

Abstract
We previously identified FOXL2 as a critical component in FSHβ gene transcription. Here, we show that mice deficient in FOXL2 have lower levels of gonadotropin gene expression and fewer LH- and FSH-containing cells, but the same level of other pituitary hormones compared to wild-type littermates, highlighting a role of FOXL2 in the pituitary gonadotrope. Further, we investigate the function of FOXL2 in the gonadotrope cell and determine which domains of the FOXL2 protein are necessary for induction of FSHβ transcription. There is a stronger induction of FSHβ reporter transcription by truncated FOXL2 proteins, but no induction with the mutant lacking the forkhead domain. Specifically, FOXL2 plays a role in activin induction of FSHβ, functioning in concert with activin-induced SMAD proteins. Activin acts through multiple promoter elements to induce FSHβ expression, some of which bind FOXL2. Each of these FOXL2-binding sites is either juxtaposed or overlapping with a SMAD-binding element. We determined that FOXL2 and SMAD4 proteins form a higher order complex on the most proximal FOXL2 site. Surprisingly, two other sites important for activin induction bind neither SMADs nor FOXL2, suggesting additional factors at work. Furthermore, we show that FOXL2 plays a role in synergistic induction of FSHβ by GnRH and activin through interactions with the cJUN component of the AP1 complex that is necessary for GnRH responsiveness. Collectively, our results demonstrate the necessity of FOXL2 for proper FSH production in mice and implicate FOXL2 in integration of transcription factors at the level of the FSHβ promoter.
Follicle-stimulating hormone (FSH), secreted by the pituitary gonadotrope, is necessary for mammalian reproductive fitness. Its concentration is tightly regulated, fluctuating only 2–3-fold during the course of the menstrual or estrous cycle (1, 2). Both excess and deficiency of FSH cause reproductive problems in females. Low FSH levels impede follicular growth, while high levels are associated with premature ovarian failure (3). Since FSH can be secreted constitutively, the major regulatory step controlling its concentration in the circulation is at the transcriptional level. FSH is a heterodimer of a common α-glycoprotein subunit (αGSU) and a unique β-subunit. In addition to providing biological specificity, β-subunit gene expression is the limiting factor in FSH synthesis. FSH β-subunit gene transcription is induced primarily by GnRH and activin (4–6). GnRH and activin, in addition to having independent transcriptional effects on the FSHβ promoter, interact and cause higher than additive, synergistic induction, which is specific for FSHβ, and is postulated to contribute to differential regulation of the gonadotropin subunits (7).
Activin plays an important role in the regulation of FSH concentration, increasing the release of FSH from the pituitary (8) and inducing FSHβ gene expression in gonadotrope cells (6). Following binding and activation of its receptor, activin phosphorylates SMAD2 and SMAD3. In particular, phosphorylated SMAD3 binds to SMAD4 and translocates into the nucleus to activate transcription of mouse FSHβ, by binding the consensus site located at −267 in the mouse FSHβ promoter (9–11). However, this site does not account for complete activin responsiveness of the rodent promoter (7, 12), raising the question of what additional binding elements contribute to induction of FSHβ by activin.
Further analysis of the regulation of the FSHβ promoter demonstrated that FOXL2 is essential for activin responsiveness (13–15). FOXL2 is a member of the forkhead family of transcription factors, which share a conserved DNA-binding domain (16). FOXL2 is expressed in gonadotrope cells (17, 18) and mediates activin induction of the FSHβ gene, through several forkhead sites in the proximal mouse FSHβ promoter (14, 15). Additionally, FOXL2 protein has been shown to cooperate with either SMAD3 or SMAD4 to induce this gene (14, 15, 19, 20). However, it has not been determined which of the additional activin-responsive sites in the promoter, besides the forkhead sites, are also FOXL2-dependent sites, and which are SMADs sites or other, as yet unidentified, activin-inducible elements. In addition to FSHβ, FOXL2 augments transcription of both follistatin and GnRH receptor in the gonadotrope (17, 18, 21). Of interest to our investigation, FOXL2-mediated induction of these genes is dependent on SMAD sites adjacent to the forkhead elements, with FOXL2 functioning in complex with SMAD3. Yet, it is not known if interaction with SMAD3 activates FOXL2 or whether activin can directly activate FOXL2 via non-SMAD signaling pathways.
Besides the pituitary, FOXL2 has an important reproductive role in the ovary as well, particularly in granulosa cell proliferation and differentiation. FOXL2 functions as a transcriptional repressor of several key genes involved in steroidogenesis (22, 23). Evidence that FOXL2 negatively regulates SMAD3-dependent, GDF9, and activin-stimulated follistatin transcription in the ovary, through both forkhead and SMAD elements, provides support for the potential interaction between these elements (24). In the ovary, function of the FOXL2 protein can be regulated via non-SMAD signaling pathways, as demonstrated by LATS1 kinase phosphorylation of FOXL2 (25), providing a potential mechanism for direct FOXL2 protein activation. Understanding whether interaction with SMAD3 is necessary to activate FOXL2 in the gonadotrope cell or if FOXL2 itself can be activated by activin, such as by LATS1, is of great interest considering the pathophysiological effects that occur in the presence of disrupted FOXL2 function.
Various FOXL2 mutations have been identified in patients with Blepharophimosis Ptosis Epicanthus Inversus Syndrome (BPES). BPES is an autosomal dominant disorder that is characterized by distinctive eyelid abnormalities that result from mutations in the FOXL2 gene. Two clinical subtypes have been described and most BPES occurrences are classified Type I, which is associated with premature ovarian failure (26). In particular, patients with mutations in the FOXL2 gene that lead to a truncation of the FOXL2 protein are at high risk of developing premature ovarian failure (27). Considering that premature ovarian failure is not only associated with mutations of the FOXL2 gene in BPES patients, but that individuals with this reproductive defect exhibit higher FSH levels in the circulation (3), it is intriguing to postulate that mutations of FOXL2 that mimic those found in the BPES patients may underlie the dysregulation of FSHβ transcription, as a pituitary contribution to the development of premature ovarian failure.
Studies of FOXL2-null mice confirmed a plethora of physiologic problems in vivo (28, 29). In addition to craniofacial defects, FOXL2-null mice exhibit disruption of granulosa cell differentiation and fail to form secondary follicles within the ovary. Following these initial studies, two other reports analyzed the in vivo role of FOXL2 in the pituitary gland. A complete FOXL2-null mouse (30) was shown to have lower FSHβ and αGSU levels, while LHβ expression was not affected. Furthermore, both basal FSH and activin-stimulated FSH secretion were reduced, while neither basal LH, nor GnRH-stimulated LH secretion, were altered. In support of a pituitary role in the reproductive phenotype of the FOXL2-null mouse, mice with gonadotrope-specific deletion of FOXL2 using GnRH-receptor-mediated CRE expression, exhibited subfertility and lower FSH levels (31). These initial studies lay the foundation for investigating the molecular mechanism whereby FOXL2 contributes to maintenance of gonadotropin gene expression.
Given the role of FOXL2 in activin induction of FSHβ, the question remains whether activin can induce FOXL2 protein itself or whether FOXL2 is activated by protein-protein interaction with SMAD proteins at the level of the promoter. Several putative FOXL2 sites are found in the proximal FSHβ promoter, most with adjacent or overlapping SMAD elements. In this manuscript, we determine which of these putative forkhead elements are functional FOXL2 sites, and which are not FOXL2 sites, although they are activin-responsive sites. Furthermore, we demonstrate that SMAD4 proteins and FOXL2 form a higher-order complex on the most proximal forkhead element, which has the largest effect on activin-responsiveness of the FSHβ promoter. Interestingly, we show that mutations in FOXL2 that lead to truncated FOXL2 proteins analogous to those found in BPES, elicit higher induction of the FSHβ promoter. Additionally, we determine that FOXL2 plays a role in synergistic induction of FSHβ reporter activity by GnRH and activin, integrating their pathways at the level of the FSHβ promoter.
Materials and Methods
FOXL2-null mice and timed matings
The FOXL2-null mice were a generous gift from Louise Bilezikjian at the Salk Institute and have been previously described (30). Animals were maintained under a 12-h light, 12-h dark cycle and received food and water ad libitum. All experiments were performed with approval from the University of California San Diego Animal Care and Use Committee and in accordance with the National Institutes of Health Animal Care and Use Guidelines. Briefly, genomic DNA was extracted from toe biopsies and analyzed for the presence of the insertion of a Neomycin gene by PCR amplification using primers specific to FOXL2 and Neo: Neo-forward 5′-CTTGGGTGGAGAGGCTATTC-3′ and Neo-reverse 5′-AGGTGAGATGACAGGAGATC −3′, FOXL2wt-forward 5′-CACGGGAAAGCAGAGGCCGC −3′ and FOXL2wt-reverse 5′-GGATCTCTGAGTGCCAACGC −3′. Heterozygous males and females were used for timed mating experiments. Males and females were paired in the afternoon, and the following morning, females were checked for vaginal plugs with that time designated as e0.5. At the desired stage of embryonic development, females were sacrificed and the embryos were harvested. Biopsies of embryos were taken before embryo fixing and the genotype was determined.
