

Abstract
Specific isotopic labeling at the residue or substituent level extends the scope of different spectroscopic approaches to the atomistic level. Here we describe 13C isotopic labeling of the methyl and methoxy ring substituents of ubiquinone, achieved through construction of a methionine auxotroph in Rhodobacter sphaeroides strain BC17 supplemented with l-methionine with the side chain methyl group 13C-labeled. Two-dimensional electron spin echo envelope modulation (HYSCORE) was applied to study the 13C methyl and methoxy hyperfine couplings in the semiquinone generated in situ at the Qi site of the bc1 complex in its membrane environment. The data were used to characterize the distribution of unpaired spin density and the conformations of the methoxy substituents based on density functional theory calculations of 13C hyperfine tensors in the semiquinone of the geometry-optimized X-ray structure of the bc1 complex (Protein Data Bank entry 1PP9) with the highest available resolution. Comparison with other proteins indicates individual orientations of the methoxy groups in each particular case are always different from the methoxy conformations in the anion radical prepared in a frozen alcohol solution. The protocol used in the generation of the methionine auxotroph is more generally applicable and, because it introduces a gene deletion using a suicide plasmid, can be applied repeatedly.
The biosphere is driven by membrane proteins that participate in protonic circuits in which redox or photoredox reactions drive the generation of the proton gradient used to sustain the cell through a variety of direct and indirect pumps and nanomachines.1−3 As structures have become available, and as molecular engineering approaches have facilitated direct tests of the structural underpinnings, our understanding of mechanism has expanded from the “black-box” view provided by simple physicochemical approaches to the molecular level. Advances in spectroscopy have supplemented this broad front to provide atomistic detail. These methods can be amplified by specific isotopic labeling. However, the enzymes of interest are often studied in a context where such labeling is not available. Specific labeling requires the use of auxotrophic strains and in the past has entailed expression of the protein of interest in a prokaryotic background in which auxotrophic strains have been developed, most commonly in Escherichia coli (cf. refs (4) and (5)) The photosynthetic bacterium Rhodobacter sphaeroides has become a test vehicle for mechanistic studies of a number of respiratory and photosynthetic membrane proteins. Under different growth conditions, it expresses a remarkable metabolic versatility, for example, a “mitochondrial” respiratory chain under aerobic conditions and a photosynthetic chain under anaerobic illumination. Because for many of the enzymes of interest the functional complex cannot be expressed in E. coli, it has not been possible to take advantage of specific isotopic labeling except by in vitro substitution. Auxotrophic strains have been developed in Rb. sphaeroides by MacKenzie and colleagues,6 but in the wild-type 2.4.1 background, where they were generated by transposon mutagenesis, which is a random technique. Most studies of particular enzymes are performed in a modified genetic background specifically developed to facilitate mutagenesis, protein purification, deletion of extraneous genes for functions that interfere with measurement, etc., and in these cases, it is of interest to generate auxotrophic strains by deletion of a particular gene. Here we demonstrate a general method for achieving this end through construction of a methionine auxotroph in Rb. sphaeroides.
Central to all the major electron transfer chains are enzymes of the cytochrome (cyt) bc1 complex family,7−9 which under many physiological conditions control the flux. In the mitochondria that power eukaryotic cells, the cyt bc1 complex is known as complex III of the respiratory chain, but the simpler enzymes of bacteria perform the same enzymatic function and are more accessible to the experimental spectroscopic and kinetic approaches exploited in previous work.
The Q cycle mechanism of the cyt bc1 complex8,10−13 results in oxidation of two quinol (QH2) molecules with release of four protons to the positive side of the membrane (P phase, the intermembrane space in mitochondria, the periplasm of bacteria), reduction of one quinone (Q) with uptake of two protons from the negative side (N phase, the matrix in mitochondria, the cytoplasm in bacteria), and the transfer of two electrons between these phases, in an overall reaction for turnover given by
| 1 |
In the forward chemistry of the Q cycle, electrons from the intermediate semiquinone generated at the Qo site (SQo) upon oxidation of QH2 are delivered to the Qi site by the low-potential chain (hemes bL and bH) to reduce Q to QH2 in a two-electron gate mechanism.13 With the quinone pool initially oxidized, the electron from the first turnover of the Qo site (using QH2 generated in the photochemistry) crosses the membrane to reduce Q at the Qi site to SQ, while that from the second turnover reduces SQ to QH2. The SQ intermediate at the Qi site (SQi) is relatively stable14,15 and can be generated by forward chemistry, by reversal of the second step upon addition of a reductant (ascorbate or QH2) to the complex with heme bH oxidized, or by redox titration. The physical chemistry of the reaction can be studied via the kinetics of electron transfer using spectrophotometry of the heme centers, and the thermodynamics by taking advantage of the paramagnetic properties of the SQ. The redox potentials of the SQ couples can be gleaned from the bell-shaped titration curve, which when studied as a function of pH also maps out pK values important for the mechanism.14,15 Current controversies discuss how these mechanistic considerations relate to structures and atomistic detail. Several structures show a quinone species bound at the Qi site; that from Protein Data Bank (PDB) entry 2QJY(16) shows the Rb. sphaeroides structure, which is most pertinent to this study (Figure 1). Specific mutagenesis coupled with kinetic, spectroscopic, and thermodynamic measurements to explore functional consequences had indicated that the three potential H-bonding partners shown in Figure 1 were all important.10,17−19 More recently, high-resolution EPR approaches have been used to explore the interaction between the SQi paramagnet and nuclear spins in the immediate environment.20−24 Our own results were quite coherent with the mutagenesis studies and the structure shown; they revealed spin interaction with a N atom, with characteristics compatible with the direct H-bond to Nε of His-217 of the cyt b subunit, additional 1H or 2H interactions indicating a second H-bond, likely to either a water or Asp-252, and no other strong spin interaction with protein or solvent.20−22 However, structural interpretations from both crystallographic25−30 and spectroscopic approaches23 have been controversial, with several other suggestions for H-bonding partnerships.
Figure 1.

Qi site topology. Liganding of ubiquinone at the Qi site of the Rb. sphaeroides bc1 complex, showing potential hydrogen bonding partners. Also shown is the network of hydrogen bonds connecting Asn-221 and other residues to the heme bH propionates and His-111, one of the heme Fe ligands. The stereopair is for crossed-eye viewing: structural data from PDB entry 2QJY, chain D. Residues discussed herein are shown as wireframe models, labeled by residue number; UQ and heme bH are shown as ball-and-stick models, and crystallographic waters are represented by spheres showing their O atoms (atom coloring is CPK). Bonds of interest are shown as magenta lines, labeled by lowercase italic letters, with details in Table 1. The hydrogen bond network involving crystallographically defined waters (bonds d–m) is discussed in the text.
Detailed information about the influence of the protein environment on the electronic structure and distribution of the unpaired spin density can be obtained from analysis of 13C couplings of carbon nuclei in the quinone ring and substituents.5,31 However, as noted above, these data are currently not available for the SQi in the Qi site, because approaches for selective isotopic labeling of proteins and cofactors have not been developed in Rb. sphaeroides. In this work, we demonstrate isotopic labeling of the ring substituents of ubiquinone, achieved through construction of a methionine auxotroph in Rb. sphaeroides strain BC17. We extend our studies by 13C labeling of the methyl and methoxy substituents of the ubiquinone ring using 13C-labeled l-methionine, which in the synthetic pathway provides the methyl groups. 13C hyperfine couplings for the methyl and two methoxy groups of SQi generated in situ in the native complex in its membrane environment were determined by 2D ESEEM (HYSCORE). Analysis of the data with the aid of DFT calculations has provided a first look at the distribution of the unpaired spin density over the ring and conformations of methoxy groups in SQi.
