

Abstract
One of the master regulators of both glucose and lipid cellular metabolism is 5′-AMP-activated protein kinase (AMPK). As a metabolic pivot that dynamically responds to shifts in nutrient availability and stress, AMPK dysregulation is implicated in the underlying molecular pathology of a variety of diseases, including cardiovascular diseases, diabetes, cancer, neurological diseases, and aging. Although the regulation of AMPK enzymatic activity by upstream kinases is an active area of research, less is known about regulation of AMPK protein stability and activity by components of the ubiquitin-proteasome system (UPS), the cellular machinery responsible for both the recognition and degradation of proteins. Furthermore, there is growing evidence that AMPK regulates overall proteasome activity and individual components of the UPS. This review serves to identify the current understanding of the interplay between AMPK and the UPS and to promote further exploration of the relationship between these regulators of energy use and amino acid availability within the cell.
The survival of every entity depends on the ability to quickly recognize and adapt to changing environments. Within all eukaryotic cells, a key mediator of these responses to variations in energy availability is the 5′-AMP-activated protein kinase (AMPK) holoenzyme (also known as protein kinase, AMP activated) (see Table 1 for all abbreviations). Although it had been recognized for many years that a kinase negatively regulated both 3-hydroxy-3-methyl-glutaryl-coenzyme A (CoA) reductase and acetyl-CoA carboxylase, which are the rate-limiting enzymes in cholesterol and fatty acid synthesis, respectively (1, 2), it was eventually determined that a single kinase, AMPK, regulated both these enzymes (3). Since that time, a huge variety of AMPK substrates have been discovered, suggesting a general model in which AMPK acts as a fuel sensor within the cell, exerting kinase activity to activate energy-conserving (catabolic) pathways and simultaneously inhibit energy-consuming (anabolic) pathways (4).
Table 1.
Nomenclature and Abbreviations
| Genes/Gene | Products | Genes/Gene | Products |
|---|---|---|---|
| Akt | Protein kinase B | Snf1 | Sucrose nonfermenting protein kinase 1 |
| AMPK | AMP-activated protein kinase | SUMO | Small ubiquitin-like modifier |
| APC/C | Anaphase-promoting complex | TAK1 | TGFβ-activated kinase-1 |
| Atrogin-1 | F-box protein 32 (FBXO32) | TNFα | Tumor necrosis factor α |
| BCA2 | Breast cancer-associated gene 2 | Trim32 | Tripartite motif containing 32 |
| BRSK2 | Brain-selective kinase 2 | Ubp8 | Ubiquitin carboxy-terminus hydrolase 8 |
| c-Cbl | Casitas B-lineage lymphoma protooncogene | USP9X | Ubiquitin specific peptidase 9, X linked |
| CaMKKβ | Calcium/calmodulin-dependent protein kinase kinase β | WWP1 | WW domain-containing protein 1 |
|
Molecules |
|||
| CBS | Cystathione β-synthase | ADP | Adenosine diphosphate |
| CHIP | C terminus of Hsp70-interacting | AMP | Adenosine monophosphate |
| protein (STUB1) | ATP | Adenosine triphosphate | |
| Cidea | Cell death-inducing DFF45-like effector A | cAMP | 3′-5′-cyclic adenosine monophosphate |
| CRBN | Cereblon-A | Coa | Coenzyme A |
| FoxO | Forkhead box O family | HDL | High-density lipoprotein |
| IGF | Insulin-like growth factor | HMG-CoA | 3-Hydroxy-3-methyl-glutaryl-CoA |
| LKB1 | Liver kinase B1 |
Chemicals |
|
| MARK4 | Microtubule affinity-regulating kinase 4 | AICAR | 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside |
| MEF-2 | Myocyte enhancer factor 2A | ZMP | AICAR monophosphate |
| MuRF1 | Muscle ring finger1 (TRIM63) |
Cells, Domains, and Enzymes |
|
| Nedd4 | Neural precursor cell expressed, | AID | Autoinhibitory domain |
| developmentally down-regulated 4 | C2C12 | Mouse myoblast/myotube cell line | |
| NUAK1 | Nua (novel) kinase family 1 | CBM | Carbohydrate binding motif |
| OGT | O-linked N-acetylglucosamine transferase | DUB | Deubiquitinase |
| PKA | Protein kinase A | HUVEC | Human umbilical vein endothelial cells |
| PP2 | Protein phosphatase 2 | KD | Kinase domain |
| PPARγ | Peroxisome proliferator-activated receptor γ | RING | Really interesting new gene |
| PSMD11 | Proteasome 26S subunit non-ATPase regulatory subunit 11 | SBS | Subunit binding domain |
| UPS | Ubiquitin proteasome system | ||
| PTG | Protein targeting to glycogen | VSMC | Vascular smooth muscle cells |
| SAGA | Spt-Ada-Gcn5-acetyltransferase | ||
| Skp2 | S-phase kinase associated protein 2 | ||
AMPK is implicated as therapeutic target in a wide variety of diseases. From a clinical standpoint, AMPK clearly has an important role in promoting energy balance that is disrupted in metabolic diseases, such as metabolic syndrome and type 2 diabetes (5). Emerging research also demonstrates that AMPK activation is important in cardiovascular disease (6) and cancer (5). AMPK also demonstrates neuroprotective effects (7). Therefore, understanding the regulation of AMPK expression, activity, and protein stability are paramount therapeutic goals.
