 5E,F), whereas kindlin-3 and the non-targeting K2232 chimera bound very little. Thus, we have identified a kindlin-isoform-specific determinant that dictates both FA targeting and ILK binding.
Fig. 5.

Kindlin-2 and kindlin-3 differ in ILK binding. (A,B) Pulldown of GFP–kindlin-2-F2PH or GFP–kindlin-3-F2PH from CHO cell lysates by immobilized GST or GST–ILK-KD + parvin-CH2 was assessed by immunoblotting (A) and quantified in (B) (n = 3; mean±s.e.m.). (C,D) Pulldown of GFP–kindlin-1, -2 and -3 and chimeras by immobilized GST or GST–ILK-KD + parvin-CH2 was assessed by immunoblotting (C) and quantified (D) (n≥3; mean±s.e.m.). (E,F) Pulldown of GFP-tagged kindlins from CHO cell lysates using immobilized GST or minimal IPP was assessed by immunoblotting (E) and quantified in (F) (n = 5; mean±s.e.m.). *P<0.05, **P<0.01, ***P<0.001 (Student's t-test). Loading of GST proteins was assessed by Ponceau S staining; inputs (3%) are also shown.
A conserved leucine residue within the kindlin F2PH region is required for ILK binding
To more precisely map the ILK-binding site within kindlin, the GST–ILK-KD complex was immobilized on beads and incubated with lysates from cells expressing previously generated GFP–kindlin-1 fragments (Goult et al., 2009; Yates et al., 2012). As expected, the N-terminal F0F1 subdomains did not interact with the ILK-KD complex, whereas the C-terminal F2F3 subdomains did (Fig. 6A). As the kindlin PH domain is implicated in kindlin function (Qu et al., 2011), we then tested two different kindlin-1 deletion constructs: one lacking all residues that separated the split F2 subdomains including the PH domain (Δ326–495) and another smaller deletion lacking only the structurally defined PH domain (Yates et al., 2012) (Δ363–499). We found that the 38-amino-acid linker preceding the PH domain (present in K1 Δ363–499 but not in K1 Δ326–495) was required for ILK binding, whereas the PH domain itself was dispensable (Fig. 6A).
Fig. 6.

Fine mapping of the ILK–kindlin-2 interaction and identification of a leucine residue crucial for binding. (A,B) Binding of GFP–kindlin-1 or kindlin-2 deletions and/or truncations from CHO cell lysates to immobilized GST or GST–ILK-KD + parvin-CH2 was assessed by immunoblotting. GST protein loading was assessed by Ponceau S staining. (C) Alignment of the amino acid sequence of the F2 linker region in the three human kindlins and the C. elegans ortholog UNC-112. The F2 subdomain (left) and inserted PH domain (right) are shaded. UNC-112 D382 and human kindlin-2 L357 are highlighted. (D,E) Immunoblot analysis of binding of GFP-kindlin-2, and kindlin-2 truncations/mutants to immobilized GST or GST-ILK-KD + parvin-CH2 (D) or to immobilized GST or minimal IPP (E). (F) Co-immunoprecipitation (IP) of endogenous ILK and FLAG-tagged kindlin isoforms, mutants and chimeras from HEK293T cell lysates. 3%, 3% input; IB, immunoblot. (G) Quantification of the amount of endogenous ILK co-immunoprecipitated with FLAG-tagged kindlins. Results are normalized to the amount of ILK co-immunoprecipitated with FLAG–kindlin-2 in each experiment (mean±s.e.m., n≥3). ***P<0.001 (Student's t-test). (H) Quantification of GFP–kindlin-2 or kindlin-2 mutants binding to purified integrin tails (mean±s.e.m., n≥5).
In kindlin-2, this linker is also necessary and sufficient for ILK binding, as the GST–ILK-KD complex pulled down the GFP–kindlin-2 linker alone, but not GFP–kindlin-2 lacking this linker (Fig. 6B). A sequence alignment of this previously uncharacterized linker revealed a middle region conserved between species and isoforms (Fig. 6C). The C-terminal ∼25 amino acids, including the more conserved region, were sufficient to bind the ILK-KD complex, whereas the N-terminal region did not bind (Fig. 6B). Interestingly, an aspartic acid residue in this region of the C. elegans kindlin otholog UNC-112 (D381 in UNC-112) has been reported to be key for binding the C. elegans ILK ortholog PAT-4 (Qadota et al., 2012). This residue is a serine in all three human kindlins and a valine substitution at this site (S351V in human kindlin-2) did not inhibit ILK binding to the kindlin-2 linker or full-length kindlin-2 (Fig. 6D). We therefore attempted to map the interaction between mammalian kindlin-2 and ILK by mutating additional residues in this region. Introducing a L357A mutation within the GFP–kindlin-2 linker or within full-length GFP–kindlin-2 resulted in a dramatic loss of ILK binding (Fig. 6D). Moreover, the L357A mutation abrogated GFP–kindlin-2 binding to minimal IPP complex (Fig. 6E), underscoring its importance for interaction with full-length ILK.
Consistent with its importance in ILK binding, kindlin-2 L357 is conserved across isoforms and species (Fig. 6C). Notably, it is also conserved in kindlin-3 (L334). To test whether the low ILK binding seen for kindlin-3 nonetheless depends on L334 we performed a series of co-immunoprecipitation experiments. FLAG-tagged kindlins were overexpressed in HEK293T cells, immunoprecipitated from cell lysate using anti-FLAG M2-coated Sepharose beads, and co-precipitating endogenous ILK was detected by immunoblotting. ILK co-immunoprecipitated with FLAG–kindlin-2 and (to a much lesser extent) with FLAG–kindlin-3 (Fig. 6F,G). In both cases, replacement of the conserved leucine (L357 in kindlin-2, L334 in kindlin-3) with an alanine residue strongly inhibited ILK co-immunoprecipitation (Fig. 6F,G). Moreover, co-immunoprecipitation of endogenous ILK with the loss-of-function K2232 and gain-of-function K3323 chimeras recapitulated what we had previously observed in pulldown assays with purified ILK (Fig. 6F,G). Thus, both kindlin-2 and kindlin-3 bind ILK in cells and the interaction involves the same conserved leucine residue, but kindlin-2 binds ILK better than kindlin-3 does and the F2PH is responsible for this difference.