Experimental Procedures
Construction of the Methionine Authotroph of Rb. sphaeroides
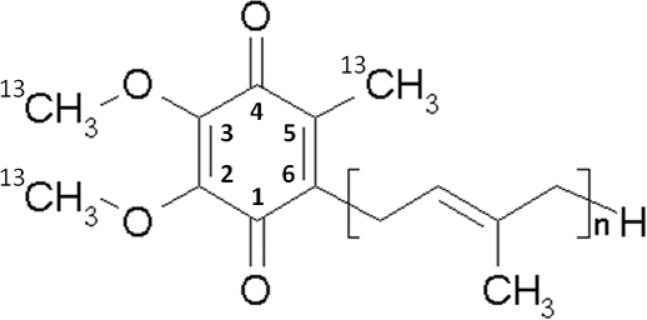
A methionine (Met) auxotroph of Rb. sphaeroides was generated by replacing the metA gene in the chromosome with a cat gene that provides chloramphenicol resistance. For the generation of the auxotroph, the chloramphenicol resistance cassette flanked by ∼500 bp regions homologous to the upstream and downstream regions of metA was constructed by three-step polymerase chain reactions (PCRs). The double-stranded linear PCR product was then inserted into suicide vector pLO1 containing the sacB gene that kills Gram-negative cells when expressed in the presence of sucrose.32,33 The recombinant pLO1 derivative was transformed into E. coli S-17 and then mobilized into Rb. sphaeroides BC17, a bc1-deficient strain,34 by biparental conjugation.35 The Rb. sphaeroides conjugants were selected by their newly acquired chloramphenicol resistance, and the cells containing the pLO1 derivatives among the conjugants were deselected by culture in the presence of sucrose. The deletion of metA in the finally selected strains (metA::cat) was verified by the Met auxotrophic phenotype. Growth of this strain in the presence of 13C methylmethionine results in 13C labeling of the methoxy and methyl carbons of the substituents at positions 2, 3, and 5 on the ring of ubiquinone-10 (UQ-10) (Scheme 1). Strain BC17 was derived from strain Ga, in which the carotenoid synthetic chain of the parental 2.4.1 wild type was truncated by random mutagenesis. In the BC17 strain, the fbc operon encoding the bc1 complex was deleted from the chromosome to generate a background for experiments in which expression of the catalytic subunits of the bc1 complex from the plasmid-borne operon in strains with specific mutations has been exploited in the exploration of details of the mechanism. This library of mutant strains from previous work is now available for further study using isotopic labeling.
Scheme 1.
Preparation of Chromatophores
Chromatophores were prepared according to established methods.36 Chromatophores are sealed vesicles formed upon mechanical disruption of cells; inner membrane invaginations break off and reseal, and they contain the periplasmic medium (including cyt c2) and the entire membrane-bound apparatus for photosynthetic phosphorylation. The bc1 complex is therefore in its native state and reacts with its native partners. The composition of the chromatophore membrane depends on growth conditions, strain, etc., and can vary quite substantially. In the Ga parental strain, when cells are grown under standard laboratory conditions and harvested in late log phase, the ratio of bacterial reaction center to bc1 complex is approximately 2:1; however, the components are likely distributed at random, and the ratio in individual vesicles is stochastic within normal limits. However, the mean ratio can vary over a wide range.37
Characterization of the Qi Site Semiquinone
Redox titrations were performed in a potentiometric cell maintained under anaerobic conditions using argon gas. Chromatophores were suspended in 100 mM KCl and 50 mM MOPS (pH 7.0) or 100 mM KCl and 50 mM CHES (pH 9.2) to a reaction center concentration of ∼0.7 mM, and the redox potential was adjusted by adding small aliquots of potassium ferricyanide or sodium dithionite. The following redox mediators were present: 4 μM each of 2-OH-1,4-naphthoquinone and pyocyanin; 8 μM each of 3,6-diaminodurene, phenazine ethosulfate, and phenazine methosulfate; and 40 μM each of p-benzoquinone, 1,2-naphthoquinone, 1,4-naphthoquinone, and duroquinone. The samples were taken in the dark at the desired Eh values using an airtight syringe, placed in argon-flushed EPR tubes, frozen rapidly, and stored in liquid nitrogen until the samples were measured. At each point shown in the titration, data were obtained in the absence and presence of antimycin, and the value shown represents the difference, giving the antimycin-sensitive component (see Figure S1 of the Supporting Information and its legend), contributing ∼87% of the overall signal at the peak of the curve. However, because the antimycin-insensitive component showed essentially the same amplitude over the range of the titration, the fractional contributions varied.
EPR and ESEEM Measurements
The CW EPR measurements were performed on an X-band Varian EPR-E122 spectrometer, with the sample at 100 K. The pulsed EPR experiments were conducted using an X-band Bruker ELEXSYS E580 spectrometer equipped with an Oxford CF 935 cryostat. All pulsed EPR measurements were taken at 80 K. The 2D ESEEM, four-pulse experiment (π/2−τ–π/2–t1–π–t2–π/2−τ–echo, also called HYSCORE), with a magnetic field of 343.0 mT, a time between the first and second pulses (τ) of 136 ns, and a microwave frequency of 9.619 GHz,38 was employed with appropriate phase cycling schemes to eliminate unwanted features from the experimental echo envelopes. The intensity of the echo after the fourth pulse was measured with t2 and t1 varied and a constant τ. The length of a π/2 pulse was 16 ns and that of a π pulse 32 ns. HYSCORE data were collected in the form of 2D time domain patterns containing 256 × 256 points with steps of 20 or 32 ns. Spectral processing of ESEEM patterns, including subtraction of the relaxation decay (fitting by polynomials of three to six degrees), apodization (Hamming window), zero filling, and fast Fourier transformation (FT), were performed using Bruker WIN-EPR version 2.22 revision 10. Processed data were then imported into Matlab R2010a via the EasySpin package39 either to be simulated by EasySpin version 4.5.5 or to be analyzed by a homemade script for fitting data in (ν1)2 versus (ν2)2 coordinates. After the HYSCORE had been plotted as (ν1)2 versus (ν2)2, ridges were fit via a linear regression with each point on the ridge weighted according to its HYSCORE intensity (see below). The peculiarities of powder 13C HYSCORE spectra and all details of the analysis of the 13C cross-peaks from methyl and methoxy substituents are described in the recent study of the 13C-labeled SQs in the QA and QB sites of the bacterial reaction center (see also the Supporting Information).31
Computational Methods
All density functional calculations were performed using Gaussian 09.40 Geometries were partially optimized at the B3LYP/6-31G(d) level, and hyperfine couplings were calculated at the B3LYP/EPR-II level.
Results
Physicochemical Properties of the Qi Site Semiquinone
The antimycin-sensitive semiquinone generated in this work was similar to those reported earlier in chromatophores from Rb. sphaeroides(15) or in submitochondrial particles14 (Figure 2). The two values of pH at which measurements were taken preclude detailed discussion of Em–pH relationships, but our results are consistent with the previous work. The 13C labeling of the methyl groups does not influence the SQ line width, indicating that it is still dominated by the g-tensor anisotropy, and likely in the anionic form. The 1H–14N ESEEM spectra were identical with the spectra of unlabeled UQ-10 SQs measured in the isolated bc1 complex,20,21 thus confirming similar H-bond patterns, independent of isolation procedures, membrane location, or 13C labeling. This similarity also shows that there is no significant contribution to the spectra from SQ species at catalytic sites outside the bc1 complex.