Molecular Structure of AMPK
The general model of AMPK activation requires a combination of functioning as an “energy sensor” through allosteric changes in response to free AMP:ATP ratios, as well as posttranslational modification of the AMPK holoenzyme. Under energy-restricted conditions, the AMP:ATP ratio increases, promoting AMPK activation. The AMPK holoenzyme is a heterotrimer comprised of 3 subunits encoded by unique genes, the catalytic α-subunit (α1 and α2 isoforms) is paired with a scaffolding β-subunit (β1 and β2 isoforms) and a regulatory γ-subunit (γ1, γ2, and γ3 isoforms). The α-subunit contains a kinase domain, autoinhibitory domain, and a β-subunit binding sequence (SBS) (8). In cell culture models, the 2 α-isoforms appear to localize to different cellular compartments; α1 appears to be predominantly cytosolic, whereas α2 is localized to the nucleus in a stress-dependent manner (9). The β-subunit contains a carbohydrate binding motif (10) and an α/γ-SBS (11); outside of differential tissue expression, it is unclear whether there is a functional difference between β1 and β2. The γ-subunit contains a β-SBS and 2 nucleotide-binding Bateman domains, which consist of a total of 4 cystathione β-synthase motifs (12, 13). The γ-isoforms have the most structural variability within the subunit isoforms, which likely contributes to the degree of AMP-mediated activation seen in some rodent tissues, including rat brain and heart (14–16). Structural schematics and relative tissue expression of the isoforms in rodents are shown in Figure 1.
Figure 1.
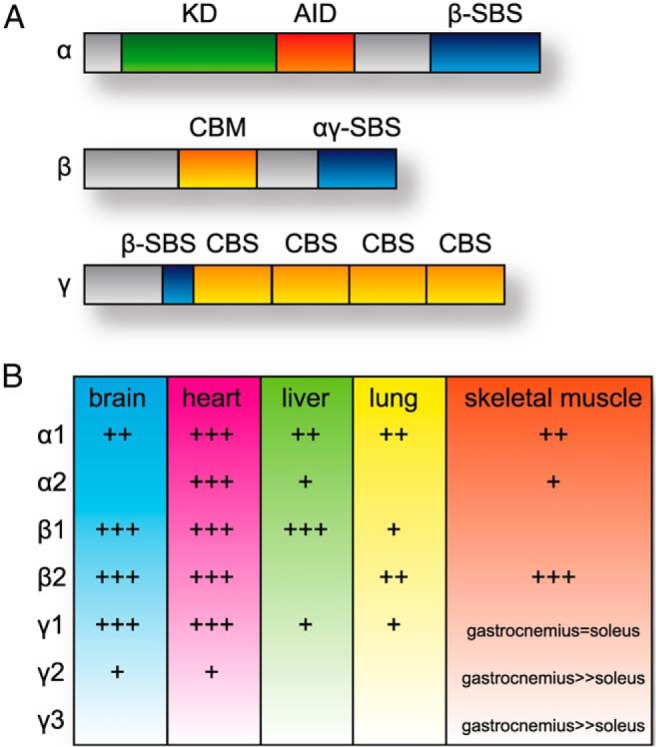
Protein domains and tissue expression of AMPK subunits. A, The linear domain structure of the α, β, and γ subunits is shown. KD, kinase domain; AID, autoinhibitory domain; SBS, subunit binding sequence; CBM, carbohydrate binding motif; CBS, cystathione β-synthase motif. B, The relative protein expression of α, β, and γ isoforms in various organs is provided. Data are summarized from rodent models as previously described (11, 16, 157).