It has been reported that in C. elegans, PAT-4/ILK binding to UNC-112/kindlin facilitates UNC-112/kindlin interaction with PAT-3/β integrin (Qadota et al., 2012). We therefore compared the binding of wild-type and L357A mutant kindlin-2 to β-integrin tails. As previously reported (Harburger et al., 2009; Ma et al., 2008; Montanez et al., 2008), the W615A mutation within the kindlin-2 F3 clearly inhibited binding to β1 tails, but the L357A mutation that abrogates ILK binding had no effect (Fig. 6H). We therefore conclude that disruption of ILK binding does not negatively impact the mammalian kindlin-2–integrin interaction.
ILK binding aids kindlin-2 FA targeting
To evaluate whether ILK binding is involved in kindlin-2 FA targeting, we expressed the ILK-binding defective GFP–kindlin-2-L357A in CHO cells. FA targeting of GFP–kindlin-2-L357A was strongly impaired (Fig. 7A), indicating that ILK binding is important for targeting of kindlins to FAs. Furthermore, introducing the L357A mutation into the K3323 chimera abolished its targeting (Fig. 7A), illustrating that ILK binding is important for the gain-of-targeting effect.
Fig. 7.

ILK binding is required for kindlin-2 to efficiently target to FAs. (A) Subcellular localization of endogenous vinculin and GFP-tagged kindlins, mutants and chimeras in CHO cells plated on fibronectin. (B) Immunoblot (IB) analysis of endogenous ILK, kindlin-2, parvin, PINCH and vinculin in ILKfl/fl and ILK−/− fibroblasts. (C) Subcellular localization of GFP-tagged kindlins and chimeras in ILKfl/fl and ILK−/− fibroblasts plated on fibronectin. When specified, ILK−/− cells were co-transfected with FLAG–ILK. Vinculin and ILK were visualized by immunofluorescence. (D) Subcellular localization of endogenous vinculin and GFP-tagged kindlins, mutants and chimeras in ILK−/− fibroblasts plated on fibronectin. (E) Detection of endogenous kindlin-2 and vinculin by immunofluorescence in ILKfl/fl and ILK−/− cells, 4 h and 21 h after plating on fibronectin. Scale bars: 10 µm.
As a further test of the importance of ILK in the gain-of-targeting effect, we assessed the localization of K3323 in ILK-deficient cells. We generated several ILK-knockout (ILK−/−) clonal lines by infecting immortalized newborn ILKfl/fl mouse fibroblasts with adeno-Cre recombinase. Immunoblotting confirmed ILK knockout and, as previously reported (Fukuda et al., 2003; Gkretsi et al., 2007; Wang et al., 2008), this also led to loss of the ILK-interacting proteins PINCH1 and α-parvin (Fig. 7B). In the absence of ILK, endogenous kindlin-2 levels also diminished by ∼50% (Fig. 7B). Morphologically, ILK-null cells spread and formed vinculin-rich FAs when plated on fibronectin. However, in agreement with other reports (Elad et al., 2013), we noticed that ILK−/− cells had mostly peripheral FAs and lacked the centrally located fibrillar adhesion seen in ILKfl/fl cells. As observed in CHO cells, GFP-tagged kindlin-2 and K3323 targeted to FAs in ILKfl/fl fibroblasts, whereas GFP-kindlin-3 did not (Fig. 7C). However, K3323 targeting was entirely lost in ILK−/− cells (Fig. 7C). Similar results were obtained in two other independent ILK−/− lines. Importantly, FA targeting of GFP–K3323 was restored in ILK−/− cells by co-expressing FLAG–ILK, whereas the point-mutated K3323 L357A remained defective in FA targeting even in the presence of FLAG–ILK (Fig. 7C). Taken together, our data establish that ILK binding is responsible for the gain-of-function conferred by introducing kindlin-2 F2PH into kindlin-3.
In the course of our studies of ILK−/− cells, we made the initially surprising observation that GFP–kindlin-2 exhibited weak, but clear, targeting to FAs in the absence of ILK (Fig. 7C). We also noticed that, in contrast with what we observed in CHO cells, GFP–kindlin-2-L357A displayed some weak FA targeting in ILK−/− cells, similar to that of wild-type GFP–kindlin-2 (Fig. 7D). Thus, although ILK binding aids FA targeting, GFP–kindlin-2 can accumulate in an ILK-independent manner in fibroblasts, albeit at a much reduced level. Consistent with this, although endogenous kindlin-2 was always detected in FAs in ILKfl/fl cells, it was hardly detectable in ILK−/− cells 4 h after plating but was detected in FAs 21 h post-plating (Fig. 7E). The reduction of kindlin-2 levels in ILK−/− cells (Fig. 7B) might account for some of this reduction in kindlin-2 localization but the fact that some endogenous kindlin-2 was found in the FAs of ILK−/− cells suggests that, although ILK is important for efficient localization of kindlin-2 to adhesions, in certain circumstances, kindlin-2 can target to FAs in an ILK-independent manner.
Reduction of endogenous kindlin-2 levels permits kindlin-3 targeting to FAs
As kindlin-2 binds ILK more efficiently than kindlin-3 does, we aimed to test whether knocking down endogenous kindlin-2 in NIH3T3 fibroblasts (Brahme et al., 2013) would permit GFP–kindlin-3 localization in FAs. Our knockdown fibroblasts exhibited only ∼15% residual kindlin-2 (Fig. 8A). Unfortunately, as previously reported (Montanez et al., 2008), loss of kindlin-2 impairs cell spreading making it difficult to assess FA targeting in knockdown cells. However, we noted that within the knockdown cell population some cells exhibited partial spreading with very weak kindlin-2 staining in adhesions suggesting that they retain low levels of kindlin-2 (Fig. 8B). Weak ILK localization to adhesions in these cells could also be detected (data not shown). Focusing on this partially spread population allowed us to assess GFP–kindlin localization in cells with low endogenous kindlin-2 levels. As expected, GFP–kindlin-2 targeted to FAs in these cells but, for the first time, we saw clear GFP–kindlin-3 targeting (Fig. 8C). Thus, reduction of endogenous kindlin-2 facilitates FA targeting of kindlin-3. Furthermore, GFP–kindlin-2-L357A also targets to FAs in these cells (Fig. 8C) confirming that ILK binding is not essential for FA targeting in the absence of competing endogenous kindlin-2. To our surprise, GFP–kindlin-2-W615A also targeted in kindlin-2-knockdown cells; however, we did not observe FA targeting of the double-mutant GFP–kindlin-2-L357A,W615A which is impaired in both integrin and ILK binding (Fig. 8C). These data suggest that both ILK and integrin binding contribute to the FA localization of kindlins.