Figure 2.
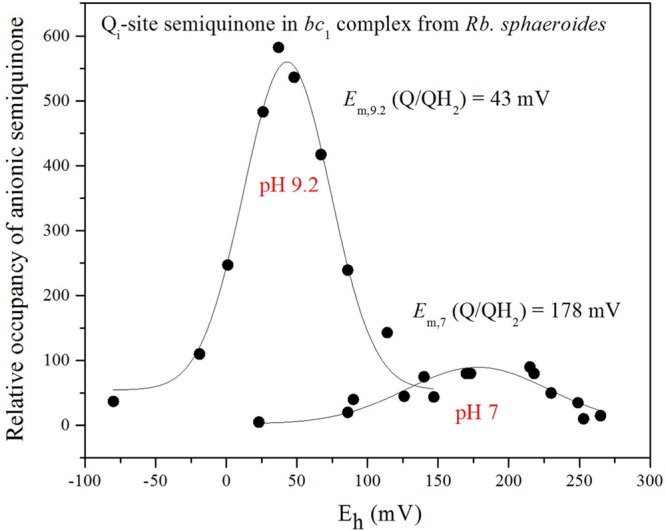
Potentiometric titration curves of the SQi EPR signals at pH 7.0 and 9.2. The spectra were obtained in the range of redox potentials between −80 and 265 mV vs the standard hydrogen electrode (SHE). Redox potentiometry was performed under argon with redox mediators, and the semiquinone radical was titrated with sodium dithionite. EPR spectra were obtained using a Varian E-112 X-band spectrometer with a magnetic field center of 325.0 mT and a field sweep width of 20.0 mT. The semiquinone bound at the Qi site was assayed from the antimycin-sensitive component by measuring the peak to trough amplitude of the g = 2.005 signal obtained at 100 K with a microwave power of 0.2 mW, with occupancy shown in arbitrary units.
Spectroscopic Properties
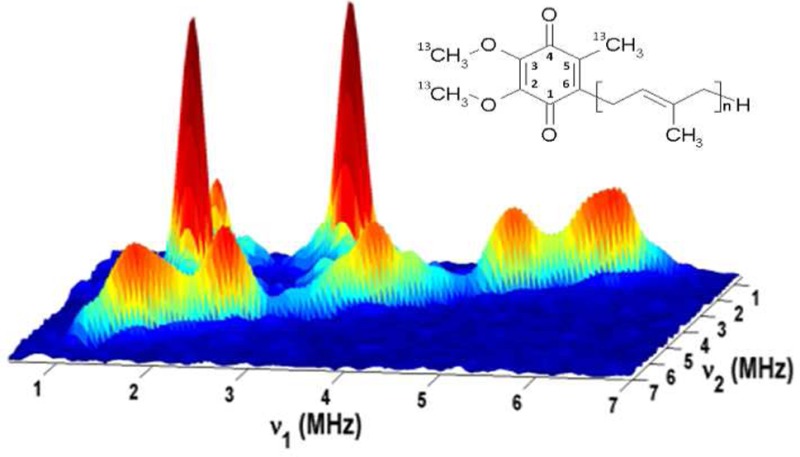
The 13C hyperfine couplings in SQi were probed by HYSCORE experiments. Figure 3 (left) shows representative HYSCORE spectra for SQi in the frequency interval from 0 to 7 MHz for both axes. The spectrum of SQi contains the lines from 14N and 13C nuclei. 14N cross-peaks with coordinates (3.1, 1.7) MHz produced by Nε of H217 were previously analyzed in detail.20,21 Here we focus on the analysis of the 13C lines. The 13C cross-features are located along the antidiagonal, symmetrically relative to the diagonal point (νC, νC) where νC is ∼3.7 MHz, in the applied magnetic field. Those include peak 1C with extended shoulders around the diagonal point (νC, νC) and two pairs of cross-peaks 2C and 3C. Peak 1C has resolved shoulders with a splitting of ∼1 MHz (see also Figure S3 of the Supporting Information). Cross-peaks 2C and 3C are partially overlapped but possess well-separated maxima at (5.0, 2.4) MHz (2C) and (5.9, 1.6) MHz (3C), which correspond to first-order estimated hyperfine couplings of 2.6 and 4.3 MHz, respectively.
Figure 3.
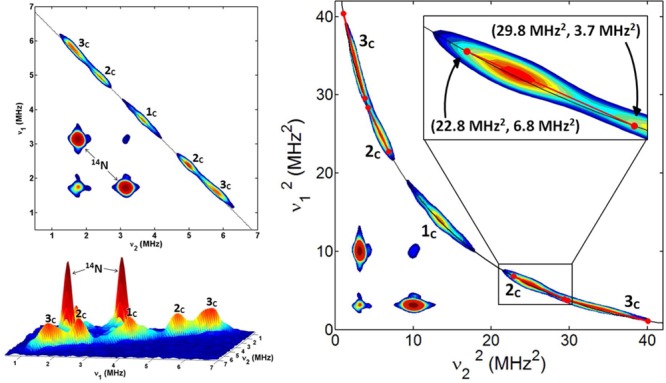
Contour (left, top) and stacked (left, bottom) HYSCORE spectra of SQi in the bc1 complex [magnetic field of 343.0 mT, time between first and second pulses (τ) of 136 ns, and microwave frequency of 9.619 GHz]. Contour presentation of the HYSCORE spectra of SQi in (ν1)2 vs (ν2)2 coordinates (right). The curved line is defined by |ν1 + ν2| = 2νC. The inset shows the linear regression fit for selected cross-peak 2C. A graph showing the insert for the fitted ridge 3C is included in the Supporting Information.
Hyperfine tensors of the 13C nuclei contributing to the spectrum of SQi can be obtained from the analysis of the contour line shape of the cross-peaks. An analytical estimate of the 13C hyperfine tensors, i.e., a (isotropic coupling constant) and T (anisotropic coupling constant), was performed by plotting the HYSCORE spectrum in (ν1)2 versus (ν2)2 coordinates41 (Figure 3, right). As a result, cross-peaks that are approximately axial appear as linear segments that can be fit to a linear regression. Intersection points of the linear fit with the line |ν1 + ν2| = 2νC define the principal values of the hyperfine tensor. There are two possible assignments to (να⊥, νβ⊥) or (να∥, νβ∥) for each crossing point and, consequently, two solutions, one for each assignment. Linear regression analysis of cross-peaks 2C and 3C and calculation of the tensors are discussed in the Supporting Information (Table S1 and Figure S2).
To choose between the two sets of axial tensors from the analysis of the 13C HYSCORE plotted as (ν1)2 versus (ν2)2, the 13C ridges for SQi were simulated with EasySpin.39 Simulations were done starting from the squared-frequency analysis and then further optimized by adjusting the values and introducing rhombicity factor δ into the anisotropic hyperfine tensor (−T(1 + δ), −T(1 – δ), 2T). Simulated spectra were found to be essentially independent of the relative Euler angles between the 13C hyperfine tensors, possibly because of the low level of hyperfine anisotropy. Thus, calculations were performed without adjusting the Euler angles. Spectra for two possible tensors for cross-features 2C and 3C and their comparison with the experiments are provided in the Supporting Information (Table S2 and Figure S3).