Molecular Activation of AMPK
AMPK activation is primarily regulated by 2 events: allosteric activation via binding of AMP, which competes with ATP binding to the 4 cystathione β-synthase domains in the γ-subunit (17), and the phosphorylation of Thr172 within the kinase domain of the α-subunit (18), mediated by the upstream kinases liver kinase B1 (LKB1) (19), calcium/calmodulin-dependent protein kinase kinase-β (20, 21), and TGFβ-activated kinase-1 (22, 23), which compete with the phosphatases protein phosphatase 2 (PP2)A and PP2C (24). The binding of AMP also positively regulates AMPK activity through the promotion of LKB1-mediated Thr172 phosphorylation, which simultaneously inhibits dephosphorylation of Thr172 (25). In addition to activation by AMP, AMPK is activated physiologically by calcium influx (20), high-density lipoprotein cholesterol (26), leptin (27), adiponectin (28), and other stimuli that signify increased energy demand; conversely, AMPK is inhibited by factors associated with energy-abundant situations, including Akt (29), amino acids (30), and TNFα (31).
Pharmacological Manipulation of AMPK
Experimentally, AMPK can be activated in cells through depletion of ATP using 2-deoxyglucose, a nonmetabolizable glucose analog that inhibits glycolysis. Used widely in rodent studies, pharmacologic activation of AMPK is achieved with 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR), which is metabolized to ZMP (AICAR monophosphate), an AMP-mimicking molecule (32). However, this approach lacks AMPK specificity, because AICAR also activates other AMP-sensitive enzymes. Metformin, a biguanidine widely used in patients with diabetes to improve insulin sensitivity and decrease circulating lipids, increased AMPK phosphorylation (33) only when the cells tested (Chinese hamster ovary fibroblasts, and H4IIE, rat hepatoma derived) remained intact, and did not affect the ADP:AMP ratio (34). However, recent studies have questioned whether the action of metformin is dependent on AMPK (35). In wild-type primary mouse hepatocytes, biguanidines increase AMP and decrease cAMP and protein kinase A (36). However, it appears that biguanidines exert homeostatic energy effects by antagonizing glucagon-induced cAMP production and downstream glucagon signaling pathways, because primary hepatocytes derived from mice lacking AMPKα1/α2-subunit expression demonstrated a similar decrease in cAMP with biguanidine administration (36). Thiazolidinediones are synthetic ligands for peroxisome proliferator-activated receptor-γ that also increase AMPK phosphorylation in cells by increasing the AMP:ATP ratio (37) and may exert actions in vivo by stimulating adiponectin signaling (38). Several natural compounds also indirectly increase AMPK activity, and designing nonnucleotide analog compounds that activate AMPK remains a goal within the pharmaceutical industry (39). The small molecule compound C can be used to inhibit AMPK (33) and has been used in numerous cell-based studies examining AMPK function, but because this molecule broadly inhibits protein kinases (40), cellular phenotypes associated with compound C treatment may not be AMPK dependent.
Molecular Effects of AMPK Activation
Upon activation, AMPK exerts kinase activity on a variety of substrates involved in pathways that increase ATP generation and limit ATP-consuming processes. More specifically, AMPK promotes ATP generation through kinase activity directed toward proteins involved in glucose (41) and fatty acid (42) uptake, glycolysis (43), fatty acid oxidation (44), and autophagy (45) and inhibits synthesis of glycogen (46), triglycerides (47), fatty acids (48), and mammalian target of rapamycin signaling-related protein translation (49–51). The specific substrates of AMPK are myriad and an active area of research (52), and current knowledge of AMPK substrates is discussed in several excellent reviews (39, 53–55). Although the regulation of AMPK activation has been widely studied, less discussed are studies involved in AMPK regulation either at a transcriptional (for example, Ref. 56) or posttranslational level. In this article, we discuss recent studies demonstrating an interplay between AMPK and the posttranslational regulation of AMPK by the ubiquitin-proteasome system (UPS), as well as the new evidence that demonstrates that AMPK is involved in the regulation of the UPS.
Regulation of AMPK by the UPS
Because of the many signaling pathways that converge on AMPK and the multiple metabolic pathways affected by AMPK activity, the regulation of AMPK by adenine nucleotides, upstream kinases, and pharmacological agents is a popular research topic. Less studied is the regulation of AMPK by the UPS. The UPS is divided into 2 major components: 1) a host of enzymes involved in the modification of proteins by the covalent addition or removal of the small 9-kDa protein ubiquitin via ubiquitin ligases or deubiquitinases (DUBs), and 2) a large protein complex collectively called the proteasome that recognizes polyubiquitinated proteins and mediates the enzymatic degradation of substrates (Figure 2). Understanding how the UPS regulates AMPK may lead to strategies that could modify AMPK posttranslational stability.
Figure 2.