Immunohistochemistry
Embryos were fixed (10% acetic acid, 30% formaldehyde, and 60% ethanol) overnight at 4°C and dehydrated in ethanol/water washes before embedding in paraffin. Fixed embryos were embedded in paraffin by the University of California, San Diego, Histology Core. Embedded embryo heads were cut into 14-μm sagittal sections with a microtome and floated onto SuperFrost Plus slides (Fisher Scientific) and dried overnight at room temperature. Slides were incubated at 60°C for 30 min, deparaffinized in xylene washes, and rehydrated in ethanol/water washes. Antigen unmasking was performed by heating for 10 min in a Tris-EDTA-Tween20 mixture. After cooling and washing two times in water, endogenous peroxidase was quenched by incubating for 10 min in 0.3% hydrogen peroxide. After washing in phosphate-buffered saline (PBS), slides were blocked in PBS with 5% goat serum and 0.3% Triton X-100 for 45 min, then incubated with primary antibodies against LH, FSH, αGSU, TSH, ACTH, or GH (1:1000, obtained from National Hormone and Peptide Program, NIDDK) in PBS with 5% serum overnight at 4°C. After washing three times in PBS, slides were incubated with biotinylated goat antichicken or goat antirabbit IgG (Vector Laboratories) diluted 1:300 in PBS, for 30 min, then washed 3 times in PBS. The Vectastain ABC elite kit (Vector Laboratories) was used per manufacturer's instructions and incubated for 30 min. After washing, the VIP peroxidase kit was used for colorimetric staining for 3 min. Slides were dehydrated in an ethyl alcohol series and xylene, then coverslips were mounted with Vectamount (Vector Laboratories). Sixty-four sections were obtained per pituitary and stained alternatingly for two hormones. To quantify the number of gonadotropin subunit-staining cells, five sections per animal were counted from the middle of the pituitary, approximately 50 μm apart. Counting was performed blinded to the genotype, using ImageJ software from NIH, after which the count and cell delineation were confirmed visually. Statistical differences between genotypes were determined using a t-test and JMP software (SAS Institute).
qPCR analysis of embryo pituitary at e18.5
Adult FOXL2 heterozygous mice were set up in timed matings. On e18.5, the pregnant females were euthanized and the embryos were extracted. Pituitaries were dissected and placed individually in tubes on dry ice. The embryos were genotyped from tail biopsies using primers for the Neomycin selective marker and FOXL2 wild-type gene. The pituitaries were pooled into wild-type and FOXL2-null groups. Each group contained five individual embryonic pituitaries. RNA was isolated from the pituitaries using a QIAshredder and RNeasy mini kit (QIAGEN Sciences), as directed by the manufacturers. Total RNA was reverse transcribed using an iScript cDNA Synthesis kit (Bio-Rad Laboratories). qPCR was performed using an iQ SYBR Green supermix and an IQ5 real-time PCR machine (Bio-Rad Laboratories), with the following primers: LHβ forward: CTGTCAACGCAACTCTGG, LHβ reverse: ACAGGAGGCAAAGCAGC; GAPDH forward: TGCACCACCAACTGCTTAG, GAPDH reverse: GGATGCAGGGATGATGTTC; FSHβ forward: GCCGTTTCTGCATAAGC, FSHβ reverse: CAATCTTACGGTCTCGTATACC; αGSU forward: ATTCTGGTCATGCTGTCCATGT, αGSU reverse: CAGCCCATACACTGGTAGATGG; GH forward: CCTCAGCAGGATTTTCACCA, GH reverse: CTTGAGGATCTGCCCAACAC; under the following conditions: 95°C for 15 min, followed by 40 cycles at 95°C for 20 s, 56°C for 30 s, and 72°C for 30 s. Three samples with five pooled animals each was assayed. A standard curve with dilutions of 10 pg/well, 1 pg/well, 100 fg/well, and 10 fg/well of a plasmid containing LHβ, FSHβ, or GAPDH cDNA, was generated in each run with the samples. The amount of LHβ was calculated by comparing threshold cycle obtained for each sample with the standard curve generated in the same run. Replicates were averaged and divided by the mean value of GAPDH in the same sample. After each run, a melting curve analysis was performed to confirm that a single amplicon was generated in each reaction.
Cell culture and transient transfections
The expression vectors for SMAD3, SMAD4, and FOXL2 were kindly provided by Dr J. Massague (Memorial Sloan-Kettering Cancer Center), Dr D. Bernard (McGill University), and Dr L. Bilezikjian (Salk Institute), respectively. The human FOXL2 expression vector and its mutations were a gift from Dr. J. Bae (Pochon CHA University), while LATS1 kinase and its mutant were kindly provided by Dr M. Pisarska (UCLA). The mouse FSHβ-luciferase reporter vectors were published previously (7, 32). Mutagenesis was performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's protocol, using the mouse 1 kb FSHβ promoter in the pGL3 luciferase vector as a template. Mutated residues are indicated in Figure 6A below. Mutations were confirmed via dideoxyribonucleotide sequencing.
Figure 6.

The role of FOXL2 binding sites in FSHβ induction. A, To delineate critical elements within the individual FOXL2 sites, several separate mutations were created in the 1 kb mouse FSHβ-luciferase reporter. Forkhead elements are indicated with a solid underline while adjacent SMAD sites are delineated with the dashed underline on the wild-type sequence. Mutations are listed below and residues that differ from wild-type sequence illustrated with lower case letters. db, indicates double mutations; sm, mutation in a SMAD element; fh, mutation in a forkhead element; and, both, mutation in the overlapping sequence that affects both the SMAD and FOXL2 binding sites. B, Basal expression of the mutants was compared to the basal expression of the wild-type reporter; * indicates statistically significant difference as determined by ANOVA and Tukey's post hoc test. C, LβT2 cells were transfected with various reporters were treated with vehicle or activin for 5 h, as described in Figure 4. Fold induction by activin over vehicle-treated control for each reporter is presented and * indicates significantly lower induction of the mutant compared to the wild type. D, Mouse FOXL2 or vector control were overexpressed with each reporter. * indicates decrease in fold induction by FOXL2 of the mutant reporter compared to the wild-type. E, F, G, SMAD3, SMAD4, and a combination of SMAD4 and FOXL2, respectively, or their vector control, were overexpressed in the same manner.
LβT2 cells were cultured at 37°C in DMEM (Cellgro, Mediatech, Inc.) containing 10% fetal bovine serum (Omega Scientific Inc.) and penicillin. Cells were split into 12-well plates one day prior to transfection and transfected using the Fugene 6 reagent (Roche Molecular Biochemicals) in accordance with the manufacturer's protocol. Wells were transfected with 500 ng of FSHβ-luciferase reporter plasmids, 100 ng of the β-galactosidase reporter plasmid driven by the Herpes virus thymidine kinase promoter to serve as an internal control for transfection efficiency, and 200 ng of expression vectors, as indicated in the figures and legends. Cells were incubated in serum-free DMEM containing 0.1% BSA and antibiotics overnight prior to hormone treatment with 10 ng/mL activin (Calbiochem), or 10 nM GnRH (Sigma). Subsequent to treatment, cells were washed with 1X phosphate buffer saline (PBS) and lysed with 60 μL of 0.1 M potassium-phosphate buffer pH 7.8 with 0.2% Triton X-100. A 96-well luminometer plate was loaded with 20 μL of each of the lysates and luciferase activity was measured after injection of a buffer containing 100 mM Tris-HCl with pH 7.8, 15 mM MgSO4, 10 mM ATP, and 65 μM luciferin per well, using a Veritas Microplate Luminometer (Turner Biosystems). The Galacto-Light Assay (Tropix) was performed according to the manufacturer's protocol to measure galactosidase activity. All experiments were performed a minimum of three times and in triplicates within each experiment. Luciferase values were normalized to β-galactosidase for each sample, relative to the empty vector pGL3 luciferase reporter activity. Statistical significance was determined with analysis of variance (ANOVA) and significance was set at P ≤ .05 represented by an asterisk.
EMSA
In vitro transcribed and translated FOXL2 and SMAD proteins were synthesized using the TnT kit from Promega (Coupled Reticulocyte Lysate System, Promega Corporation) according to the manufacturer's instructions. Two hours following activin treatment of LβT2 cells, nuclear extracts were obtained by swelling the cells with hypotonic buffer [20 mM Tris pH 7.4, 10 mM NaCl, 1 mM MgCl2, 1 mM PMSF, protease inhibitor cocktail (Sigma-Aldrich), 10 mM NaF, 0.5 mM EDTA, 0.1 mM EGTA]. Cells were broken by passing through a 255/8 G needle, three times. Samples were centrifuged at 4000 rpm for 4 min and the nuclear pellets were resuspended in hypertonic buffer [20 mM Hepes pH 7.8, 20% glycerol, 420 mM KCl, 1.5 mM MgCl2, 1 mM PMSF, protease inhibitor cocktail (Sigma-Aldrich), 10 mM NaF, 0.5 mM EDTA, and 0.1 mM EGTA]. Protein determination was performed using the Bradford reagent (Bio-Rad). The following oligonucleotides were used as 30-bp probes, encompassing the sites of interest: FBE1: AATTAAGACATATTTTGGTTTACCTTCGCA, 202: CATATCAGATTCGGTTTGTACAGAAACCAT, FBE2: CTCTGTGGCATTTAGACTGCTTTGGCGAGG, and FBE3: CTCCCTGTCCGTCTAAACAATGATTCCCTT. Oligonucleotides were annealed and labeled with γ32P ATP using T4 Polynucleotide Kinase (New England Biolabs, Inc). Binding reactions contained 2 μg of nuclear proteins in a total volume of 20 μL containing the following: 10 mM Hepes pH 7.8, 50 mM KCl, 0.5 mM MgCl2, 10% glycerol, 0.1% NP-40, 0.25 μg dIdC, 5 mM DTT, and 5 fmol of labeled probe. Reactions were loaded onto a 5% nondenaturing polyacrylamide gel and ran in 0.25X Tris-borate-EDTA buffer. Gels were run at 250 V/cm2 constant voltage and dried. Autoradiography was performed to identify complexes.
Western blot
Cells were rinsed with 1× PBS and lysed with a buffer containing: 20 mM Tris pH 7.4, 140 mM NaCl, protease inhibitors (Sigma), 1 mM PMSF, 10 mM NaF, 1% NP-40, 0.5 mM EDTA, and 1 mM EGTA to obtain whole cell lysates. Nuclear extracts were obtained as described above. Bradford reagent was used to determine protein concentrations, calculated using a standard curve. Equal amounts of protein were loaded on the gel, resolved by gel electrophoresis and transferred to a polyvinylidene fluoride (PVDF) membrane. The membranes were blocked with 10% milk in wash buffer (20 mM Tris 7.4, 0.1% tween, 150 mM NaCl, and 0.5% BSA) and then probed with antibodies to cMYC. Proteins were detected with an Enhanced Chemiluminescence (ECL) Western Blotting Detection Reagent (GE Healthcare). To assure equal loading, membranes were stripped at 60°C for 1 h with a strip buffer (50 mM Tris pH 6.8, 5% SDS, and 100 mM β-mercaptoethanol and re-exposed to ECL and autoradiography to ensure complete removal of the antibody and then blocked again with milk and reprobed for lamin B or β-tubulin).
GST interaction assay
The Glutathione S-Transferase (GST)-FOXL2 in the pGEX vector was kindly provided by Dr L. Bilezikjian. The SMAD3 expression vector was obtained from J. Massague, cJUN from M. Birrer, and cFOS from Dr Tulchinsky. 35S-labeled proteins were produced using the TnT T7 Coupled Reticulocyte Lysate System (Promega Corporation). Bacteria transformed with the pGEX vectors were grown to an OD of 0.6, upon which protein expression was induced by addition of 0.25 mM isopropyl-β-D-thiogalactosidase (IPTG). Bacterial pellets were sonicated in PBS with 5 mM EDTA and 0.1% Triton X-100, centrifuged and the supernatant was bound to glutathione sepharose beads (Amersham Pharmacia). Beads were washed four times with a sonication buffer followed by equilibration in the binding buffer (below), and split equally between different samples and the control. 35S-labeled proteins were added to the beads and bound for 1 h at 4°C in 20 mM Hepes (pH 7.8), with 50 mM NaCl, 10 mg/ml BSA, 0.1% NP-40, and 5 mM DTT. After extensive washing, samples were eluted from the beads by boiling in an Leammli sample buffer and subjected to SDS-PAGE. Afterwards, the gels were dried and autoradiographed.