Special comment is needed with regard to the analysis of feature 1C. It did not fit well with a single set of a and T. This suggested that 1C results from a distribution of methoxy orientations, and therefore, a distribution of a and T may be necessary. However, even after an approximate distribution of hyperfine tensors had been implemented, the simulations were unable to match the spectrum, giving a simple featureless peak centered on the diagonal. As seen in the spectrum, peak 1C instead clearly has symmetrical shoulder features with a splitting of ∼1.0 MHz. A reasonable fit for the 1C line shape was achieved using a two-state model. T is not expected to vary significantly from 0.5 MHz following the data listed in Table 2. Thus, it was fixed to that value, and spectra were simulated with two unique isotropic coupling constants a corresponding to two different methoxy orientations contributing to 1C. Optimization of the rhombicity for 1C (which is fixed along with T for both methoxy orientations in the simulations), the actual values of the two a’s, and the relative populations of the two states has given a surprisingly agreeable simulation when ∼60% is due to a methoxy conformation giving a = 0.2 MHz and 40% for a = 1.1 MHz (Figure S3 of the Supporting Information). The final results of the simulations are summarized in Table 2 for comparison with the similar 13C tensors for SQA and SQB in bacterial reaction centers.31
Table 2. 13C Hyperfine Tensors Determined from the HYSCORE Spectral Simulations for SQi in Comparison with SQA and SQB.
Discussion
Structural Considerations
High-resolution structures of mitochondrial (bovine, chicken, and yeast at ≥2.1 Å) and bacterial complexes (at ≥2.6 Å) show different configurations of the quinone species bound at the Qi site,16,26,27,30,42−44 but all have the same two residues close enough to the quinone to allow formation of either direct or H2O-mediated H-bonds, H217 and D252 (in Rb. sphaeroides numbering). In a model structure,21 N221 was positioned as a potential ligand to one of the methoxy O atoms, but the distance seen in crystallographic structures is not compatible with such a role (Figure 1). In the bovine mitochondrial complex (italic numbering), the corresponding residues are H201 and D228, with N221 replaced by S205. However, we note that in relation to this discussion, the data from crystallographic structures relate to an unknown redox state of the Qi site occupant. Also, the B factors of the occupant, even for the highest-resolution structures, show more disorder than the best defined regions, and there is considerable disagreement between different structures with respect to the configuration. Because of these ambiguities, interpretation with respect to semiquinone binding and selection of models for quantum calculation cannot be unequivocal. However, mutagenesis studies provided strong evidence of a functional role for each of these residues.10,17−19
DFT Computations of 13C Tensors
For the selectively 13C-labeled UQ-10 used here, three 13C hyperfine couplings are expected corresponding to the two methoxy groups and the 5-CH3 group carbon nuclei. The HYSCORE spectra of Figure 3 clearly illustrate these three 13C features for SQi. Analysis of the spectra shows that for SQi one of these carbons has a small hyperfine coupling of ∼0–1 MHz, whereas values with magnitudes of 2.5 and 4.4 MHz can be estimated for the other two. For the assignment of these measured tensors to the 13C nuclei of the methyl or methoxy groups, it is necessary to compare with previous experimental results in vitro and in vivo or with values calculated using DFT approaches.
The DFT computational approach extensively tested on the QA and QB sites in the bacterial reaction center from Rb. sphaeroides(45−47) has also been applied to the Qi site. We have chosen to use the highest-resolution bovine mitochondrial structure available (PDB entry 1PP9 at ≥2.1 Å resolution)26 as the basis for our model, although the bacterial structure (PDB entry 2QJY at ≥2.6 Å resolution)16 might seem to be the obvious choice. The rationale for this choice is as follows. (i) The quantum chemical models contain only D228 and H201 as contributions from the protein, and these same side chains (D252 and H217) contribute in the Rb. sphaeroides structures. (ii) Neither of the other potential ligands, S205 in mitochondria or N221 in Rb. sphaeroides, is within H-bonding distance of the quinone species in the structures. Other groups undoubtedly contribute to the site, but we have no justification for preferring the lower-resolution structure unless experimental data are available to show specific parameters that would affect the EPR data. We have analyzed two cases for the Qi site of the bovine bc1 complex structure (PDB entry 1PP9), with H201 either deprotonated (a) or protonated (b) at Nδ (Figure S4 of the Supporting Information). Both models indicate that the COOH group of D228 and the NεH group of H201 are hydrogen bonded to the carbonyls of the semiquinone. Geometry optimization of both models shows that when Nδ of H201 is protonated, the proton from NεH transfers to quinone O4 and the SQ is a neutral free radical with its OH group acting as a donor of a hydrogen bond to Nε of H201. For the deprotonated Nδ model, the SQ is an anion radical with both its O1 and O4 atoms accepting hydrogen bonds from D228 and H201, respectively. Previous experimental data for H-bond protons and Nε20−22 are more consistent with the anion radical and rule out the neutral radical. The DFT-calculated 13C hyperfine couplings for the anion radical model are listed in Table 3, where we compare these with the experimental determinations. The DFT-calculated values strongly support the assignment of the largest-magnitude 4.4 MHz value to the 5-CH3 carbon, the 2.5 MHz value to the 3-methoxy carbon, and the smaller-magnitude 0.2 or 1.0 MHz value to the 2-methoxy carbon.
Table 3. Calculated 13C, Isotropic Constant (a), and Anisotropic Hyperfine Tensor Components (Tnn) (in megahertz) for the Anion Radical Model of SQia.
| position | a | |
|---|---|---|
| 13C–CH3 (5) | 0.8 (T11) | –4.8 (|4.4|) |
| –0.4 (T22) | ||
| –0.4 (T33) | ||
| 13C–CH3O (3) | 0.8 (T11) | 2.0 (|2.5|) |
| –0.2 (T22) | ||
| –0.6 (T33) | ||
| 13C–CH3O (2) | 0.6 (T11) | 0.5 (|0.2|, |1.0|) |
| –0.1 (T22) | ||
| –0.6 (T33) |
The magnitudes of the experimental isotropic hyperfine couplings are given in parentheses.
Spin populations calculated previously for the ubisemiquinones in model systems (QA and QB quinone sites31 and the high-affinity QH site of cytochrome bo3 ubiquinol oxidase and its D75H mutant5,48) are shown in Table 4. The last row contains spin populations for the anion radical in the Qi site. Our assignment of the largest hyperfine coupling to the 5-CH3 group in SQi is also consistent with experimental and calculated isotropic hyperfine couplings (Table 5) for the ubisemiquinones from Table 4.
Table 4. Mülliken Spin Populations in Selected SQs.
| semiquinone | C2 | C3 | C5 |
|---|---|---|---|
| SQAa | 0.11 | –0.01 | 0.05 |
| SQBa | 0.09 | 0.03 | 0.07 |
| SQM1a,c | 0.07 | 0.07 | 0.09 |
| SQM2a,c | 0.11 | 0.00 | 0.07 |
| SQHb | –0.02 | 0.16 | 0.14 |
| SQH (D75H)b | 0.00 | 0.16 | 0.13 |
| SQi | 0.06 | 0.05 | 0.11 |
From ref (31).
From ref (48).
SQM1, 6-methyl-UQ with four H2O molecules; SQM2, 6-methyl-UQ with one H2O (see Figure S5 of the Supporting Information).
Table 5. Comparison of Experimental and Calculated 13C Methyl (5′) Isotropic Couplings in SQs.
The spin populations in Table 4 demonstrate the asymmetry of the spin density distribution in different SQs resulting from different interactions with their protein environment. Spin densities obtained for SQH arise from stronger hydrogen bonding with O1. In contrast, the opposite asymmetry of spin density distribution in the QA site suggests stronger hydrogen bonding at O4. The asymmetry in spin density distribution observed for the SQi radical might indicate that a slightly stronger hydrogen bond is formed at O1 with D252. A notable peculiarity of the unpaired spin density distribution for SQi is the similarity of values for the spin populations on C2 (0.06) and C3 (0.05). This is in contrast with the QA and QB sites where the C2 population is much larger than C3 or with the QH site where the opposite relationship occurs.