Fig. 8.

Kindlin-2-ILK interaction is required for β1 integrin activation. (A) Immunoblot (IB) analysis of kindlin-2 and vinculin in parental, control (scrambled, SC) and kindlin-2-knockdown (K2 shRNA) NIH3T3 cells. (B) Immunofluorescence staining of endogenous kindlin-2 and vinculin in scrambled control and kindlin-2-knockdown NIH-3T3 fibroblasts. Spread kindlin-2-knockdown cells were selected for imaging. (C) Subcellular localization of GFP or GFP-tagged kindlins, and of endogenous vinculin in K2 shRNA cells plated on fibronectin for 5 h. (D) β1 integrin surface levels measured by flow cytometry. (E) Integrin α5β1 activation index, measured by flow cytometry and normalized to β1 staining (n = 4). (F–H) Scramble control or kindlin-2-knockdown fibroblasts were transfected with GFP or GFP-tagged kindlins and α5β1 integrin activation and β1 surface levels was assessed in transfected cells. Bar graphs represent mean±s.e.m.; n≥3; *P<0.05; **P<0.01; ***P<0.001; ns, not significant (Student's t-test). Scale bars: 10 µm.
ILK binding is important for kindlin-2-mediated β1 integrin activation
The preceding data demonstrate a role for ILK binding in kindlin FA targeting and reveal that isoform-specific differences in ILK binding contribute to the differential kindlin targeting to FAs. Although all three kindlins play key roles in integrin activation, isoform-specific differences in activation have also been described (Calderwood et al., 2013). We therefore compared kindlin-2 and -3 rescue of α5β1 activation in kindlin-2-knockdown NIH3T3 fibroblasts (Brahme et al., 2013). The ability of integrins to bind soluble ligand provides a widely used functional readout of integrin activation (Bouaouina et al., 2012b; Calderwood, 2004b). We assessed α5β1 integrin activation using a well-established flow-cytometry-based assay, which measures binding of a soluble biotinylated fragment of the endogenous α5β1 ligand fibronectin (FN9-11) (Bouaouina et al., 2012b). To compare activation across experimental conditions, we calculated an ‘activation index’ that is normalized for β1 integrin surface levels. In agreement with published data (Montanez et al., 2008), we observed a significant decrease in surface β1 integrins in kindlin-2 shRNA cells (Fig. 8D). Even allowing for this reduction, we see a significant decrease in β1 integrin activation in kindlin-2-knockdown cells (Fig. 8E). Importantly, reconstitution of the kindlin-2-knockdown cells with GFP–kindlin-2 restored integrin activation, whereas overexpression of GFP–kindlin-3 did not (Fig. 8F). This is not due to differences in GFP–kindlin expression as rescue populations were all gated to have the same GFP levels and the activation index takes into account any changes in β1 surface expression. Thus, kindlin-3 is unable to substitute for kindlin-2 in supporting activation of fibroblast α5β1.
To test whether ILK binding is important for kindlin-2-mediated integrin activation, we reconstituted kindlin-2-knockdown cells with point-mutated kindlins. GFP–kindlin-2 and GFP–kindlin-2-L357A rescued integrin surface levels, whereas GFP–kindlin-2-W615A did not (Fig. 8G), showing that the effect of kindlin-2 on integrin surface levels relies on binding integrins but not ILK. In contrast, GFP–kindlin-2 increased β1 integrin activation, whereas the L357A and W615A kindlin-2 mutants did not (Fig. 8H), indicating that binding to both ILK and integrin are necessary for kindlin-mediated integrin activation. This provides the first evidence that the kindlin-2–ILK interaction is important for integrin activation. Furthermore, the ILK-binding K3323 kindlin chimera also restored integrin activation to levels seen with GFP-kindlin-2, whereas GFP–K3323-L357A did not (Fig. 8H). Taken together, our data show that ILK binding is important for kindlin-2-mediated integrin activation, and suggest that differences in ILK binding contribute to functional differences between kindlin-2 and kindlin-3.
DISCUSSION
Kindlins are essential integrin regulatory proteins (Calderwood et al., 2013). Each of the three mammalian kindlins is implicated in integrin activation but kindlins also have isoform-specific functions. Kindlins cooperate with talin to trigger integrin activation (Calderwood et al., 2013) and they might do so by clustering αIIbβ3 integrins (Feigelson et al., 2011; Ye et al., 2013). Kindlins also play key roles in integrin signaling downstream of ligand binding, and kindlin recruitment to integrin-rich adhesive structures is likely important for these processes. However, the interactions that control kindlin recruitment and/or retention at adhesion sites and the mechanisms underlying the roles of kindlins in integrin activation have remained poorly understood. Here, combining chimeric mapping of the kindlin subdomain responsible for isoform-specific localization, biochemical analysis of the ILK–kindlin interaction and functional studies in ILK-knockout and kindlin-2-knockdown cells we report that: (1) optimal kindlin-2 accumulation at FAs requires binding to not only integrin, through its F3 subdomain, but also to the ILK complex, through a linker region located within its F2PH subdomain; (2) binding to both integrin and ILK–parvin is crucial for kindlin-2 to exert its role on integrin activation; and (3) kindlin isoforms exhibit marked differences in integrin and ILK binding and these differences impact on localization and function.
An ILK–kindlin interaction was first identified in a yeast two-hybrid screen with C. elegans proteins, and has been validated by co-immunoprecipitation in mammalian cells (Mackinnon et al., 2002; Montanez et al., 2008). We now show direct mammalian kindlin binding to the ILK-KD–parvin-CH2 complex. Parvin-CH2 alone cannot support kindlin binding and, together with published yeast two-hybrid data (Mackinnon et al., 2002; Qadota et al., 2012), this suggests that there is a direct kindlin–ILK-KD interaction. On kindlin, ILK–parvin complex binding occurs at a previously uncharacterized linker between the N-terminal split F2 and the PH subdomains of kindlin-2. Notably, although all three kindlins bind ILK, kindlin-3 binds less well. Our inability to produce soluble recombinant fragments of kindlin-3 prevented us comparing the affinity of kindlin-2 and -3 for ILK–parvin-CH2, but pulldown assays with a variety of ILK constructs, as well as co-immunoprecipitation with endogenous ILK, consistently demonstrated less ILK binding to kindlin-3 than to kindlin-2. We identified a conserved leucine residue in the F2-PH linker (L357 in kindlin-2 and L334 in kindlin-3) that is required for ILK complex binding, and inhibition of kindlin-3 binding to ILK by an L334A mutation demonstrates that, although weak, kindlin-3 ILK interactions are specific. Our initial mutational studies targeting the F2 linker residues conserved in kindlin-1 and -2, but not in kindlin-3, did not yield kindlins with altered ILK-binding properties (data not shown). Therefore, it is still unclear which kindlin residues are responsible for isoform specificity.