Results
FOXL2 is required for Fshb and Cga gene expression in vivo
Using the LβT2 gonadotrope cell model, we and others have identified FOXL2 as a key player in activin induction of FSHβ transcription (13, 15). These findings were followed by confirming the role of FOXL2 in vivo, using genetically modified mice (30, 31). However, in the first study using whole-body, germline FOXL2 ablation, the absence of FOXL2 within the ovary may compromise the effects observed in the pituitary of complete null, adult female mice, due to the lack of steroid feedback. In the second model, incomplete recombination in the CRE-loxP animals may have resulted in a knock down rather than complete deletion in the gonadotrope, since the animals maintained some fertility. Based on these concerns, we analyzed gonadotropin gene expression during the late embryonic stage, ie, prior to the influence of steroid feedback. In our initial assessment, we performed immunohistochemistry to detect gonadotropin hormones in the pituitary of wild-type and FOXL2-null mice at embryonic day 18.5 (e18.5), the earliest developmental stage that FSH hormone can be detected in the fetal pituitary. LH is present in the wild-type pituitaries at the ventral surface (Figure 1A), as shown previously (33). Pituitary glands from the FOXL2-null mice also exhibited LH staining, but at a lower level than wild-type littermates. Likewise, FSH-expressing cells were present in wild-type animals, yet, they were undetectable in the null animals. Meanwhile, αGSU-containing cells were present in both wild-type and null mice. Since FOXL2 is specifically expressed in gonadotrope and thyrotrope lineages within the pituitary, we examined TSH staining and found that TSHβ-containing cells are present at the same level in both genotypes, consistent with a previous report (30). We also examined specification of other pituitary cells that do not express FOXL2, except for prolactin, which cannot be detected at this stage of development. Growth hormone (GH) and adrenocorticotropin hormone (ACTH) cells were present equally in both wild-type and FOXL2-null animals. We counted the number of cells per section to quantify differences in gonadotropin hormone containing cells observed with staining. The number of either LH- or FSH-containing cells were significantly reduced in the null embryos, while αGSU cell numbers were similar in FOXL2-deficient mice and wild-type littermates (Figure 1B), demonstrating a critical role of FOXL2 in gonadotropin synthesis.
Figure 1.

FOXL2 is necessary for FSH gene expression in vivo. A, Immunohistochemistry of sectioned pituitary glands collected from wild-type (WT) and FOXL2-null mice (KO) at embryonic 18.5 (e18.5) for luteinizing hormone (LH), follicle-stimulating hormone (FSH), common α-subunit αGSU, growth hormone (GH), thyroid-stimulating hormone (TSH), and adrenocorticotropin (ACTH) with antibodies obtained from the National Hormone and Peptide program, reveals reduced levels of gonadotropin hormones in FOXL2-null mice. B, To determine cell numbers, five sections per animal and three animals per group were counted and the difference in cell numbers between genotypes was analyzed by the t-test.
To determine whether the decrease in gonadotropins is due to lower expression of their genes, we performed quantitative PCR (qPCR) analysis of the e18.5 pituitaries (Figure 2). Lhb mRNA in the FOXL2-null pituitary exhibited 74% lower expression compared to wild-type littermates. Expression of Fshb mRNA was 98% lower in the FOXL2-null pituitary, and the expression of Cga (αGSU) subunit mRNA was reduced by 55%. The difference in Gnrhr mRNA did not reach statistical significance, although null animals exhibited 43% lower expression. As expected, Gh mRNA in the pituitary was equivalent in wild-type and null animals. Foxl2 mRNA was below the detection level in the null animals, confirming deletion in the pituitary gland of FOXL2 knock-out animals. Collectively, these findings identify FOXL2 as an important factor in mature gonadotropin hormone production and focus our attention on the molecular mechanism whereby FOXL2 mediates gonadotropin gene expression.
Figure 2.
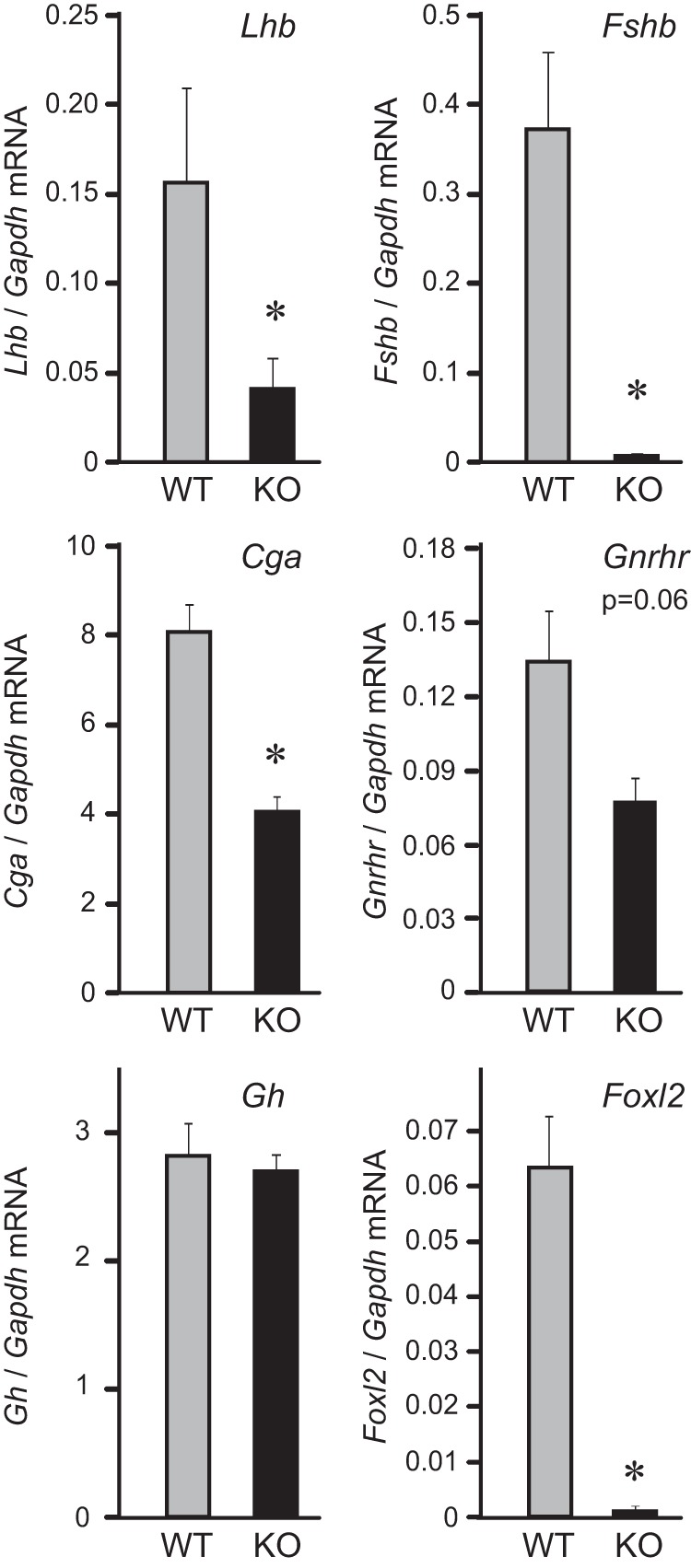
FOXL2-null mice have lower expression of gonadotrope-specific mRNAs. Quantitative PCR of e18.5 pituitaries shows that gonadotropin gene expression is significantly reduced in FOXL2-null mice. Total RNA was purified from three samples per WT or KO group, each with five pooled pituitaries, reverse transcribed, and the level of hormone expression assayed by real-time PCR. In each sample, the amount of hormone mRNA, calculated from the standard curve, was compared to the amount of Gapdh, and presented as a ratio. * indicate significant difference in the expression in the FOXL2-null animals from the wild-type animals.
Higher induction of FSHβ transcription by truncated FOXL2
FOXL2 protein contains a forkhead domain (FH, Figure 3A), which is critical for DNA binding, and an alanine-rich domain (A, Figure 3A), common to homeodomain proteins. We obtained expression vectors that contain deletions of the critical FH or A domains and vectors containing several truncations of the FOXL2 protein that mimic mutations found in the BPES patients (34), and co-transfected each of them along with a 1kb mouse FSHβ-luciferase reporter into LβT2 cells, to investigate the importance of these protein domains for activation of FSHβ gene expression (Figure 3B). Overexpression of wild-type FOXL2 (WT) induced FSHβ-luciferase. In contrast, a FOXL2 expression vector with deletion of the forkhead domain (dFH) failed to significantly induce reporter activity. Surprisingly, deletion of the alanine-rich domain (dAla) or truncations that mimic mutations in BPES patients (t274 and t218) caused higher induction of the FSHβ reporter. Since mutations can alter the expression and the half-life of the protein and therefore its function, we analyzed the quantity of FOXL2 mutant proteins produced by overexpression in LβT2 cells using western blotting to detect the cMYC-tag on each of the mutant proteins (Figure 3C). β-tubulin (β-tub, lower image Figure 3C) served as a loading control. Surprisingly, we determined that mutations which resulted in higher FSHβ-luciferase expression were actually expressed at a lower level than the wild-type FOXL2 protein. Given that these mutations may alter nuclear localization of FOXL2, we also assayed nuclear levels of overexpressed proteins (Figure 3D) and nucleus-restricted lamin B served as a loading control in the bottom image. All overexpressed FOXL2 proteins were found at a similar level in the nucleus. These findings suggest that mutations in the FOXL2 gene, reported in some BPES patients, may cause a pituitary phenotype (in addition to the ovarian effects), which results in higher expression of the FSHβ gene.
Figure 3.