As shown previously, the 13C isotropic hyperfine coupling of the methoxy group will be proportional to the π(p) spin population of the corresponding ring carbon and the dihedral angle of the methoxy CO bond with respect to the SQ ring plane.31 The unpaired spin density giving rise to the 13C isotropic hyperfine coupling for the methoxy group carbon atom arises from a combination of spin polarization and hyperconjugation. When the methoxy group is held in the ring plane, hyperconjugation is expected to be zero and small negative hyperfine couplings are expected for in-plane or near to in-plane orientations, due to spin polarization by the methoxy oxygen spin density. As the methoxy orientation is moved progressively out of plane, a positive contribution from hyperconjugation with the ring carbon π(p) spin density arises.31 The agreement with experiments observed in Table 3 for the geometry-optimized isolated SQi model (starting from the crystal structure coordinates) suggests that the methoxy groups in the Qi site are close to the minimal energy conformation of an isolated SQ. The conformations for the minimal energy model are 3-methoxy (CM2–O2–C2–C1) = −68° and 2-methoxy (CM3–O3–C3–C4) = 56°. This is in contrast to the QA and QB site semiquinones in bacterial photosynthetic reaction centers, which deviate significantly from the optimal angles for an isolated model.
Influence of the Protein Environment on Methoxy Conformations
HYSCORE spectra of the SQ in the QA, QB, and Qi sites resolve all three cross-peaks from the three 13C site-specifically labeled nuclei in the SQ structure, thus suggesting significant constraints on the methoxy group conformation by the protein environment. The 2-methoxy groups in SQA and SQB were shown to give rise to quite distinct 13C isotropic hyperfine couplings, with the magnitude of SQB exceeding that of SQA (Table 2). The larger value for SQB could be qualitatively explained by a more out-of-plane orientation of the 2-methoxy group compared with SQA. DFT calculations demonstrated a strong dependence of the electron affinity of the quinone on the methoxy dihedral angles,31 and the higher redox potential of the QB ubiquinone was rationalized on this basis. A similar comparative analysis for the 3-methoxy orientation was however precluded by the negligible unpaired spin density on the adjusted C3 atom in SQA (Table 4). In contrast to SQA and SQB, direct computation of the SQi optimized structure and distribution of the unpaired spin density allows us to assign the observed 13C couplings to particular substituents and gives an estimated conformation of 3-methoxy that possesses a larger isotropic coupling (and larger deviation) than 2-methoxy. On the other hand, this approach leaves open the interpretation of the complex line shape of peak 1C assigned to the 2-methoxy group and suggests a behavior more complex than a single conformation with a small strain. Analysis of the bovine mitochondrial complex (PDB entry 1PP9) shows a different conformation of the methoxy group for the quinone in the Qi site that may correlate with the two-conformation model giving a best fit of 1C peaks with the simulated spectra. This structural difference, however, was not found in lower-resolution bacterial structures.
In contrast to the spectra in proteins, the HYSCORE spectrum for the 13C methyl, methoxy-labeled anion radical of UQ-10 in alcohol consists of two broad cross-peaks extending along the antidiagonal (Figure S6 of the Supporting Information) and does not show significant intensity around the diagonal point. This spectrum may indicate substantial discrete line widths of three individual couplings contributing to these cross-peaks. In addition, the similarity between the spin density on C2 and C3 for the anion radical (model SQM1 in Table 4) suggests that its spin density distribution is described well by the model and indicates a significant average deviation from the in-plane configuration for both methoxy groups in an alcohol solution. The spin density at C2 and C3 in SQi is also similar, but the spectra are quite different, suggesting that a dynamic situation in solution is replaced by a more constrained configuration in the protein environment.
For the QA and QB sites, it has been suggested that modification of the methoxy orientation fine-tunes the redox potentials to ensure the transfer of an electron from QA to QB. However, the operational ranges at both QA and QB sites (Em,7 values of −100 and 0 mV, respectively) are at redox potentials much lower than that of the Q pool (Em.7 ∼ 90 mV), while both the Qi site (Em,7 ∼ 150 mV) and the Qo site (Em,7 ∼ 120 mV) function at a higher potential.8,53 This substantial difference must be considered in discussing any such control. The higher redox potential for Qi would at least qualitatively be in accord with having methoxy groups near the minimal energy conformations for the SQ form. This stabilizes the semiquinone, increasing both the electron affinity and the redox potential. However, the high potential for the Q/QH2 couple at both these Qi and Qo sites is achieved through preferential binding of QH2. Although both sites operate in the high-potential range, their mechanism and function are very different. The difference in function comes in part from the wide difference in SQ stability and in part from mechanistic differences. The higher stability of SQi allows it to function as an intermediate, storing the first electron in a two-electron gate mechanism. This stability might reflect the minimal energy configuration but certainly also reflects the two H-bonds shown by our earlier ESEEM work.20−22 In the bifurcated reaction at the Qo site,53 the very unstable SQo allows a thermodynamic coupling between two electron transfer pathways, so that the work derived from oxidation of QH2 to SQ by the high-potential chain (ISP, heme c1, and cyt c2) generates a strong reductant in the Q/SQ couple to drive reduction at the Qi site, and maintenance of the proton gradient by electrogenic electron transfer across the membrane. Clearly, factors other than methoxy orientation are in play in the mechanisms of both sites.
The apparent influence of the protein environment on the conformation of the methoxy groups in each particular case suggests that the role of the methoxy conformations in the Qi site of the bc1 complex could be better understood from experiments with Qi site mutants. A strategy exploiting mutagenesis in the context of the synergy between spectroscopy and specific isotopic labeling made possible with auxotrophic strains, and used with great success in studies of the SQH species of the bo3 oxidase of E. coli,(5,48) can now be applied to the bc1 complex, where the richness of structural information and kinetic studies opens wider horizons.