In addition to characterizing the kindlin–ILK interaction, we provide the first evidence for its functional significance in FA localization and integrin activation. Our FA-targeting studies with chimeric kindlins, and with ILK-binding defective mutants, strongly support a role for ILK in localizing or retaining kindlins at FAs. As yet, no kindlin-binding-defective ILK mutants have been identified, preventing us from testing whether such mutants recapitulate the FA localization phenotypes we observe when diminishing the ability of kindlin to bind ILK. However, additional evidence supports the importance of kindlin–ILK interactions in FA targeting: (1) targeting of endogenous and exogenously expressed kindlin-2 is impaired in ILK−/− fibroblasts, (2) the gain of function observed with K3323 is not observed ILK−/− cells, and (3) re-expression of ILK in ILK−/− cells restores FA targeting of wild-type but not L357A mutant kindlins. Nonetheless, at least in some cells, ILK binding is not absolutely required and the residual FA localization of kindlin-2 in ILK-knockout cells is likely due to the integrin-binding site in the F3 subdomain. Indeed, in the absence of competing endogenous kindlin-2, ILK-binding-defective kindlins can target to FAs, providing that their integrin-binding site is intact.
Based on the inability of kindlin-3, ILK-binding-defective mutant kindlins and some kindlin chimeras to rescue the integrin activation defect in kindlin-2-knockdown cells, we suggest that ILK binding is also important for integrin activation. Kindlin is a well-established regulator of integrin activation and ILK is known to be required for normal integrin activation (Friedrich et al., 2004; Honda et al., 2009; Tucker et al., 2008). Consistent with this, our ILK-knockout fibroblasts have an ∼75% reduction in active cell-surface integrins (not shown). Thus, our data establishing the importance of ILK–kindlin interactions for integrin activation are in keeping with known kindlin- and ILK-deficient phenotypes. It is unclear how ILK binding triggers kindlin-mediated integrin activation or kindlin retention in FAs. Kindlin–integrin interactions are also important for both integrin activation and FA targeting, and it has been proposed that in C. elegans PAT-4/ILK binding to UNC-112/kindlin regulates UNC-112/kindlin binding to PAT-3/β integrin (Qadota et al., 2012). However, although we observe isoform-specific differences in integrin binding, we find that integrin binding is determined by the F3 subdomain and is unaffected by ILK binding. Thus, at least in mammals, ILK regulates kindlin-2 FA localization and integrin activation through a mechanism distinct from an increase in integrin binding. ILK also contributes to FA targeting of β-parvin (Stiegler et al., 2012) suggesting that ILK might recruit or stabilize multiple FA proteins. This scaffolding action might also play into the ability of ILK to impact on integrin activation.
It is currently unclear how to reconcile the lack of effect of kindlin-3 on integrin activation in fibroblasts with the broad evidence that kindlin-3 is required for β1, β2 and β3 integrin activation in hematopoietic cells (Moser et al., 2009; Moser et al., 2008). However, as ILK is also important for activation of platelet integrins (Tucker et al., 2008) and we find that kindlin-3 binds weakly but specifically to ILK, it might be that kindlin-3 requires additional hematopoietic-specific interactions in addition to that with ILK to efficiently trigger integrin activation. Alternatively, considering the difference in phenotypes of kindlin-3-knockout (Moser et al., 2008) and blood-cell-specific ILK-knockout platelets (Tucker et al., 2008), kindlin-3 might not require interactions with the IPP complex to regulate hematopoietic integrins.
In summary, we have shown that ILK-complex binding is important for kindlin-2 FA targeting, and establish for the first time that kindlin–ILK interactions are required for integrin activation. Furthermore, we reveal that differences in ILK binding contribute to functional differences between kindlin isoforms. The mechanisms by which ILK regulates kindlin-mediated integrin activation and influences kindlin FA accumulation, and the signaling pathways in which the ILK–kindlin complex participates at adhesions will be the subject of future studies.
MATERIALS AND METHODS
Antibodies
Primary antibodies against vinculin (Sigma), GFP (Rockland), kindlin-2 (ProteinTech and AbCam), ILK (Cell Signaling), PINCH1 (Proteintech Group) and α-parvin (Cell Signaling), and Alexa-Fluor-conjugated (Invitrogen) and IRDye-conjugated (Li-Cor) secondary antibodies were purchased.
Constructs
Vectors encoding N-terminally GFP-tagged human kindlin-1 (amino acids 1–677) and human kindlin-2 (amino acids 1–680) (Harburger et al., 2009) and the GST-tagged, His-tagged or His-FLAG-tagged α-Parvin CH2 domain, ILK and ILK-ARD, and the PINCH1-LIM1 domain (Chiswell et al., 2008; Stiegler et al., 2012; Stiegler et al., 2013) were as previously described. The human kindlin-3 short form DNA (encoding amino acids 1–663; IMAGE clone 3677274) was purchased from the ATTC, amplified by PCR and subcloned into the pEGFP-C1 vector (Invitrogen). Kindlin chimeras were generated by splice overlap PCR using the following amino acid boundaries: kindlin-1, F0F1 1–275, F2PH 276–567, PH 366–498, F3 568–677; kindlin-2, F0F1 1–277, F2PH 278–570, PH 369–501, F3 571–680; kindlin-3, F0F1 1–254, F2PH 255–551, PH 346–478, F3 552–663. Point mutations were introduced by QuikChange site-directed mutagenesis (Stratagene). For bacterial expression, kindlins were cloned into pGEX-4T3 or a modified pET32 vector (pET32-TT) with an N-terminal GST tag. GST-tagged ILK-KD (residues 175–452) was generated by PCR and subcloned into pET-32-TT. All inserts were verified by DNA sequencing. TRC library-based lentiviral scramble (catalog no. SHC002) and kindlin-2 short hairpin (sh)RNA (TRCN000019185) plasmids were purchased from Sigma and lentivirus was produced as previously described (Bandyopadhyay et al., 2012; Brahme et al., 2013).