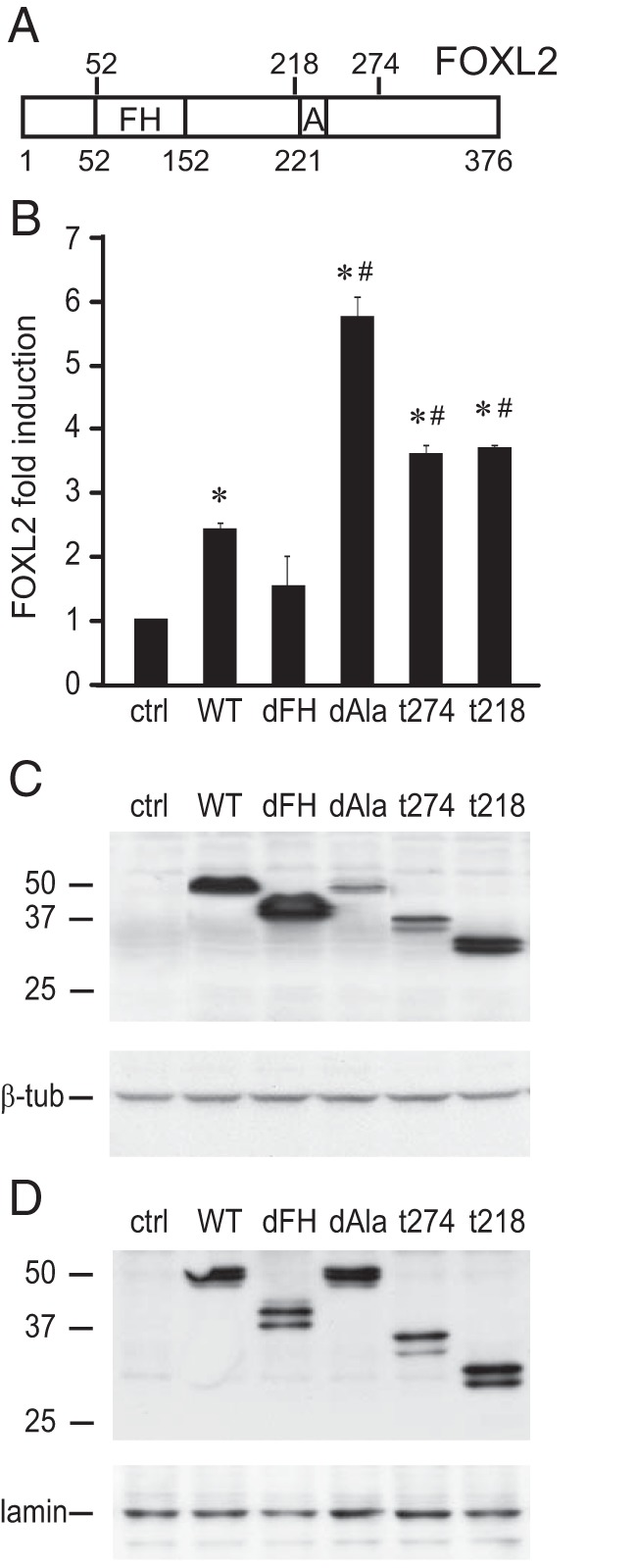
Higher induction of FSHβ by C-terminal truncations of the FOXL2 protein. Human FOXL2 was overexpressed with the mouse FSHβ-luciferase reporter in LβT2 cells. A, Schematic presentation of the FOXL2 protein and its functional domains, forkhead domain (FH) and alanine-rich domain (A). The numbers above the bar represent residues after which truncations used in B were made, while numbers below the bar show domain positions. B, Induction of the reporter by each FOXL2 mutation was compared to the induction by the wild-type FOXL2: deletions of the two domains found in the FOXL2 protein, forkhead domain (dFH), and alanine-rich domain (dAla); and truncations, t274 and t218, which mimic mutations that occur in BPES patients due to insertion of premature stop codons. * indicate significant induction of the reporter by FOXL2, as determined by one-way ANOVA followed by Tukey's post hoc test, while # indicates a significant increase in induction compared to the wild-type FOXL2. C, Whole cell lysates were obtained from LβT2 cells transfected with the same MYC-tagged FOXL2 expression vectors as in B, and western blots performed with anti-MYC-tag to analyze the amount of FOXL2 protein. β-tubulin (β-tub) serves as a loading control. D, Western blot of the nuclear lysate was performed to determine nuclear localization of various mutations. Nuclear lamin B1 serves as a loading control presented at the bottom panel.
FSHβ promoter elements, FBE1 at −350 and FBE3 at −120, are FOXL2 sites
Using the LβT2 cell model, we previously determined that the distal FOXL2 site plays a role in activin induction of FSHβ, but not in the basal expression of this gene (15). However, FOXL2 binding to this site in the FSHβ promoter is not different between nuclear extracts from LβT2 cells with or without activin treatment (15) and it is not clear how activin activates the FOXL2 protein to induce FSHβ transcription. We could not detect a change in FOXL2 migration following activin treatment using high resolution western blots, which would have indicated the possibility of post-translational modification (data not shown). In the ovary, FOXL2 plays a role in granulosa cell gene expression and is activated by phosphorylation by LATS1 kinase (25). In the gonadotrope, however, overexpression of LATS1 or its dominant-negative mutant did not affect activin induction of the FSHβ reporter (data not shown). As an alternative approach, we performed a detailed analysis of FOXL2-binding sites present in the mouse FSHβ promoter, since FOXL2 may be activated by protein-protein interaction at the DNA level.
There are four putative FOXL2 binding sites in the mouse FSHβ promoter (Figure 4A), three that we identified in Corpuz et al [ -350, −206 and −155; (15)], and one identified by Lamba et al [ -114, (13)]. In a later publication, Tran et al (20), named the −350 site FBE1, the −155 site FBE2, and the −114 site FBE3, and for ease of comparison, we will employ the same nomenclature, and retain the numbering for the −206 site only, which that study (20) did not analyze. We compared all of these sites in parallel, using mutations of the residues underlined in Figure 4A to determine the contribution of each site in activin and FOXL2 induction of the FSHβ promoter. Each individual mutation significantly lowered induction by activin, suggesting that each of these sites plays a role in activin induction of FSHβ-luciferase (Figure 4B). To determine which of these function as FOXL2 responsive sites, we overexpressed FOXL2, using an expression vector, together with a reporter containing the wild-type FSHβ promoter or individual FSHβ reporters containing these selective mutations in the putative FOXL2 sites. Mutation of either FBE1 (-350 site) or FBE3 (-114 site) lowered the induction by FOXL2 compared to the induction of the wild-type reporter (Figure 4C), implicating these sites as critical for specific regulation by FOXL2. The other two sites, FBE2 and −206, may involve different activin-regulated transcription factors, as mutation in these sites did not alter the ability of FOXL2 to induce FSHβ-luciferase expression.
Figure 4.
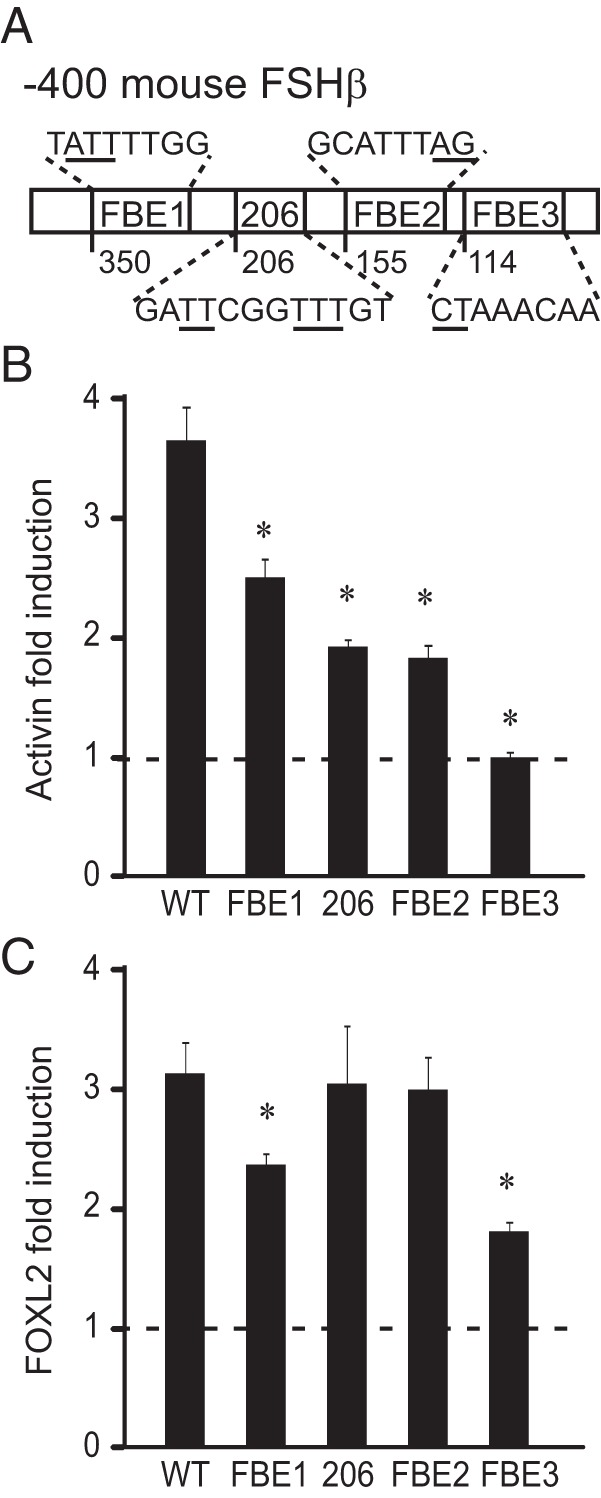
FBE1 and FBE3 are FOXL2 responsive sites. A, Schematic presentation of the putative FOXL2 sites, whose 5′ location is indicated with the number below the bar. The sequence for each site is listed and mutated residues indicated with an underline. Mouse FSHβ promoter mutations in the putative FOXL2 sites (illustrated in A), were tested for their responsiveness to activin treatment (B) and FOXL2 overexpression (C) and compared to the wild-type reporter. B, Following transfection of the wild-type (WT) 1 kb FSHβ reporter or a reporter containing mutation listed below the corresponding bars, LβT2 cells were treated with 10 ng/ml activin or vehicle control for 5 h. The results are presented as fold induction, ie, for each reporter. Activin treatment was normalized to vehicle control to account for changes in basal expression. * indicates a significant decrease in fold induction in the mutant reporter compared to the wild-type. C, Mouse FOXL2 was overexpressed with wild-type or mutant FSHβ reporters. * indicates a statistically significant decrease in fold induction when induction by FOXL2 was normalized to empty vector control for each reporter.