Mechanism of Formation of SQ at the Qi Site
In earlier models, formation of the SQ was considered in the context of the disproportionation equilibria, determined by KS = ([SQ]2)/([Q][QH2]). An anomalous feature was the low occupancy (<0.5/heme bH), accounted for either in terms of a dimeric model,14 in which only one monomer could be occupied at a time, or by quenching of the EPR signal by ferriheme bH in a fraction of the centers,15,50 or by invoking limitations arising from the dissociation state of a protein group.15 A third class of mechanism involved the “cyt b-150” phenomena.19,51 Redox titration of heme bH in the presence of antimycin shows a component usually analyzed as a single species with an Em,7 of ∼40 mV. However, in the absence of antimycin, the titration is biphasic, showing the same overall amplitude, but with a fraction (dependent on pH) titrating at Em,7 ∼ 150 mV. Addition of antimycin induced a loss of this component to give an antimycin-induced oxidation (see ref (52) for discussion). The pH dependence of the 150 mV component correlated well with the pH dependence of SQ formation, suggesting participation of heme bH in the formation of SQ through reversal of the normal forward chemistry. As structures became available, and more detailed spectroscopic observation addressed the binding of the SQ more directly, additional complicating features were identified. In particular, as discussed in ref (20), it seems probable that the pattern of H-bonding with the protein must change with the redox state of the quinone species involved, and with the dissociation state of H217 and D252. The line width of the SQ signal indicates the anionic semiquinone over a wide range of pH, so the pH dependence has to be attributed to dissociable group(s) of the protein rather than of the SQ.15 These questions have not recently been revisited, and detailed discussion in the present context would be out of place. However, because the quantum chemical calculations suggest that the anionic semiquinone is associated with a protonated H217, it seems likely that this residue is the dissociable group needed to explain the pH dependence of SQ stability15 and identified in this role in ref (20). The other direct ligand included in the quantum chemical calculations is D252. It is likely that the pKa of this group in some configurations (for example, a vacant site) would be influenced by interaction with K251, not yet included in the model. In previous reports of the effects of mutation at N221,10,22 we had discounted any importance of H-bonding because the N221P strain showed kinetic behavior and thermodynamic properties similar to those of the wild type. Although we could not exclude a weak interaction of N221 with the methoxy O on the basis of 1H, 2H, 14N, or 15N proton spin interactions,20−22 the high activity of the N221P strain and the similarity of the kinetic behavior to that of the wild type seemed to argue strongly against any H-bonding interaction. More recent experiments have failed to reproduce this result. Unfortunately, the original N221P strain was lost, but analysis of cells recovered from frozen stocks of chromatophore preparations used in previous experiments showed the wild-type sequence. We therefore believe our earlier results represent a wild-type strain obtained by either reversion or contamination. We have reconstructed an authentic N221P strain, but all attempts to grow this strain under anaerobic photosynthetic conditions, which require an active bc1 complex, have failed. The failure of the authentic N221P strain to grow would then indicate an important H-bonding role for the native asparagine side chain, supported by the low rates seen in N221I mutants.22 This necessitates a revision of our earlier conclusions, with respect to both a general H-bonding role and a more specific side chain-dependent interaction. In structures, N221 is too distant to form a direct H-bond through the side chain to the methoxy O (Figure 1 and Table 1), but this would be pertinent to the data presented here only if the X-ray beam had induced reduction of the SQ. The side chain is involved in a network of H-bonded groups that includes waters and the heme propionates, and MD simulations (see ref (53)) suggest additional waters that might provide a proton channel to the N phase. The strong inhibition of transfer of an electron from heme bH to the Qi site upon mutation at N221 could then indicate that the equilibration of the heme propionates, and possibly of H111 and the SQ, with the pH of the N phase is important in facilitating this electron transfer step. This brings up the question of how heme bH is involved in the formation of the SQ. The “cyt b-150” model had suggested that the SQ is formed via reversal of the forward reaction:
These reactions account well for the “cyt b-150” phenomena, and the oxidation of heme b–H induced upon addition of antimycin,19,51 but a simple thermodynamic model does not explain the SQ occupancy or the shape of the titration curve (which in the model was pH-dependent but experimentally shows the same shape at all pH values). No detailed model including specification of the dissociable groups involved in the equilibration of protons with the “visible” reactants is yet available.
Table 1. Parameters for Bonds Shown in Figure 1.
| label | bond distance (Å) | atom 1 | atom 2 |
|---|---|---|---|
| a | 2.26 | H217 NE2 | UQ2 O4 |
| b | 4.78 | D252 OD2 | UQ2 O1 |
| c | 4.56 | N221 ND2 | UQ2 O3 |
| d | 2.73 | N221 OD1 | H2O 1163 |
| e | 2.51 | H2O 1163 | heme O2A |
| f | 2.59 | H2O 1161 | heme O1A |
| g | 3.35 | R114 NH1 | H2O 1161 |
| h | 2.85 | H111 ND | H2O 1161 |
| i | 3.13 | R114 NH2 | heme O2A |
| k | 3.5 | R114 NH2 | H2O 1162 |
| l | 2.47 | heme O1D | H2O 1162 |
| m | 3.64 | H2O 1162 | H2O 1163 |
| n | 2.64 | heme O2D | H2O 1115 |
| o | 2.67 | R125 NE | heme O2D |
Bond a is the primary bond stabilizing Q.
Bond b is likely an indirect H-bond through water, as seen in some structures.
Bond c is included to show distance, but no evidence for bond.
The set of H-bonds d through o connects the heme ligand, His-111, and the heme propionates, to crystallographically defined waters and the protein, to form a network. It seems likely that this network is more extensive and links to proton conduction pathways to the aqueous phase through which protons equilibrate with the heme, and possibly with changes in redox state of the quinone, during the Qi-site reaction.
A more detailed examination of these different possibilities must await further work on characterization of strains with mutations at key residues involved in liganding the SQ or protonic equilibration of the reaction components, more detailed MD and quantum chemical models, and spectroscopic studies exploiting specific isotopic labeling that will be possible with extensions of this work to other auxotrophic strains.
Conclusion
Specific isotopic labeling at the residue or substituent level extends the scope of several important spectroscopic approaches to structure at the atomistic level. Here we demonstrate isotopic labeling of the ring substituents of ubiquinone, achieved through construction of a methionine auxotroph in Rb. sphaeroides strain BC17, and used to explore the question of electronic structure and geometry of substituents at the Qi site of the bc1 complex. This topic has been controversial, with different results reported from crystallography and from pulsed EPR studies in mitochondrial and bacterial complexes. We extend our studies of the complex in Rb. sphaeroides by isotopic labeling of the methyl and methoxy substituents of the ubiquinone ring using 13C and determination of the interaction of the nuclear spins with the electron spin of the semiquinone generated in situ in the complex in its membrane environment. By using HYSCORE spectroscopy, we have measured the isotropic and anisotropic spin couplings to allow comparison with properties previously estimated in the QA and QB sites of the bacterial reaction center, and the QH site of the cyt bo3 oxidase of E. coli. Differences between these sites, and the strong dependence of isotropic couplings on the dihedral angles of the substituents, allow an exploration of the orientation of the semiquinone methoxy groups in the catalytic site. The protocol used in generation of the methionine auxotroph is applicable more generally and, because it introduces a gene deletion using a suicide plasmid, can be applied repeatedly. In particular, because auxotrophy often will require deletion of a single gene, it is convenient when a highly engineered background has been developed for mutational engineering in the context of a specific protein, because previous manipulations are undisturbed. In our case, construction of an auxotroph in the BC17 strain allows conclusions from the spectroscopic studies to be directly compared with experimental data derived from in situ assays of function through kinetic spectrophotometry using an extensive collection of mutant strains.
Acknowledgments
We thank Professors Sam Kaplan, Ronald MacKenzie, and Jesus Eraso for making available their set of auxotrophic strains and for advice on engineering gene deletions.
Glossary
Abbreviations
- 2D
two-dimensional
- CW
continuous-wave
- DFT
density functional theory
- B3LYP
Becke3 Lee–Yang–Parr
- SQ
semiquinone
- EPR
electron paramagnetic resonance
- ESEEM
electron spin echo envelope modulation
- HYSCORE
hyperfine sublevel correlation
- UQ-10
ubiquinone-10.
Supporting Information Available
Relevant methodological details and Figures S1–S6. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# Departamento de Química Inorgânica, Instituto de Química, Universidade Federal Fluminense, Campus do Valonguinho, Centro, Niterói, RJ, CEP: 24020-141, Brazil
The authors declare no competing financial interest.