Cell culture and transfection
CHO, HEK293T, NIH3T3, EA.hy926 and CMK cells (from Seiji Tadakoro, Osaka University, Japan) were grown as previously described (Brahme et al., 2013; Liu et al., 2013; Nakazawa et al., 2013). Cells were transfected with PEI (Linear Polyethylenimine, MW 25,000, Polysciences, Inc.), or nucleofected (Amaxa, Lonzabio). Kindlin-2-knockdown and control NIH-3T3 cells were generated as previously described (Brahme et al., 2013).
Generation of ILK-knockout fibroblasts
Fibroblasts were isolated from the skin of a new-born ILK flox/flox mouse (Terpstra et al., 2003) and immortalized through repeated passaging. Cells were then infected with an adenovirus co-expressing Cre Recombinase and GFP under the same promoter (a gift from Xiaoyong Yang, Yale University, New Haven, CT). After 48 h, GFP-expressing cells were single-cell sorted and more than 10 clonal cell lines were established. All animal experiments were performed according to approved guidelines.
Immunofluorescence and scoring of FA targeting
Transfected or nucleofected cells were plated on glass coverslips coated with 10 µg/ml bovine plasma fibronectin (Sigma) or 10 µg/ml bovine plasma fibrinogen (Sigma), and blocked with 2% BSA. When fibrinogen was used, cells were plated in medium containing 0.1% FBS. At 24 h after plating, cells were simultaneously fixed and permeabilized with cold 4% paraformaldehyde containing 0.1% Triton X-100 for 30 min, quenched and blocked with 50 mM NH4Cl, 0.2% BSA and 0.1% Triton X-100 in PBS, and stained using standard immunofluorescence methods and mounted in ProLong Gold anti-fade reagent (Invitrogen). Cells were visualized using a Nikon TI microscope equipped with a ×100 immersion oil objective. Images displayed in figures are inverted to highlight FAs.
The percentage of GFP-positive cells showing FA targeting of GFP-tagged proteins was scored blindly, using GFP–kindlin-2 and -3 as controls in all experiments. At least 40 well-spread, non-dividing transfected cells with moderate-to-low cytoplasmic fluorescence were scored in each condition. Each construct was tested in at least three experiments, and final results reflect scoring of 120 to 1000 cells per construct. Data are shown as proportions with the 95% confidence interval. Intensity of GFP signal in FAs was also assessed blindly. The overall intensity of FA targeting for a given construct varied little from cell to cell in a given experiment so a representative score (in arbitrary units) was given blindly for each construct in each experiment. The score was 0 for no targeting, 1 for low targeting, 2 for medium targeting (like that for GFP–kindlin-1), 3 for strong targeting (like that for GFP–kindlin-2), and 4 for very strong targeting. Scores for each construct were then averaged from at least three experiments to generate a FA targeting index.
Protein production and purification
Recombinant His-tagged integrin cytoplasmic tails, GST, GST–kindlin-1, GST–α-parvin-CH2, and the GST–ILK-ARD–PINCH1-LIM1 complex and minimal IPP complex were produced and purified as previously described (Chiswell et al., 2008; Harburger et al., 2009; Lad et al., 2007; Stiegler et al., 2012; Stiegler et al., 2013). GST–kindlin-2-F2PH was produced in E. coli Rosetta cells and GST–ILK-KD in complex with α-parvin-CH2 was produced in E. coli De3 BL21 cells. Both were purified on glutathione–Sepharose beads (GE Healthcare). Proteins were kept immobilized on beads or the GST tag was removed with the Tobacco Etch virus protease.
Protein-binding assays
Pulldown assays were performed from CHO cell lysates as previously described (Brahme et al., 2013; Lad et al., 2007). Bound proteins were fractionated by SDS-PAGE in reducing conditions, immunoblotted, imaged on an Odyssey Infrared Imaging System (Li-Cor) and analyzed using ImageJ or Image Studio Lite (Li-Cor). For quantification of binding, the intensity of the bound protein of interest was expressed as a percentage of the total input (based on the band intensity of the ‘input’ lane) or as a percentage of binding relative to an internal control. Bead loading was verified by staining the relevant portion of the gel with Coomassie Blue protein stain or by staining the nitrocellulose membrane with Ponceau S.
Co-immunoprecipitation was performed as follows. HEK293T cell pellets were lysed in buffer X (1 mM NaVO4, 50 mM NaF, 40 mM sodium pyrophosphate, 50 mM NaCl, 150 mM sucrose, 10 mM Pipes pH 6.8) containing 0.5% Triton X-100, 0.2% deoxycholic acid and EDTA-free protease inhibitor cocktail (Roche), and incubated with anti-FLAG M2 beads (Sigma) in buffer X-T (buffer X with 0.05% Triton X-100). Beads were washed and bound proteins fractionated by reducing SDS-PAGE and analyzed by immunoblotting.
The purified protein-binding assay was performed as follows. 8 µg of soluble protein was diluted in buffer X-T and incubated with beads coated with GST or GST fusion protein. Beads were washed, and bound proteins were fractionated by reducing SDS-PAGE and analyzed in gel by Coomassie Blue staining or by immunoblotting as described above.
Kinase domain measurements were performed as follows. GST–kindlin-2-F2PH or GST alone were prepared at 0.05 mg/ml in assay buffer (50 mM HEPES pH 7.5, 150 mM NaCl) and captured on the anti-GST surface of BLItz GST Biosensors (Pall Life Sciences). The ILK-KD–α-parvin-CH2 complex, or α-parvin-CH2 alone as negative control, were prepared in assay buffer in dilution series ranging from ∼0.5–150 µM. Binding responses to GST–kindlin were measured at room temperature with the BLItz instrument and referenced against the signal from binding to GST. The dissociation constant was calculated in GraphPad Prism in the nonlinear regression ‘equilibrium binding – specific binding’ mode.
Analysis of integrin activation
The activation state of endogenous α5β1 integrins was assessed by flow cytometry using biotinylated recombinant GST–FN9-11 as previously described (Bouaouina et al., 2012b). Briefly, cells were suspended 24 h post-transfection and FN9-11 binding in the presence or absence of EDTA was assessed on an LSRII instrument (BD Biosciences). β1 integrin surface levels were assessed in parallel by staining with biotinylated anti-CD29 antibody (Biolegend). The activation index (AI) of cells gated to have equivalent mean GFP intensity was calculated as AI = (F−Fo)/Fintegrin, where F is the geometric mean fluorescence intensity (MFI) of FN9-11 binding, Fo is the MFI of FN9-11 binding in presence of inhibitor, and Fintegrin is the normalized MFI of CD29 binding to transfected cells. Cytometry data were analyzed using FlowJo software.