We compared the binding of FOXL2 to its putative sites using EMSA. Using control and activin-treated nuclear extracts from LβT2 cells, gel shift assays showed that FOXL2 binds to FBE1 and FBE3, but not to FBE2 and −206 (Figure 5A; C, control; A, activin; αL2, FOXL2 antibody; Ig, IgG control antibody; and data not shown). We previously determined that FBE3 is proximal to the Pbx/Prep complex binding site (35), and that these proteins can be seen binding to FBE3 in LβT2 nuclear extracts (Figure 5A). Therefore, to eliminate the influence of potential protein-protein interaction through which FOXL2 may be recruited to the promoter, and to analyze binding of only FOXL2 protein to DNA, in vitro translated and transcribed FOXL2 protein was also utilized and showed similar results (Figure 5B, V, vector control; F, FOXL2 expression vector). Thus, FOXL2 binds FBE1 and FBE3 and mutation of these sites decreases induction by FOXL2, while the FBE2 and −206 site mutations do not affect induction by FOXL2, suggesting that these sites are not functional FOXL2 sites, but may bind different activin-regulated proteins.
Figure 5.
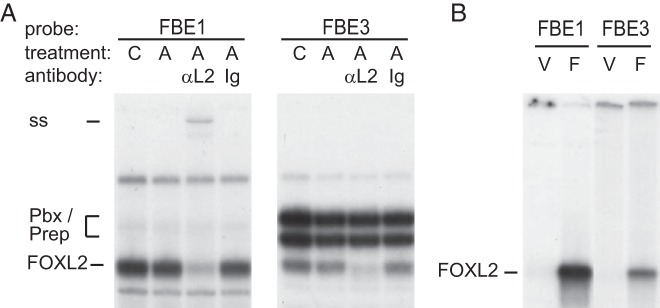
FBE1 and FBE3 bind FOXL2. EMSA was used to analyze the binding of FOXL2 to elements FBE1 and FBE3, which contribute to the induction by FOXL2. A, EMSA with nuclear extracts using 30 bp probes that encompass FBE1 and FBE3. “C” above the lanes designates extracts from control, vehicle-treated LβT2 cells, while “A” designates 2-h activin treatment. Antibodies to FOXL2 “αL2” were included in the binding reaction to identify FOXL2 containing complex (supershift, ss), and specificity determined by comparison to nonspecific IgG antibody (Ig). B, EMSA with in vitro transcribed and translated vector control (V) and FOXL2 (F) with 30 bp probes that encompass FBE1 or FBE3.
FBE3 requires both the SMAD and FOXL2 elements
Due to the possibility that FOXL2 binding may be affected by protein-protein interaction while bound to DNA, we performed a detailed analysis of the FOXL2 binding sites present in the mouse FSHβ promoter. The FBE2 and FBE3 sites were originally identified as activin-responsive elements due to encompassing a SMAD element, AGAC or GTCT (dashed underline in the wild-type sequence for each site; Figure 6A) (35). Contained within each of the FBE2 and FBE3 sites is a SMAD element, which overlaps the binding site for FOXL2, a forkhead element: CTAAACAC (solid underline). Unintentionally hindering our ability to distinguish between actions through either site, our original publication describing the FBE2 and FBE3 sites (35) established their role in activin induction using specific 2-base-pair mutations of the overlapping two residues shared by the SMAD and forkhead sites (“FBE2 both” and “FBE3 both,” mutations in both the SMAD and forkhead elements; Figure 6A). FBE1, the −350 FOXL2 site (solid line), was identified with truncation/mutation analysis (15), and it also is juxtaposed to a SMAD site (dashed underline). Since the mechanisms of activation of FOXL2 by activin signaling remain unclear, yet SMAD activation is considered a prototypical phosphorylation target of the activin receptor serine/threonine kinase, we asked whether elements for either of these proteins contribute to activin induction of the FSHβ promoter. For the sake of completeness, we also analyzed the −206 site, since the putative −206 site was identified using in silico analysis. Furthermore, the original report (15) assessed whether the double mutation (db), mutating both T repeats, affected activin induction of FSHβ [Figure 4B, and (15)]. Here, we created two separate mutations in the −206 element: 206 fh that encompasses mutations of the T repeat in the forkhead element, and 206 x that mutates the two T residues 5′ of the forkhead element. For the three FBE sites, we created mutations specifically in the SMAD sites (sm) or forkhead elements (fh), in addition to double (db) mutations for FBE1, and single mutations in the overlapping FOXL2 and SMAD elements that affect both elements (both), for FBE2 and FBE3 (Figure 6A).
Basal expression was lower with all three mutations in the most proximal FBE3 site, while other mutations did not significantly affect basal expression of the reporter (Figure 6B). To analyze the effect of activin, FOXL2 or SMAD proteins, we present data as fold induction (Figure 6, C-G), in which each reporter treated with activin or with overexpressed proteins, is normalized to its own vehicle-treated or empty expression vector control, which takes into account these changes in basal expression. Induction by activin was reduced by 41% with mutation of the −350 forkhead element, FBE1 fh, but not with mutation of the adjacent SMAD site, FBE1 sm (Figure 6C). The double mutation of the SMAD and forkhead sites, FBE1 db, had the same effect as mutation of the forkhead element alone, indicating that the adjacent SMAD site is not functional. Surprisingly, mutation of the −206 forkhead element, 206 fh, did not reduce FSHβ induction, but the mutation 5′ to it, 206 x, reduced activin induction by 73%, to the same level as double mutation, 206 db (Figure 6C). This result, combined with the gel shift assays, strongly indicates that −206 is not a FOXL2 site. As reported previously, mutation of the FBE2 element that affected both the SMAD and FOXL2 sites (FBE2 both) diminished activin induction by 74%. Unexpectedly, mutation in either the SMAD (FBE2 sm) or FOXL2 (FBE2 fh) elements alone did not significantly lower activin induction, implying that FBE2 is neither a SMAD, nor FOXL2 site, and activin responsiveness is conferred by a presently unidentified protein that requires the residues mutated in the “both” mutant. Any mutation of the FBE3 site that affected either the SMAD (FBE3 sb) or FOXL2 elements (FBE3 fh) or the two elements together (FBE3 both) eliminated induction by activin. Collectively, our findings are consistent with previous data (Figure 4B) (13, 15), indicating that the FBE1 and FBE3 sites are critical for activin responsiveness of the FSHβ promoter. Moreover, these results show that −206 and FBE2 are not FOXL2 sites, although they are critical for activin responsiveness through, as of yet, unidentified proteins.
The same mutations were used to assess the contribution of each site to overall induction by FOXL2. Wild-type FOXL2 protein was overexpressed with these reporters and established that the base pairs mutated in FBE1 db, FBE1 fh, FBE3 both, and FBE3 fh all play roles in induction by FOXL2 (Figure 6D), since mutation of these sites lowered induction by half. However, the adjacent and overlapping SMAD sites in FBE1 and FBE3 do not play roles in induction by FOXL2, and, as shown previously (Figure 4C), neither do −206 or FBE2. Thus, FOXL2 induces FSHβ through the FBE1 and FBE3 forkhead elements.
SMAD3, which is a major effector of activin signaling in the induction of FSHβ, was overexpressed with these mutant reporters, to determine the roles of these sites in SMAD3 induction of FSHβ (Figure 6E). As reported previously, the mutation FBE1 fh reduced SMAD3 induction by 66%, while mutation of the adjacent SMAD site had a minor effect. Thus, the FBE1 FOXL2 site plays a role in SMAD3 induction, presumably by recruiting SMAD3 to the promoter through a FOXL2-SMAD3 interaction (18). Surprisingly, the 206 fh mutation did not have an effect, while the 206 x mutation reduced SMAD3 induction by 67%, as did the double (206 db) mutation. Thus, 206 fh does not contribute to activin, FOXL2 or SMAD3 induction of FSHβ, while the 206 x element contributes to the activin and SMAD3 induction, which again may indicate that another, as yet unidentified, factor, which may interact with SMADs, binds to this sequence. Similar to activin induction of the FBE2 mutations, SMAD3 induction was reduced with the mutation of both elements (FBE2 both) by 64%, while mutations of the individual elements, sm and fh, did not have an effect. This again points out that SMAD3 may function in cooperation with an as yet unidentified protein through protein-protein interaction at the FBE2 site. As with activin, within the FBE3 site, both the forkhead and the SMAD elements play roles in SMAD3 induction of FSHβ-luciferase. Mutation of either of these elements diminished the induction by SMAD3, indicating that this site, encompassing both SMAD and forkhead elements, is critical for FSHβ regulation. Similar results were obtained with overexpression of the SMAD3 binding partner, SMAD4, with one interesting exception (Figure 6F). SMAD-specific mutation of the FBE2 site lowered induction by SMAD4, but not by SMAD3. This indicates that the putative protein binding to the FBE2 contributes to activin induction of the promoter through interaction specifically with SMAD4, which may bind the SMAD-binding site, while SMAD3 may be recruited through protein-protein interaction. Induction by co-expression of SMAD4 and FOXL2 (Figure 6G), SMAD3 and FOXL2 or all three proteins (data not shown) was lowered by the double mutation of FBE1, but not the single mutation, indicating that both sites were necessary for synergistic induction by SMADs and FOXL2. Induction of FSHβ reporter was also lowered by the FBE2 both mutation and all of the mutations of the FBE3, again indicating the critical role of these sites.
To further examine whether SMADs, known to interact with FOXL2, can form complexes on the FBE sites, we included increasing amounts of overexpressed SMAD3, SMAD4, or SMAD3+4 with FOXL2 and analyzed their binding in gel shift assays. As stated previously, we were unable to detect FOXL2 or SMAD3 binding to either the −206 site or the FBE2, so we analyzed two 30 base-pair probes encompassing either the FBE1 (Figure 7A) or the FBE3 (Figure 7B) site. We demonstrate that FOXL2 alone binds either the FBE1 or FBE3 site, but SMAD proteins alone do not bind either site (Figure 7A). SMAD3 is recruited to either the FBE1 or FBE3 site in the presence of FOXL2. In contrast, SMAD4 protein is recruited to only the FBE3 in the presence of FOXL2, but not FBE1, and results in formation of a high-order complex (Figure 7, A and B). This higher order complex formed only on the wild-type promoter, and was not observed when mutations of either the SMAD or FOXL2 sites were introduced into the probe (data not shown). Whether or not SMAD proteins are also binding DNA directly or only being recruited to the complex by FOXL2 cannot be distinguished here. Collectively, our data indicate that FOXL2 and SMAD4 proteins can form a complex only on the FBE3 site and this interaction may explain why the FBE3 mutation has a larger effect on activin induction of FSHβ compared to mutation of the FBE1.
Figure 7.