This research was supported by Grants DE-FG02-08ER15960 (S.A.D.) and DE-FG02-87ER13716 (R.B.G.) from the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Sciences, U.S. Department of Energy, National Institutes of Health (NIH) Grant GM062954 (S.A.D.), and National Center for Research Resources Grants S10-RR15878 and S10-RR025438 for pulsed EPR instrumentation. P.J.O. acknowledges the use of computer resources granted by the EPSRC UK national service for computational chemistry software (NSCCS). A.T.T. gratefully acknowledges support as a NIH trainee of the Molecular Biophysics Training Program (5T32-GM008276). W.B. De Almeida would like to thank FAPEMIG (Fundação de Amparo a Pesquisa no Estado de Minas Gerais) for financial support during his stay at The University of Manchester, UK, and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) for a research grant. We also thank the University of Manchester Computer Share Facilities (CSF) for providing computational resources to develop this work.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Nicholls D. G., and Ferguson S. J. (2013) Bioenergetics, 4th ed., Academic Press, Elsevier, Amsterdam. [Google Scholar]
- Mitchell P. (1966) Chemiosmotic Coupling in Oxidative and Photosynthetic Phosphorylation, Glynn Research Ltd., Bodmin, Cornwall, U.K. [DOI] [PubMed] [Google Scholar]
- Mitchell P. (1968) Chemiosmotic Coupling and Energy Transduction, Glynn Research Ltd., Bodmin, Cornwall, U.K. [Google Scholar]
- Lin I.-J.; Chen Y.; Fee J. A.; Song J.; Westler W. M.; Markley J. L. (2006) Rieske Protein from Thermus thermophilus: 15N NMR Titration Study Demonstrates the Role of Iron-Ligated Histidines in the pH Dependence of the Reduction Potential. J. Am. Chem. Soc. 128, 10672–10673. [DOI] [PubMed] [Google Scholar]
- Lin M. T.; Shubin A. A.; Samoilova R. I.; Narasimhulu K. V.; Baldansuren A.; Gennis R. B.; Dikanov S. A. (2011) Exploring by pulsed EPR the electronic structure of ubisemiquinone bound at the QH site of cytochrome bo3 from Escherichia coli with in vivo13C-labeled methyl and methoxy substituents. J. Biol. Chem. 286, 10105–10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie C.; Simmons A. E.; Kaplan S. (1999) Multiple chromosomes in bacteria: The Yin and Yang of trp gene localization in Rhodobacter sphaeroides 2.4.1. Genetics 153, 525–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer W. A.; Zhang H.; Yan J.; Kurisu G.; Smith J. L. (2004) Evolution of photosynthesis: Time-independent structure of the cytochrome b6f complex. Biochemistry 43, 5921–5929. [DOI] [PubMed] [Google Scholar]
- Crofts A. R. (2004) The Q-cycle: A personal perspective. Photosynth. Res. 80, 223–243. [DOI] [PubMed] [Google Scholar]
- Kramer D. M., Nitschke W., and Cooley J. W. (2009) The cytochrome bc1 and related bc complexes: The Rieske/cytochrome b complex as the functional core of a central electron/ proton transfer complex. The Purple Phototrophic Bacteria (Hunter C. N., Daldal F., Thurnauer M. C., and Beatty J. T., Eds.) pp 451–473, Springer Science, New York. [Google Scholar]
- Crofts A. R.; Holland J. T.; Victoria D.; Kolling D. R.; Dikanov S. A.; Gilberth R.; Lhee S.; Kuras R.; Kuras M. G. (2008) The Q-cycle reviewed: How well does a monomeric mechanism of the bc1 complex account for the function of a dimeric complex?. Biochim. Biophys. Acta 1777, 1001–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P. (1976) Possible molecular mechanisms of the protonmotive function of cytochrome systems. J. Theor. Biol. 62, 327–367. [DOI] [PubMed] [Google Scholar]
- Trumpower B. L. (1981) Function of the iron-sulfur protein of the cytochrome b-c1 segment of the respiratory chain. Biochim. Biophys. Acta 639, 129–155. [DOI] [PubMed] [Google Scholar]
- Crofts A. R.; Meinhardt S. W.; Jones K. R.; Snozzi M. (1983) The role of the quinone pool in the cyclic electron-transfer chain of Rhodopseudomonas sphaeroides: A modified Q-cycle mechanism. Biochim. Biophys. Acta 723, 202–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries S., Berden J. A., and Slater E. C. (1982) Oxidation-reduction properties of an antimycin-sensitive semiquinone anion bound to QH2:cytochrome c oxidoreductase. Function of Quinones in Energy Conserving Systems (Trumpower B. L., Ed.) pp 235–246, Academic Press, New York. [Google Scholar]
- Robertson D. E.; Prince R. C.; Bowyer J. R.; Matsuura K.; Dutton P. L.; Ohnishi T. (1984) Thermodynamic properties of the semiquinone and its binding site in the ubiquinol: Cytochrome c (c2) oxidoreductase of respiratory and photosynthetic systems. J. Biol. Chem. 259, 1758–1763. [PubMed] [Google Scholar]
- Esser L.; Elberry M.; Zhou F.; Yu C.-A.; Yu L.; Xia D. (2008) Inhibitor complexed structures of the cytochrome bc1 complex from the photosynthetic bacterium Rhodobacter sphaeroides. J. Biol. Chem. 283, 2846–2857. [DOI] [PubMed] [Google Scholar]
- Gray K. A.; Dutton P. L.; Daldal F. (1994) Requirement of histidine-217 for ubiquinone reductase activity (Qi-site) in the cytochrome-bc1 complex. Biochemistry 33, 723–733. [DOI] [PubMed] [Google Scholar]
- Brasseur G.; Sami Saribas A.; Daldal F. (1996) A compilation of mutations located in the cytochrome b subunit of the bacterial and mitochondrial bc1 complex. Biochim. Biophys. Acta 1275, 61–69. [DOI] [PubMed] [Google Scholar]
- Hacker B.; Barquera B.; Crofts A. R.; Gennis R. B. (1993) Characterization of mutations in the cytochrome b subunit of the bc1 complex of Rhodobacter sphaeroides that affect the quinone reductase site (Qc). Biochemistry 32, 4403–4410. [DOI] [PubMed] [Google Scholar]
- Kolling D. R. J.; Samoilova R. I.; Holland J. T.; Berry E. A.; Dikanov S. A.; Crofts A. R. (2003) Exploration of ligands to the Qi-site semiquinone in the bc1 complex using high resolution EPR. J. Biol. Chem. 278, 39747–39754. [DOI] [PubMed] [Google Scholar]
- Dikanov S. A.; Samoilova R. I.; Kolling D. R. J.; Holland J. T.; Crofts A. R. (2004) Hydrogen bonds involved in binding the Qi-site semiquinone in the bc1 complex, identified through deuterium exchange using pulsed EPR. J. Biol. Chem. 279, 15814–15823. [DOI] [PubMed] [Google Scholar]
- Dikanov S. A.; Holland J. T.; Endeward B.; Kolling D. R.; Samoilova R. I.; Prisner T. F.; Crofts A. R. (2007) Hydrogen bonds between nitrogen donors and the semiquinone in the Qi-site of the bc1 complex. J. Biol. Chem. 282, 25831–25841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMillan F.; Lange C.; Bawn M.; Hunte C. (2010) Resolving the EPR spectra in the cytochrome bc1 complex from Saccharomyces cerevisiae. Appl. Magn. Reson. 37, 305–316. [Google Scholar]
- Salerno J. C.; Osgood M.; Liu Y.; Taylor H.; Scholes C. P. (1990) Electron nuclear double resonance (ENDOR) of the Qc-ubiSQ radical in the mitochondrial electron transport chain. Biochemistry 29, 6987–6993. [DOI] [PubMed] [Google Scholar]