Supplementary Material
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
C.H.C. and N.N.B. designed and performed experiments, analyzed results and wrote the paper. N.K. and A.L.S. helped perform experiments and analyze data. A.L.S., S.R. and T.J.B. provided essential reagents and/or expertise and edited the paper. D.A.C. designed experiments, analyzed data and wrote the paper.
Funding
This work was supported by the National Institutes of Health [grant numbers T32GM007223, R01GM068600 and R01GM088240]; and by the American Cancer Society [grant number RSG-12-053-01]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.155879/-/DC1
References
- Bandyopadhyay A., Rothschild G., Kim S., Calderwood D. A., Raghavan S. (2012). Functional differences between kindlin-1 and kindlin-2 in keratinocytes. J. Cell Sci. 125, 2172–2184 10.1242/jcs.096214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialkowska K., Ma Y. Q., Bledzka K., Sossey-Alaoui K., Izem L., Zhang X., Malinin N., Qin J., Byzova T., Plow E. F. (2010). The integrin co-activator Kindlin-3 is expressed and functional in a non-hematopoietic cell, the endothelial cell. J. Biol. Chem. 285, 18640–18649 10.1074/jbc.M109.085746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaouina M., Goult B. T., Huet-Calderwood C., Bate N., Brahme N. N., Barsukov I. L., Critchley D. R., Calderwood D. A. (2012a). A conserved lipid-binding loop in the kindlin FERM F1 domain is required for kindlin-mediated αIIbβ3 integrin coactivation. J. Biol. Chem. 287, 6979–6990 10.1074/jbc.M111.330845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaouina M., Harburger D. S., Calderwood D. A. (2012b). Talin and signaling through integrins. Methods Mol. Biol. 757, 325–347 10.1007/978-1-61779-166-6_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahme N. N., Harburger D. S., Kemp-O'Brien K., Stewart R., Raghavan S., Parsons M., Calderwood D. A. (2013). Kindlin binds migfilin tandem LIM domains and regulates migfilin focal adhesion localization and recruitment dynamics. J. Biol. Chem. 288, 35604–35616 10.1074/jbc.M113.483016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood D. A. (2004a). Integrin activation. J. Cell Sci. 117, 657–666 10.1242/jcs.01014 [DOI] [PubMed] [Google Scholar]
- Calderwood D. A. (2004b). Talin controls integrin activation. Biochem. Soc. Trans. 32, 434–437 10.1042/BST0320434 [DOI] [PubMed] [Google Scholar]
- Calderwood D. A., Campbell I. D., Critchley D. R. (2013). Talins and kindlins: partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 14, 503–517 10.1038/nrm3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiswell B. P., Zhang R., Murphy J. W., Boggon T. J., Calderwood D. A. (2008). The structural basis of integrin-linked kinase-PINCH interactions. Proc. Natl. Acad. Sci. USA 105, 20677–20682 10.1073/pnas.0811415106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiswell B. P., Stiegler A. L., Razinia Z., Nalibotski E., Boggon T. J., Calderwood D. A. (2010). Structural basis of competition between PINCH1 and PINCH2 for binding to the ankyrin repeat domain of integrin-linked kinase. J. Struct. Biol. 170, 157–163 10.1016/j.jsb.2009.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling J. J., Vreede A. P., Kim S., Golden J., Feldman E. L. (2008). Kindlin-2 is required for myocyte elongation and is essential for myogenesis. BMC Cell Biol. 9, 36 10.1186/1471-2121-9-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elad N., Volberg T., Patla I., Hirschfeld-Warneken V., Grashoff C., Spatz J. P., Fässler R., Geiger B., Medalia O. (2013). The role of integrin-linked kinase in the molecular architecture of focal adhesions. J. Cell Sci. 126, 4099–4107 10.1242/jcs.120295 [DOI] [PubMed] [Google Scholar]
- Feigelson S. W., Grabovsky V., Manevich-Mendelson E., Pasvolsky R., Shulman Z., Shinder V., Klein E., Etzioni A., Aker M., Alon R. (2011). Kindlin-3 is required for the stabilization of TCR-stimulated LFA-1:ICAM-1 bonds critical for lymphocyte arrest and spreading on dendritic cells. Blood 117, 7042–7052 10.1182/blood-2010-12-322859 [DOI] [PubMed] [Google Scholar]
- Friedrich E. B., Liu E., Sinha S., Cook S., Milstone D. S., MacRae C. A., Mariotti M., Kuhlencordt P. J., Force T., Rosenzweig A. et al. (2004). Integrin-linked kinase regulates endothelial cell survival and vascular development. Mol. Cell. Biol. 24, 8134–8144 10.1128/MCB.24.18.8134-8144.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T., Chen K., Shi X., Wu C. (2003). PINCH-1 is an obligate partner of integrin-linked kinase (ILK) functioning in cell shape modulation, motility, and survival. J. Biol. Chem. 278, 51324–51333 10.1074/jbc.M309122200 [DOI] [PubMed] [Google Scholar]
- Fukuda K., Gupta S., Chen K., Wu C., Qin J. (2009). The pseudoactive site of ILK is essential for its binding to α-Parvin and localization to focal adhesions. Mol. Cell 36, 819–830 10.1016/j.molcel.2009.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger T., Zaidel-Bar R. (2012). Opening the floodgates: proteomics and the integrin adhesome. Curr. Opin. Cell Biol. 24, 562–568 10.1016/j.ceb.2012.05.004 [DOI] [PubMed] [Google Scholar]
- Ghatak S., Morgner J., Wickström S. A. (2013). ILK: a pseudokinase with a unique function in the integrin-actin linkage. Biochem. Soc. Trans. 41, 995–1001 10.1042/BST20130062 [DOI] [PubMed] [Google Scholar]
- Gkretsi V., Mars W. M., Bowen W. C., Barua L., Yang Y., Guo L., St-Arnaud R., Dedhar S., Wu C., Michalopoulos G. K. (2007). Loss of integrin linked kinase from mouse hepatocytes in vitro and in vivo results in apoptosis and hepatitis. Hepatology 45, 1025–1034 10.1002/hep.21540 [DOI] [PubMed] [Google Scholar]