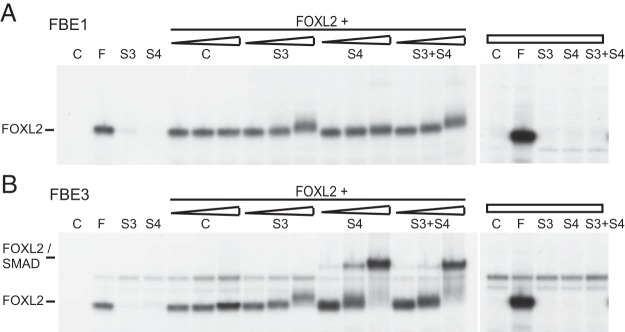
FOXL2 and SMAD proteins form complexes on FBE3. A, and B, Using FBE1 (A) or FBE3 (B) as probes in a gel shift, increasing amounts of in vitro transcribed and translated SMAD3 (S3) or SMAD4 (S4), or both (S3+S4), were incubated with in vitro transcribed and translated FOXL2 to show the appearance of a complex binding FBE3 that is not present with SMAD protein alone. C, probe only; F, FOXL2.
FOXL2 contributes to synergistic induction of FSHβ by activin and GnRH
Previously, we reported that activin and GnRH cause synergistic activation of FSHβ transcription (an induction that is higher than additive induction by each hormone alone). Indeed, the finding that endogenous activin is necessary for maximal induction by GnRH (7), provides another piece of evidence supporting this synergy, which may be critical for differential regulation of gonadotropin subunits. Here, we analyzed the FBE sites to investigate additional mechanisms underlying synergistic induction by GnRH. The 206 db, the 206 x, and the FBE3 both mutation, lowered induction by GnRH by 25%, 24%, and 27%, respectively (Figure 8A). Synergy was reduced by mutation of the same sites that lowered activin induction (compare Figures 6B and 8B), and, additionally, by mutation of the FBE2 fh forkhead element that did not have a role in activin induction alone (Figure 7B). Although FBE1 fh, FBE2 both, FBE3 fh, and FBE3 sm played roles in both activin induction and synergy, they did not contribute to the induction by GnRH alone. Together, these data indicate that there is site specificity for activin, GnRH, and synergistic responsiveness.
Figure 8.
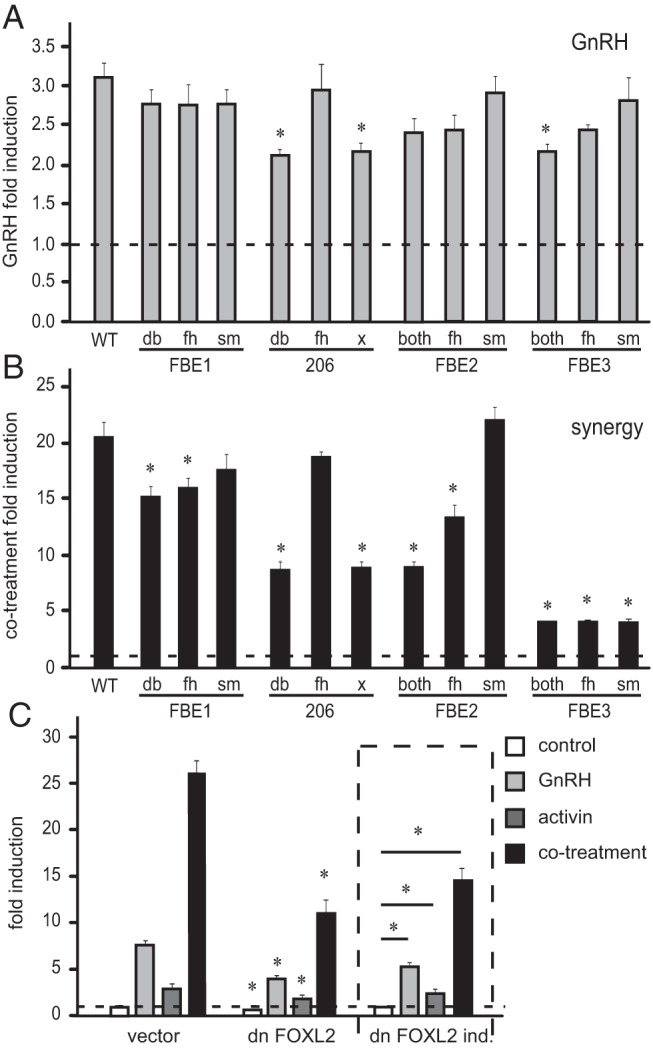
FOXL2 sites and functional FOXL2 protein are necessary for synergistic induction of FSHβ by GnRH and activin. Following transfection with reporters listed in Figure 6, LβT2 cells were treated with 10 nM GnRH (A), or activin and GnRH cotreatment (B), and the induction of the reporter was analyzed. Results represent fold induction for each reporter and * indicates statistically significant reduction in induction of the mutant reporter compared to the wild type. C, Dominant-negative (dn) FOXL2 or its control vector were overexpressed with the FSHβ-luciferase reporter then cells treated with GnRH, activin, or both for 5 h. Expression of the dn FOXL2 protein was confirmed by Western blot (data not shown). The results are normalized to control vehicle treated cells and * indicates significant reduction in the induction of the reporter co-expressed with the dn FOXL2 compared to empty vector control. On the right site, delineated with the dashed line, are the same results with dn FOXL2, this time presented as fold induction, for easier observation of the changes in induction by hormone.
To determine the contribution of the FOXL2 protein to the synergistic effect of activin and GnRH, we overexpressed a dominant-negative (dn) FOXL2 protein that contains a deletion in the forkhead domain (18, 36). Overexpression of the dn FOXL2 reduced basal expression of the FSHβ reporter by 25% (Figure 8C), as well as induction by hormone treatments. It also lowered fold induction by hormones, which is presented for clarity in the bracketed part of the Figure 8C, when induction by the hormone was normalized to the reduction in basal expression. Fold induction by GnRH was lowered by 35%, activin by 28%, and synergy by 45%. Thus, functional FOXL2 protein is necessary for FSHβ induction by GnRH, activin and the synergy between the two hormones.
We examined the mechanism by which FOXL2 contributes to synergy between GnRH and activin by analyzing whether FOXL2 protein can interact with cJUN or cFOS, immediate early proteins induced by GnRH that act to increase FSHβ transcription (32). Alternatively, the role of SMADs may be to form a bridge between FOXL2 and the cJUN/cFOS AP1 complex, since SMAD3, which is activated by activin to induce FSHβ (37), can interact with both cJUN (38) and FOXL2 (18). To differentiate between these two hypotheses, we performed GST pull-down assays in which in vitro transcribed and translated cFOS and cJUN proteins were tested for protein-protein interaction with a FOXL2-GST fusion protein. SMAD3 was included as a positive control. In this assay, cJUN interacts with the FOXL2 protein (Figure 9, middle panel). S35-labeled cJUN, but not cFOS, precipitated with glutathione beads through an interaction with GST-FOXL2. No interactions were observed using GST alone, indicating specificity of this binding (Figure 9, right panel). Thus, FOXL2 and cJUN form heteromeric complexes in vitro, through direct protein-protein interaction without the need for SMAD3.
Figure 9.
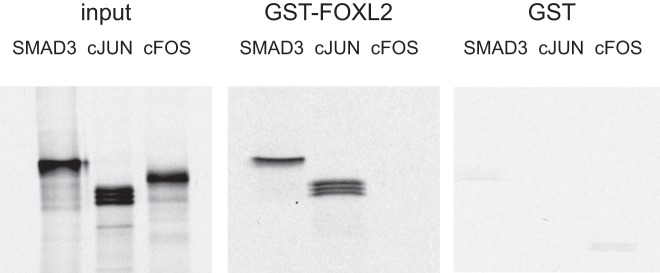
FOXL2 interacts with cJUN and SMAD3. S35 labeled proteins, SMAD3, cJUN or cFOS, indicated above the corresponding lanes over the panels, were used in a binding assay with FOXL2-GST fusion protein (middle panel). In the left panel, 1/10 of protein used in the GST pulldowns was run as input control to monitor for the migration of each protein. In the right panel, proteins were incubated with GST alone to confirm the specificity of the interaction. GST proteins were induced with IPTG overnight and the bacterial pellets were sonicated. These proteins were bound to Glutathione Sepharose beads and incubated with in vitro transcribed and translated labeled proteins. After extensive washing, the SMAD3 and cJUN are retained in the precipitate through interaction with FOXL2-GST that was visualized after running on a gel and autoradiography. The experiment was repeated three times with the same results and a representative experiment is shown.
Discussion
In this study, we determined that functional FOXL2 is necessary for FSH hormone synthesis and LHβ, FSHβ, and αGSU gene expression in the pituitary during the late stage of fetal development. Furthermore, we have substantially extended understanding of the molecular mechanisms of FOXL2 function by identifying critical elements necessary for DNA binding and transcriptional activation, and demonstrating that FOXL2 contributes to synergistic induction of FSHβ by GnRH and activin.
FOXL2 is a member of the forkhead family of transcription factors (16) that has important roles in reproductive function. In the human population, BPES is caused by mutations in the FOXL2 gene. One manifestation of BPES is premature ovarian failure, strongly implicating FOXL2 in female reproductive fitness (26). FOXL2-null mice exhibit reproductive defects including disruption of granulosa cell differentiation and failure of follicle growth (29). FOXL2 exhibits cell-specific expression in the pituitary, where it is restricted to cells of the gonadotrope and thyrotrope lineages and co-localizes with FSH (17, 18). Thus, we postulate that the reproductive effects of these mutations could be due to pituitary as well as ovarian effects. Indeed, previous reports investigating FOXL2 in vivo illustrated that 3–4 week-old FOXL2-null mice have lower levels of FSHβ, αGSU, GnRH receptor, and GH, but not LHβ (30). In our analysis at e18.5, FOXL2-null mice had lower levels of LHβ, FSHβ, and αGSU, but not TSHβ, GH, or ACTH, and the difference in GnRH receptor expression did not reach significance. Lack of an effect on GH or ACTH expression is not surprising, given that FOXL2 expression is limited to the gonadotrope and thyrotrope lineages. The difference in GH expression that Justice et al (30) observed, may manifest later in development due to the reported decrease in the size of the null pituitary that would be expected to affect somatotropes disproportionally, as this endocrine cell type is the most abundant within the anterior pituitary. In contrast to this previous report, we demonstrate a decrease in LH expression, which may stem from a developmental delay that is resolved by the age of animals studied in Justice et al (30), or reflects the possibility that the LHβ gene is a target of FOXL2. Activin does regulate LHβ, although to a lesser extent than FSHβ (39–41). Whether this reflects a developmentally dependent action of FOXL2 on LHβ remains an intriguing question. In agreement with the previous report, αGSU was expressed at a lower level, supporting a role for FOXL2 in its expression. Consistent with the fact that αGSU is expressed at a relatively high basal level and its mRNA has a long half-life, there was no difference in the number of cells containing αGSU protein, as observed in the previous report. In the conditional FOXL2 knock-out mouse using GnRH-receptor CRE, only FSHβ was lower, while the other gonadotrope-specific genes, LHβ, αGSU, and GnRH receptor were not affected (31). In contrast to the completely null mice used herein, the unchanged levels of αGSU demonstrated in the conditional knock-out mouse likely stem from αGSU expression in the thyrotrope, which was not affected by CRE expression. Different results regarding the LHβ expression may again implicate developmental effects of FOXL2. Thus, all of the in vivo models thus far, regardless of the age of the animals, confirm a role for FOXL2 in FSHβ expression and provide a strong rationale for investigating the molecular mechanism of FOXL2 function in the gonadotrope cell.