- Gao X.; Wen X.; Esser L.; Quinn B.; Yu L.; Yu C.-A.; Xia D. (2003) Structural basis for the quinone reduction in the bc1 complex: A comparative analysis of crystal structures of mitochondrial cytochrome bc1 with bound substrate and inhibitors at the Qi site. Biochemistry 42, 9067–9080. [DOI] [PubMed] [Google Scholar]
- Huang L. S.; Cobessi D.; Tung E. Y.; Berry E. A. (2005) Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: A new crystal structure reveals an altered intramolecular hydrogen-bonding pattern. J. Mol. Biol. 351, 573–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunte C.; Koepke J.; Lange C.; Roßmanith T.; Michel H. (2000) Structure at 2.3 Å resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure 8, 669–684. [DOI] [PubMed] [Google Scholar]
- Lange C.; Nett J. H.; Trumpower B. L.; Hunte C. (2001) Specific roles of protein-phospholipid interactions in the yeast bc1 complex structure. EMBO J. 23, 6591–6600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsdottir H.; Lojero C. G.; Trumpower B. L.; Hunte C. (2003) Structure of the yeast cytochrome bc1 complex with a hydroxyquinone anion Qo site inhibitor bound. J. Biol. Chem. 278, 31303–31311. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Huang L.-S.; Shulmeister V. M.; Chi Y.-I.; Kim K.-K.; Hung L.-W.; Crofts A. R.; Berry E. A.; Kim S.-H. (1998) Electron transfer by domain movement in cytochrome bc1. Nature 392, 677–684. [DOI] [PubMed] [Google Scholar]
- Taguchi A. T.; O’Malley P. J.; Wraight C. A.; Dikanov S. A. (2013) Conformational differences between the methoxy groups of QA and QB site ubisemiquinones in bacterial reaction centers: A key role for methoxy group orientation in modulating ubiquinone redox potential. Biochemistry 52, 4648–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eraso J. M.; Kaplan S. (2002) Redox flow as an instrument of gene regulation. Methods Enzymol. 348, 216–229. [DOI] [PubMed] [Google Scholar]
- Lenz O.; Schwartz J.; Eitinger M.; Friedrich B. (1994) The Alcaligenes eutrophus H16 hoxX gene participates in hydrogenase regulation. J. Bacteriol. 176, 4385–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun C.-H.; Beci R.; Crofts A. R.; Kaplan S.; Gennis R. B. (1990) Cloning and DNA sequencing of the fbc operon encoding the cytochrome bc1 complex from Rb. sphaeroides: Characterization of fbc deletion mutants, and complementation by a site-specific mutational variant. Eur. J. Biochem. 194, 399–411. [DOI] [PubMed] [Google Scholar]
- Donohue T. J.; McEwan A. G.; Van Doren S.; Crofts A. R.; Kaplan S. (1988) Phenotypic and genetic characterization of cytochrome c2 deficient mutants of Rhodobacter sphaeroides. Biochemistry 27, 1918–1925. [DOI] [PubMed] [Google Scholar]
- Bowyer J. R.; Tierney G. V.; Crofts A. R. (1979) Secondary Electron Transfer in Chromatophores of Rhodopseudomonas capsulata A1a pho–: Binary Oscillations. FEBS Lett. 101, 201–206. [DOI] [PubMed] [Google Scholar]
- Crofts A. R.; Guergova-Kuras M.; Hong S. (1998) Chromatophore heterogeneity explains effects previously attributed to supercomplexes. Photosynth. Res. 55, 357–362. [Google Scholar]
- Höfer P.; Grupp A.; Nebenführ H.; Mehring M. (1986) Hyperfine sublevel correlation (HYSCORE) spectrocopy: A 2D ESR investigation of the squaric acid radical. Chem. Phys. Lett. 132, 279–282. [Google Scholar]
- Stoll S.; Britt R. D. (2009) General and efficient simulation of pulse EPR spectra. Phys. Chem. Chem. Phys. 11, 6614–6625. [DOI] [PubMed] [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas Ö., Foresman J. B., Ortiz J. V., Cioslowski J., and Fox D. J. (2009) Gaussian 09, revision A.1, Gaussian, Inc., Wallingford, CT. [Google Scholar]
- Dikanov S. A.; Bowman M. K. (1995) Cross-peak lineshape of two-dimensional ESEEM spectra in disordered S = 1/2, I = 1/2 spin system. J. Magn. Reson., Ser. A 116, 125–128. [Google Scholar]
- Xia D.; Yu C.-A.; Kim H.; Xia J.-Z.; Kachurin A. M.; Zhang L.; Yu L.; Deisenhofer J. (1997) Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science 277, 60–66. [DOI] [PubMed] [Google Scholar]
- Iwata S.; Lee J. W.; Okada K.; Lee J. K.; Iwata M.; Rasmussen B.; Link T. A.; Ramaswamy S.; Jap B. K. (1998) Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 281, 64–71. [DOI] [PubMed] [Google Scholar]
- Berry E. A.; Huang L.-S.; Saechao L. K.; Pon N. G.; Valkova-Valchanova M.; Daldal F. (2004) X-ray structure of Rhodobacter capsulatus cytochrome bc1: Comparison with its mitochondrial and chloroplast counterparts. Photosynth. Res. 81, 251–275. [DOI] [PubMed] [Google Scholar]
- Lin T.-J.; O’Malley P. J. (2008) An ONIOM study of the QA site semiquinone in the Rhodobacter sphaeroides photosynthetic reaction centre. THEOCHEM 870, 31–35. [Google Scholar]
- Martin E.; Samoilova R. I.; Narasimhulu K. V.; Lin T.-J.; O’Malley P. J.; Wraight C. A.; Dikanov S. A. (2011) Hydrogen bonding and spin density distribution in the QB semiquinone of bacterial reaction centers and comparison with the QA site. J. Am. Chem. Soc. 133, 5525–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E.; Baldansuren A.; Lin T.-J.; Samoilova R. I.; Wraight C. A.; Dikanov S. A.; O’Malley P. J. (2012) Hydrogen bonding between the QB site ubisemiquinone and Ser-L223 in the bacterial reaction center: A combined spectroscopic and computational perspective. Biochemistry 51, 9086–9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M. T.; Baldansuren A.; Hart R.; Samoilova R. I.; Narasimhulu K. V.; Yap L.-L.; Choi S. K.; O’Malley P. J.; Gennis R. B.; Dikanov S. A. (2012) Interactions of intermediate semiquinone with surrounding protein residues at the QH site of the wild-type and D75H mutant cytochrome bo3 from Escherichia coli. Biochemistry 51, 3827–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samoilova R. I.; Gritsan N. P.; Hoff A. J.; van Liemt W. B. S.; Lugtenburg J.; Spoyalov A. P.; Tsvetkov Yu. D. (1995) ENDOR and EPR studies of highly isotopically 13C-enriched ubiquinone radicals. Part 2. J. Chem. Soc., Perkin Trans. 2 2063–2068. [Google Scholar]
- de la Rosa F. F.; Palmer G. (1983) Reductive titration of CoQ-depleted Complex III from baker’s yeast: Evidence for an exchange-coupled complex between QH• and low-spin ferricytochrome b. FEBS Lett. 163, 140–143. [DOI] [PubMed] [Google Scholar]
- Meinhardt S. W., and Crofts A. R. (1984) A new effect of antimycin on the b-cytochromes of Rps. sphaeroides. In Advances in Photosynthesis Research (Sybesma C., Ed.) Martinus Nijhoff/Dr. W. Junk Publishers, The Hague, 1984; pp 649–652. [Google Scholar]
- Crofts A. R. (2004) The cytochrome bc1 complex: Function in the context of structure. Annu. Rev. Physiol. 66, 689–733. [DOI] [PubMed] [Google Scholar]
- Crofts A. R.; Hong S.; Wilson C.; Burton R.; Victoria D.; Harrison C.; Schulten K. (2013) The mechanism of ubihydroquinone oxidation at the Qo-site of the cytochrome bc1 complex. Biochim. Biophys. Acta 1827, 1362–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.