- Goult B. T., Bouaouina M., Harburger D. S., Bate N., Patel B., Anthis N. J., Campbell I. D., Calderwood D. A., Barsukov I. L., Roberts G. C. et al. (2009). The structure of the N-terminus of kindlin-1: a domain important for alphaiibbeta3 integrin activation. J. Mol. Biol. 394, 944–956 10.1016/j.jmb.2009.09.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan G. E., Leung-Hagesteijn C., Fitz-Gibbon L., Coppolino M. G., Radeva G., Filmus J., Bell J. C., Dedhar S. (1996). Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 379, 91–96 10.1038/379091a0 [DOI] [PubMed] [Google Scholar]
- Harburger D. S., Calderwood D. A. (2009). Integrin signalling at a glance. J. Cell Sci. 122, 159–163 10.1242/jcs.018093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harburger D. S., Bouaouina M., Calderwood D. A. (2009). Kindlin-1 and -2 directly bind the C-terminal region of β integrin cytoplasmic tails and exert integrin-specific activation effects. J. Biol. Chem. 284, 11485–11497 10.1074/jbc.M809233200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda S., Shirotani-Ikejima H., Tadokoro S., Maeda Y., Kinoshita T., Tomiyama Y., Miyata T. (2009). Integrin-linked kinase associated with integrin activation. Blood 113, 5304–5313 10.1182/blood-2008-07-169136 [DOI] [PubMed] [Google Scholar]
- Karaköse E., Schiller H. B., Fässler R. (2010). The kindlins at a glance. J. Cell Sci. 123, 2353–2356 10.1242/jcs.064600 [DOI] [PubMed] [Google Scholar]
- Kogata N., Tribe R. M., Fässler R., Way M., Adams R. H. (2009). Integrin-linked kinase controls vascular wall formation by negatively regulating Rho/ROCK-mediated vascular smooth muscle cell contraction. Genes Dev. 23, 2278–2283 10.1101/gad.535409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers T. W., van de Vijver E., Weterman M. A., de Boer M., Tool A. T., van den Berg T. K., Moser M., Jakobs M. E., Seeger K., Sanal Ö. et al. (2009). LAD-1/variant syndrome is caused by mutations in FERMT3. Blood 113, 4740–4746 10.1182/blood-2008-10-182154 [DOI] [PubMed] [Google Scholar]
- Lad Y., Harburger D. S., Calderwood D. A. (2007). Integrin cytoskeletal interactions. Methods Enzymol. 426, 69–84 10.1016/S0076-6879(07)26004-5 [DOI] [PubMed] [Google Scholar]
- Lai-Cheong J. E., Ussar S., Arita K., Hart I. R., McGrath J. A. (2008). Colocalization of kindlin-1, kindlin-2, and migfilin at keratinocyte focal adhesion and relevance to the pathophysiology of Kindler syndrome. J. Invest. Dermatol. 128, 2156–2165 10.1038/jid.2008.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larjava H., Plow E. F., Wu C. (2008). Kindlins: essential regulators of integrin signalling and cell-matrix adhesion. EMBO Rep. 9, 1203–1208 10.1038/embor.2008.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Fukuda K., Xu Z., Ma Y. Q., Hirbawi J., Mao X., Wu C., Plow E. F., Qin J. (2011). Structural basis of phosphoinositide binding to kindlin-2 protein pleckstrin homology domain in regulating integrin activation. J. Biol. Chem. 286, 43334–43342 10.1074/jbc.M111.295352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Draheim K. M., Zhang R., Calderwood D. A., Boggon T. J. (2013). Mechanism for KRIT1 release of ICAP1-mediated suppression of integrin activation. Mol. Cell 49, 719–729 10.1016/j.molcel.2012.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y. Q., Qin J., Wu C., Plow E. F. (2008). Kindlin-2 (Mig-2): a co-activator of β3 integrins. J. Cell Biol. 181, 439–446 10.1083/jcb.200710196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon A. C., Qadota H., Norman K. R., Moerman D. G., Williams B. D. (2002). C. elegans PAT-4/ILK functions as an adaptor protein within integrin adhesion complexes. Curr. Biol. 12, 787–797 10.1016/S0960-9822(02)00810-2 [DOI] [PubMed] [Google Scholar]
- Malinin N. L., Zhang L., Choi J., Ciocea A., Razorenova O., Ma Y. Q., Podrez E. A., Tosi M., Lennon D. P., Caplan A. I. et al. (2009). A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat. Med. 15, 313–318 10.1038/nm.1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanez E., Ussar S., Schifferer M., Bösl M., Zent R., Moser M., Fässler R. (2008). Kindlin-2 controls bidirectional signaling of integrins. Genes Dev. 22, 1325–1330 10.1101/gad.469408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser M., Nieswandt B., Ussar S., Pozgajova M., Fässler R. (2008). Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 14, 325–330 10.1038/nm1722 [DOI] [PubMed] [Google Scholar]
- Moser M., Bauer M., Schmid S., Ruppert R., Schmidt S., Sixt M., Wang H. V., Sperandio M., Fässler R. (2009). Kindlin-3 is required for β2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 15, 300–305 10.1038/nm.1921 [DOI] [PubMed] [Google Scholar]
- Nakazawa T., Tadokoro S., Kamae T., Kiyomizu K., Kashiwagi H., Honda S., Kanakura Y., Tomiyama Y. (2013). Agonist stimulation, talin-1, and kindlin-3 are crucial for α(IIb)β(3) activation in a human megakaryoblastic cell line, CMK. Exp. Hematol. 41, 79, e1 10.1016/j.exphem.2012.09.011 [DOI] [PubMed] [Google Scholar]
- Perera H. D., Ma Y. Q., Yang J., Hirbawi J., Plow E. F., Qin J. (2011). Membrane binding of the N-terminal ubiquitin-like domain of kindlin-2 is crucial for its regulation of integrin activation. Structure 19, 1664–1671 10.1016/j.str.2011.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluskota E., Dowling J. J., Gordon N., Golden J. A., Szpak D., West X. Z., Nestor C., Ma Y. Q., Bialkowska K., Byzova T. et al. (2011). The integrin coactivator kindlin-2 plays a critical role in angiogenesis in mice and zebrafish. Blood 117, 4978–4987 10.1182/blood-2010-11-321182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadota H., Moerman D. G., Benian G. M. (2012). A molecular mechanism for the requirement of PAT-4 (integrin-linked kinase (ILK)) for the localization of UNC-112 (Kindlin) to integrin adhesion sites. J. Biol. Chem. 287, 28537–28551 10.1074/jbc.M112.354852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J., Wu C. (2012). ILK: a pseudokinase in the center stage of cell-matrix adhesion and signaling. Curr. Opin. Cell Biol. 24, 607–613 10.1016/j.ceb.2012.