BPES is characterized by eyelid dysplasia in both sexes and premature ovarian failure in females in a large proportion of affected individuals, which are distinguished as having type I BPES. More than 260 different FOXL2 mutations have been identified since the discovery that the FOXL2 gene is mutated in BPES in 2001 (42). Several of these mutations were previously tested for functionality in a granulosa cell line (34). The function of FOXL2 in granulosa cells is to negatively regulate transcriptional activation of ovarian genes, including several involved in steroidogenesis (22, 34, 43). The FOXL2 mutated proteins that are found in BPES patients lose the ability to repress these genes (34). However, the role of these FOXL2 mutations in the pituitary was not examined. Due to dysregulation of steroidogenesis and the resulting disruption in ovarian feedback which influences pituitary hormone synthesis, it is difficult to distinguish direct pituitary effects of FOXL2 in BPES patients. Thus, we employed the immortalized gonadotrope cell line, LβT2, to examine the role of FOXL2 mutations that mimic those in BPES on FSHβ reporter activity. As we showed in the previous manuscript (15), both the mouse and human FSHβ promoters, which include activin-responsive sites, are conserved in the proximal 350 base pairs from the transcriptional start site, and, thus, they are similarly regulated by FOXL2. Although the FOXL2 polyalanine repeat domain expansion is a mutational hotspot, it is found in type II BPES, which is not associated with premature ovarian failure (27). In this manuscript, we show that deletion of the polyalanine domain causes higher expression of FSHβ. It is likely that expansion may have the opposite effect of deletion. Polyalanine expansion leads to intranuclear aggregation or cytoplasmic mislocalization, thus preventing transcriptional activity of FOXL2 (42), while deletion may lead to a more functional protein. Most mutations in individuals with type I BPES create premature stop codons in FOXL2 or cause frame shifts, both resulting in truncated proteins (26, 44). Two of these truncations were used in our studies and caused higher transcriptional activation of FSHβ compared to the wild-type protein. Thus, the effects of the FOXL2 mutations in BPES type I may also be of pituitary origin, causing higher FSHβ expression and higher FSH in the circulation, in addition to the critical role for FOXL2 in the ovary.
Several putative FOXL2 binding sites were compared in this study. Of those, FBE1 and FBE3 are bona fide FOXL2 binding sites, while −206 and FBE2 are likely not. This is surprising, since the mouse FBE2 is very similar to the porcine homologous element, and this corresponding porcine element has FOXL2 and SMAD3 binding sites, playing an important role in activin regulation of the porcine promoter (13, 14). As for the FBE1 at −350 in the mouse promoter, it is a forkhead element that binds FOXL2 and plays a role in activin, FOXL2, and SMAD induction of the FSHβ promoter. Although FBE1 binds FOXL2, as does the FBE3 site, it lacks the ability to bind SMADs and is unaffected by a mutation of the adjacent SMAD half-site, providing an explanation for its lesser role in activin, FOXL2, and SMAD3/4 induction than FBE3. FBE3 is critical for activin responsiveness of the mouse FSHβ promoter. It was initially identified as a putative SMAD-binding element in the proximity of the Pbx/Prep complex with which SMADs can interact (35). Lamba et al (13) and Tran et al (20), determined that FOXL2 binds to this site to induce the murine FSHβ, and that either SMAD3 or SMAD4, and FOXL2 proteins synergistically induce FSHβ through this element. In this report, we determine that both FOXL2 and SMAD binding elements are required for induction. Of further significance, we determine that FOXL2 and SMAD4 form a higher order complex specifically on FBE3. Since we did not detect SMAD proteins binding alone to this site, our data indicate that SMAD4 can be recruited to the FBE3 through FOXL2-SMAD protein-protein interaction, and that the SMAD-binding site may contribute to the functional cooperation between these proteins.
Two other sites that contribute to activin responsiveness of the FSHβ gene examined in this study, −206 and FBE2, do not bind FOXL2 or play a role in FOXL2 induction of FSHβ. The forkhead element at the −206 site was identified with in silico analysis (15) and was thought to contain two overlapping repeats to accommodate binding of a FOXL2 dimer (13). In this report, we created separate mutations in these two regions and determined that the 3′ region that corresponds to the forkhead element (named 206 fh), does not contribute to activin, FOXL2, SMAD3, or SMAD4 induction of the mouse FSHβ promoter, while the 5′ region (named 206 x), contributes to activin, SMAD3, and SMAD4, but not FOXL2, induction. We speculate that a different, as yet unidentified factor, binds to the 206 x site. Indeed, this site is homologous to the LHX3 site in the human FSHβ promoter (45, 46). We determined that LHX3 does not bind the mouse FSHβ promoter in this region (data not shown), but it remains a possibility that a different Lim protein plays a role in mouse FSHβ expression. Since this site has a role in GnRH induction as well, it is intriguing to examine Lim proteins for a possible role in activin, GnRH, and their synergy on FSHβ. The FBE2 site within the mouse promoter, although homologous to a high affinity FOXL2 site in the porcine promoter (13), binds FOXL2 with a very low affinity (15). The overlapping SMAD/forkhead residues contributed to activin, SMAD3, and SMAD4 induction, but mutations of the individual elements, FBE2 sm and FBE2 fh, did not contribute to activin or SMAD3 induction. Surprisingly, mutation of the SMAD element contributes to induction by SMAD4. As with the 206 x site, this may mean that a different protein is involved in activin regulation of the mouse gene through this element, since we were not able to detect binding of either FOXL2 or SMAD3 to this site and it is unlikely that these proteins form a complex that requires only overlapping residues. This protein may interact with SMAD4, however. Further experiments are needed to identify new activin-regulated factors that function through these two elements in the FSHβ promoter.
GnRH and activin synergize to induce FSHβ. This synergy is specific for FSHβ and may be one mechanism of differential regulation of gonadotropin genes (7, 47). We determined previously that there is both cross-talk between GnRH and activin signaling pathways and interaction between activin-activated SMAD3 and GnRH-induced AP1 on the FSHβ promoter (7). Due to the role of FOXL2 in activin induction of the mouse FSHβ promoter and its interaction and complex formation with SMADs, we examined whether forkhead elements play a role in GnRH and activin synergy. The sole forkhead site that plays a role in GnRH induction, likely through a necessity for endogenous activin for maximal GnRH induction (7), is FBE3, the same site where FOXL2 and SMADs form a complex. The other site that contributes to GnRH induction is 206 x, which may bind an unidentified protein, as discussed above. Not surprisingly, other sites that are important for activin induction are also important for synergy. Additionally, functional FOXL2 protein is necessary for maximal GnRH induction and synergy, since introduction of a dominant-negative FOXL2 decreases the expression. This likely occurs by disrupting the function of endogenous FOXL2, since FOXL2 forms a homodimer (13). Interestingly, FOXL2 can directly interact with cJUN, and this interaction may contribute to the GnRH-activin crosstalk, in addition to SMAD3 interaction with both cJUN and FOXL2. Thus, FOXL2 may be a part of a transcriptionally active complex that specifically regulates FSHβ.
In summary, we have shown that FOXL2 is required for gonadotropin gene expression in vivo and for synergistic induction of FSHβ by GnRH and activin. Of multiple forkhead elements, we identify the most proximal site as the site most important for SMAD and FOXL2 complex formation. Additionally, we show that truncated FOXL2 proteins cause higher induction of FSHβ, while the forkhead element is necessary for FOXL2 function. Further studies are needed to establish whether these truncated FOXL2 proteins that cause higher FSHβ promoter activity, have higher affinity for interacting SMADs, resulting in either enhanced activin responsiveness, or increased recruitment of basal transcriptional machinery to the FSHβ promoter. Further experiments will also aim to determine the identity of activin-responsive proteins that function through the −206 and FBE2 sites.
Acknowledgments
We thank Dr Bilezikjian, Dr Massague, Dr Pisarska, Dr Bae, Dr Birrer, Dr Bernard, and Dr Tulchinsky for expression vectors. We are especially grateful to Dr. L. Bilezikjian for the FOXL2-null mice and the FOXL2-GST vector.
This work was supported by National Institutes of Health Grants No. R01 HD057549 and R21 HD058752 (D.C.), K99/R00 HD060947 (K.B.), and R01 HD020377, R01 HD072754, and U54 HD012303 NICHD/NIH cooperative agreement (P.L.M.). P.L.M. was supported in part by P30 DK063491, P30 CA023100, and P42 ES010337. L.L.R. and A.H. were supported by the Endocrine Society Summer Research Fellowships.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ANOVA
- analysis of variance
- BPES
- Blepharophimosis Ptosis Epicanthus Inversus Syndrome
- db
- double mutation
- ECL
- enhanced chemiluminescence
- fh
- forkhead elements
- αGSU
- α-glycoprotein subunit
- IPTG
- isopropyl-β-D-thiogalactosidase
- qPCR
- quantitative PCR
- sm
- SMAD sites.