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu H., Tu Y., Shi X., Larjava H., Saleem M. A., Shattil S. J., Fukuda K., Qin J., Kretzler M., Wu C. (2011). Kindlin-2 regulates podocyte adhesion and fibronectin matrix deposition through interactions with phosphoinositides and integrins. J. Cell Sci. 124, 879–891 10.1242/jcs.076976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T., Li S., Docheva D., Grashoff C., Sakai K., Kostka G., Braun A., Pfeifer A., Yurchenco P. D., Fässler R. (2003). Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 17, 926–940 10.1101/gad.255603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil S. J., Kim C., Ginsberg M. H. (2010). The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 11, 288–300 10.1038/nrm2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X., Ma Y. Q., Tu Y., Chen K., Wu S., Fukuda K., Qin J., Plow E. F., Wu C. (2007). The MIG-2/integrin interaction strengthens cell-matrix adhesion and modulates cell motility. J. Biol. Chem. 282, 20455–20466 10.1074/jbc.M611680200 [DOI] [PubMed] [Google Scholar]
- Siegel D. H., Ashton G. H., Penagos H. G., Lee J. V., Feiler H. S., Wilhelmsen K. C., South A. P., Smith F. J., Prescott A. R., Wessagowit V. et al. (2003). Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes Kindler syndrome. Am. J. Hum. Genet. 73, 174–187 10.1086/376609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanchi F., Grashoff C., Nguemeni Yonga C. F., Grall D., Fässler R., Van Obberghen-Schilling E. (2009). Molecular dissection of the ILK-PINCH-parvin triad reveals a fundamental role for the ILK kinase domain in the late stages of focal-adhesion maturation. J. Cell Sci. 122, 1800–1811 10.1242/jcs.044602 [DOI] [PubMed] [Google Scholar]
- Stiegler A. L., Draheim K. M., Li X., Chayen N. E., Calderwood D. A., Boggon T. J. (2012). Structural basis for paxillin binding and focal adhesion targeting of β-parvin. J. Biol. Chem. 287, 32566–32577 10.1074/jbc.M112.367342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiegler A. L., Grant T. D., Luft J. R., Calderwood D. A., Snell E. H., Boggon T. J. (2013). Purification and SAXS analysis of the integrin linked kinase, PINCH, parvin (IPP) heterotrimeric complex. PLoS ONE 8, e55591 10.1371/journal.pone.0055591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson L., Howarth K., McDowall A., Patzak I., Evans R., Ussar S., Moser M., Metin A., Fried M., Tomlinson I. et al. (2009). Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 15, 306–312 10.1038/nm.1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terpstra L., Prud'homme J., Arabian A., Takeda S., Karsenty G., Dedhar S., St-Arnaud R. (2003). Reduced chondrocyte proliferation and chondrodysplasia in mice lacking the integrin-linked kinase in chondrocytes. J. Cell Biol. 162, 139–148 10.1083/jcb.200302066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker K. L., Sage T., Stevens J. M., Jordan P. A., Jones S., Barrett N. E., St-Arnaud R., Frampton J., Dedhar S., Gibbins J. M. (2008). A dual role for integrin-linked kinase in platelets: regulating integrin function and alpha-granule secretion. Blood 112, 4523–4531 10.1182/blood-2008-03-148502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussar S., Wang H. V., Linder S., Fässler R., Moser M. (2006). The Kindlins: subcellular localization and expression during murine development. Exp. Cell Res. 312, 3142–3151 10.1016/j.yexcr.2006.06.030 [DOI] [PubMed] [Google Scholar]
- Ussar S., Moser M., Widmaier M., Rognoni E., Harrer C., Genzel-Boroviczeny O., Fässler R. (2008). Loss of Kindlin-1 causes skin atrophy and lethal neonatal intestinal epithelial dysfunction. PLoS Genet. 4, e1000289 10.1371/journal.pgen.1000289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. V., Chang L. W., Brixius K., Wickström S. A., Montanez E., Thievessen I., Schwander M., Müller U., Bloch W., Mayer U. et al. (2008). Integrin-linked kinase stabilizes myotendinous junctions and protects muscle from stress-induced damage. J. Cell Biol. 180, 1037–1049 10.1083/jcb.200707175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrle-Haller B. (2012). Assembly and disassembly of cell matrix adhesions. Curr. Opin. Cell Biol. 24, 569–581 10.1016/j.ceb.2012.06.010 [DOI] [PubMed] [Google Scholar]
- Wickström S. A., Lange A., Montanez E., Fässler R. (2010). The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase! EMBO J. 29, 281–291 10.1038/emboj.2009.376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Gao J., Hong J., Ma Y. Q. (2013). Integrity of kindlin-2 FERM subdomains is required for supporting integrin activation. Biochem. Biophys. Res. Commun. 434, 382–387 10.1016/j.bbrc.2013.03.086 [DOI] [PubMed] [Google Scholar]
- Yang Y., Wang X., Hawkins C. A., Chen K., Vaynberg J., Mao X., Tu Y., Zuo X., Wang J., Wang Y. X. et al. (2009). Structural basis of focal adhesion localization of LIM-only adaptor PINCH by integrin-linked kinase. J. Biol. Chem. 284, 5836–5844 10.1074/jbc.M805319200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates L. A., Lumb C. N., Brahme N. N., Zalyte R., Bird L. E., De Colibus L., Owens R. J., Calderwood D. A., Sansom M. S., Gilbert R. J. (2012). Structural and functional characterization of the kindlin-1 pleckstrin homology domain. J. Biol. Chem. 287, 43246–43261 10.1074/jbc.M112.422089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F., Kim C., Ginsberg M. H. (2012). Reconstruction of integrin activation. Blood 119, 26–33 10.1182/blood-2011-04-292128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F., Petrich B. G., Anekal P., Lefort C. T., Kasirer-Friede A., Shattil S. J., Ruppert R., Moser M., Fässler R., Ginsberg M. H. (2013). The mechanism of kindlin-mediated activation of integrin αIIbβ3. Curr. Biol. 23, 2288–2295 10.1016/j.cub.2013.09.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.