References
- 1. Woodruff TK, Besecke LM, Groome N, Draper LB, Schwartz NB, Weiss J. Inhibin A and inhibin B are inversely correlated to follicle-stimulating hormone, yet are discordant during the follicular phase of the rat estrous cycle, and inhibin A is expressed in a sexually dimorphic manner. Endocrinology. 1996;137:5463–5467 [DOI] [PubMed] [Google Scholar]
- 2. Besecke LM, Guendner MJ, Sluss PA, et al. Pituitary follistatin regulates activin-mediated production of follicle-stimulating hormone during the rat estrous cycle. Endocrinology. 1997;138:2841–2848 [DOI] [PubMed] [Google Scholar]
- 3. Chand AL, Harrison CA, Shelling AN. Inhibin and premature ovarian failure. Hum Reprod Update. 2010;16:39–50 [DOI] [PubMed] [Google Scholar]
- 4. Kaiser UB, Conn PM, Chin WW. Studies of gonadotropin-releasing hormone (GnRH) action using GnRH receptor-expressing pituitary cell lines. Endocr Rev. 1997;18:46–70 [DOI] [PubMed] [Google Scholar]
- 5. Vale W, Rivier C, Brown M. Regulatory peptides of the hypothalamus. Annu Rev Physiol. 1977;39:473–527 [DOI] [PubMed] [Google Scholar]
- 6. Weiss J, Guendner MJ, Halvorson LM, Jameson JL. Transcriptional activation of the follicle-stimulating hormone beta-subunit gene by activin. Endocrinology. 1995;136:1885–1891 [DOI] [PubMed] [Google Scholar]
- 7. Coss D, Hand CM, Yaphockun KK, Ely HA, Mellon PL. p38 mitogen-activated protein kinase is critical for synergistic induction of the FSH(beta) gene by gonadotropin-releasing hormone and activin through augmentation of c-Fos induction and Smad phosphorylation. Mol Endocrinol. 2007;21:3071–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ling N, Ying SY, Ueno N, et al. Pituitary FSH is released by a heterodimer of the beta-subunits from the two forms of inhibin. Nature. 1986;321:779–782 [DOI] [PubMed] [Google Scholar]
- 9. Gregory SJ, Lacza CT, Detz AA, Xu S, Petrillo LA, Kaiser UB. Synergy between activin A and gonadotropin-releasing hormone in transcriptional activation of the rat follicle-stimulating hormone-beta gene. Mol Endocrinol. 2005;19:237–254 [DOI] [PubMed] [Google Scholar]
- 10. Suszko MI, Balkin DM, Chen Y, Woodruff TK. Smad3 mediates activin-induced transcription of follicle-stimulating hormone beta-subunit gene. Mol Endocrinol. 2005;19:1849–1858 [DOI] [PubMed] [Google Scholar]
- 11. Bernard DJ. Both SMAD2 and SMAD3 mediate activin-stimulated expression of the follicle-stimulating hormone beta subunit in mouse gonadotrope cells. Mol Endocrinol. 2004;18:606–623 [DOI] [PubMed] [Google Scholar]
- 12. McGillivray SM, Thackray VG, Coss D, Mellon PL. Activin and glucocorticoids synergistically activate follicle-stimulating hormone beta-subunit gene expression in the immortalized LbetaT2 gonadotrope cell line. Endocrinology. 2007;148:762–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lamba P, Fortin J, Tran S, Wang Y, Bernard DJ. A novel role for the forkhead transcription factor FOXL2 in activin A-regulated follicle-stimulating hormone beta subunit transcription. Mol Endocrinol. 2009;23:1001–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lamba P, Wang Y, Tran S, et al. Activin A regulates porcine follicle-stimulating hormone beta-subunit transcription via cooperative actions of SMADs and FOXL2. Endocrinology. 2010;151:5456–5467 [DOI] [PubMed] [Google Scholar]
- 15. Corpuz PS, Lindaman LL, Mellon PL, Coss D. FoxL2 is required for activin induction of the mouse and human follicle-stimulating hormone beta-subunit genes. Mol Endocrinol. 2010;24:1037–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carlsson P, Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev Biol. 2002;250:1–23 [DOI] [PubMed] [Google Scholar]
- 17. Ellsworth BS, Egashira N, Haller JL, et al. FOXL2 in the pituitary: molecular, genetic, and developmental analysis. Mol Endocrinol. 2006;20:2796–2805 [DOI] [PubMed] [Google Scholar]
- 18. Blount AL, Schmidt K, Justice NJ, Vale WW, Fischer WH, Bilezikjian LM. FoxL2 and Smad3 coordinately regulate follistatin gene transcription. J Biol Chem. 2009;284:7631–7645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Y, Libasci V, Bernard DJ. Activin A induction of FSHbeta subunit transcription requires SMAD4 in immortalized gonadotropes. J Mol Endocrinol. 2010;44:349–362 [DOI] [PubMed] [Google Scholar]
- 20. Tran S, Lamba P, Wang Y, Bernard DJ. SMADs and FOXL2 synergistically regulate murine FSHbeta transcription via a conserved proximal promoter element. Mol Endocrinol. 2011;25:1170–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ellsworth BS, Burns AT, Escudero KW, Duval DL, Nelson SE, Clay CM. The gonadotropin releasing hormone (GnRH) receptor activating sequence (GRAS) is a composite regulatory element that interacts with multiple classes of transcription factors including Smads, AP-1 and a forkhead DNA binding protein. Mol Cell Endocrinol. 2003;206:93–111 [DOI] [PubMed] [Google Scholar]
- 22. Kuo FT, Fan K, Bentsi-Barnes I, Barlow GM, Pisarska MD. Mouse forkhead L2 maintains repression of FSH-dependent genes in the granulosa cell. Reproduction. 2012;144:485–494 [DOI] [PubMed] [Google Scholar]
- 23. Pisarska MD, Barlow G, Kuo FT. Minireview: roles of the forkhead transcription factor FOXL2 in granulosa cell biology and pathology. Endocrinology. 2011;152:1199–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McTavish KJ, Nonis D, Hoang YD, Shimasaki S. Granulosa cell tumor mutant FOXL2C134W suppresses GDF-9 and activin A-induced follistatin transcription in primary granulosa cells. Mol Cell Endocrinol. 2013;372:57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pisarska MD, Kuo FT, Bentsi-Barnes IK, Khan S, Barlow GM. LATS1 phosphorylates forkhead L2 and regulates its transcriptional activity. Am J Physiol Endocrinol Metab. 2010;299:E101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crisponi L, Deiana M, Loi A, et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet. 2001;27:159–166 [DOI] [PubMed] [Google Scholar]
- 27. De Baere E, Beysen D, Oley C, et al. FOXL2 and BPES: mutational hotspots, phenotypic variability, and revision of the genotype-phenotype correlation. Am J Hum Genet. 2003;72:478–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Uda M, Ottolenghi C, Crisponi L, et al. Foxl2 disruption causes mouse ovarian failure by pervasive blockage of follicle development. Hum Mol Genet. 2004;13:1171–1181 [DOI] [PubMed] [Google Scholar]
- 29. Schmidt D, Ovitt CE, Anlag K, et al. The murine winged-helix transcription factor Foxl2 is required for granulosa cell differentiation and ovary maintenance. Development. 2004;131:933–942 [DOI] [PubMed] [Google Scholar]
- 30. Justice NJ, Blount AL, Pelosi E, Schlessinger D, Vale W, Bilezikjian LM. Impaired FSHbeta expression in the pituitaries of Foxl2 mutant animals. Mol Endocrinol. 2011;25:1404–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tran S, Zhou X, Lafleur C, et al. Impaired fertility and FSH synthesis in gonadotrope-specific Foxl2 knockout mice. Mol Endocrinol. 2013;27:407–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Coss D, Jacobs SB, Bender CE, Mellon PL. A novel AP-1 site is critical for maximal induction of the follicle-stimulating hormone beta gene by gonadotropin-releasing hormone. J Biol Chem. 2004;279:152–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mollard P, Hodson DJ, Lafont C, Rizzoti K, Drouin J. A tridimensional view of pituitary development and function. Trends Endocrinol Metab. 2012;23:261–269 [DOI] [PubMed] [Google Scholar]
- 34. Park M, Shin E, Won M, et al. FOXL2 interacts with steroidogenic factor-1 (SF-1) and represses SF-1-induced CYP17 transcription in granulosa cells. Mol Endocrinol. 2010;24:1024–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bailey JS, Rave-Harel N, McGillivray SM, Coss D, Mellon PL. Activin regulation of the follicle-stimulating hormone beta-subunit gene involves Smads and the TALE homeodomain proteins Pbx1 and Prep1. Mol Endocrinol. 2004;18:1158–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ghochani Y, Saini JK, Mellon PL, Thackray VG. FOXL2 is involved in the synergy between activin and progestins on the follicle-stimulating hormone β-subunit promoter. Endocrinology. 2012;153:2023–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Coss D, Mellon PL, Thackray VG. A FoxL in the Smad house: activin regulation of FSH. Trends Endocrinol Metab. 2010;21:562–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qing J, Zhang Y, Derynck R. Structural and functional characterization of the transforming growth factor-beta -induced Smad3/c-Jun transcriptional cooperativity. J Biol Chem. 2000;275:38802–38812 [DOI] [PubMed] [Google Scholar]
- 39. Coss D, Thackray VG, Deng CX, Mellon PL. Activin regulates luteinizing hormone beta-subunit gene expression through smad-binding and homeobox elements. Mol Endocrinol. 2005;19:2610–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stouffer RL, Woodruff TK, Dahl KD, Hess DL, Mather JP, Molskness TA. Human recombinant activin-A alters pituitary luteinizing hormone and follicle-stimulating hormone secretion, follicular development, and steroidogenesis, during the menstrual cycle in rhesus monkeys. J Clin Endocrinol Metab. 1993;77:241–248 [DOI] [PubMed] [Google Scholar]
- 41. Blumenfeld Z, Ritter M. Inhibin, activin, and follistatin in human fetal pituitary and gonadal physiology. Ann NY Acad Sci. 2001;943:34–48 [DOI] [PubMed] [Google Scholar]
- 42. Caburet S, Georges A, L'Hôte D, Todeschini AL, Benayoun BA, Veitia RA. The transcription factor FOXL2: at the crossroads of ovarian physiology and pathology. Mol Cell Endocrinol. 2012;356:55–64 [DOI] [PubMed] [Google Scholar]
- 43. Pisarska MD, Bae J, Klein C, Hsueh AJ. Forkhead l2 is expressed in the ovary and represses the promoter activity of the steroidogenic acute regulatory gene. Endocrinology. 2004;145:3424–3433 [DOI] [PubMed] [Google Scholar]
- 44. De Baere E, Dixon MJ, Small KW, et al. Spectrum of FOXL2 gene mutations in blepharophimosis-ptosis-epicanthus inversus (BPES) families demonstrates a genotype–phenotype correlation. Hum Mol Genet. 2001;10:1591–1600 [DOI] [PubMed] [Google Scholar]
- 45. Benson CA, Kurz TL, Thackray VG. A human FSHB promoter SNP associated with low FSH levels in men impairs LHX3 binding and basal FSHB transcription. Endocrinology. 2013;154:3016–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. West BE, Parker GE, Savage JJ, et al. Regulation of the follicle-stimulating hormone beta gene by the LHX3 LIM-homeodomain transcription factor. Endocrinology. 2004;145:4866–4879 [DOI] [PubMed] [Google Scholar]
- 47. Thackray VG, Mellon PL, Coss D. Hormones in synergy: regulation of the pituitary gonadotropin genes. Mol Cell Endocrinol. 2010;314:192–203 [DOI] [PMC free article] [PubMed] [Google Scholar]