UPS components and activity. The UPS is comprised of ubiquitin-activating E1 enzymes, ubiquitin-conjugating E2 enzymes, and ubiquitin-ligating E3 enzymes. The different families of E3 ligases can are distinguished by the type of ubiquitin ligase domain. In the first round of ubiquitination (monoubiquitination), ubiquitin is ligated to a substrate protein at the diglycine motif at the C terminus on the ubiquitin protein (inset). Subsequent addition of ubiquitin (polyubiquitination) involves a linkage between one of the 7 available lysines of the attached ubiquitin and the diglycine motif of an adjacent ubiquitin molecule. Canonical polyubiquitin chains are linked through K48 (red) and are recognized by the proteasome and degraded into free amino acids; noncanonical polyubiquitin chains are linked through non-K48 residues (yellow) and may be branched or contain mixed linkages. Noncanonical chains serve a variety of nonproteasome-targeting roles. DUBs remove ubiquitin residues from substrates.
Direct Regulation of AMPK Via E3 Ubiquitin Ligases
Most proteins within the mammalian cell are degraded by the UPS after addition of polyubiquitin chains (57). Ubiquitin chains can be linked through any of ubiquitin's 7 lysine residues (Figure 2) or combinations therein (58). Degradation-associated polyubiquitination refers to chain linkages involving only ubiquitin lysine 48, referred to as “canonical” ubiquitination. Substrates polyubiquitinated with non-K48-linked chains are not recognized by the proteasome, referred to as “noncanonical” ubiquitination (Figure 2). Experimental evidence has demonstrated that AMPK subunits α2 (59, 60), β1 (61), and β2 (62) are polyubiquitinated and degraded by the UPS; in addition, experimental evidence suggests that AMPK subunits may also be ubiquitinated with noncanonical chains, as discussed below.
Some insight into UPS regulation of AMPK activity and stability has been gleaned from genetic knockout mouse models. Cell death-inducing DFF45-like effector A (Cidea) is a transcriptional coactivator (63) involved in lipid storage and exchange (64). Mice lacking the expression of Cidea (Cidea−/−) are lean and resistant to obesity and diabetes (65), metabolic adaptations that correlated with an increase in AMPK subunit protein levels in brown adipose tissue. Further examination of a connection between Cidea and AMPK revealed a direct interaction between Cidea and AMPKβ1 that resulted in AMPKβ1 ubiquitination and UPS-dependent degradation (61). It was speculated that Cidea regulates AMPK either by direct ubiquitin ligase activity of Cidea or through Cidea-dependent recruitment of other E3 ligases to AMPKβ1 (61). The current literature does not support other examples of Cidea functioning as a bona fide ubiquitin ligase; thus, a likely explanation for the effects of Cidea on AMPK involves other unidentified proteins that mediate ubiquitination.
Additionally, our laboratory recently reported on a novel interaction between AMPK and C terminus of heat shock protein 70-interacting protein (CHIP), an E3 ubiquitin ligase that mediates protein quality control during acute and chronic stress (66, 67). Many proteins that interact with CHIP or CHIP-containing protein complexes are substrates for ubiquitination. However, we discovered that CHIP acts as an autonomous chaperone for AMPK and increases AMPK stability, activation, and activity (68). Both in cell-free systems and in mouse heart, CHIP interacts with AMPKα1- and AMPKα2-containing holoenzymes. This interaction promotes LKB1-mediated AMPK phosphorylation and phosphorylation-independent increases in AMPK activity (68). Additionally, CHIP appears to mediate the UPS-dependent degradation of LKB1 (69), suggesting a negative feedback loop, wherein CHIP can promote AMPK activity and subsequently assist in returning AMPK activity to basal levels by degrading an important upstream activator. Therefore, CHIP mediates the activity of AMPK both directly via a physical interaction and chaperoning function directed towards AMPK, as well as indirectly affecting AMPK phosphorylation via CHIP's ubiquitin ligase activity towards LKB1, the major upstream kinase that regulates AMPK. The physiological importance of the CHIP-AMPK interaction is evident in the severe pathological remodeling in CHIP−/− mouse hearts after pressure overload, a common stimuli for pathological cardiac hypertrophy. After 1 week of pressure overload, CHIP−/− mice had 65% mortality and a 35% decrease in cardiac output, which was accompanied by increased fibrosis, lipid accumulation, decreased mitochondrial content, and decreased oxidative phosphorylation-associated gene expression, in part due to an inability to activate AMPK (68). The dual role of CHIP acting both directly on AMPK as well as on an upstream regulator suggests that CHIP is well positioned to augment and attenuate AMPK signaling. This allows cellular stress signaling usually associated with CHIP to intersect directly with cellular metabolism. The effects of CHIP on regulating AMPK either directly or through LKB1 clearly plays an important role in regulating cardiac metabolism in a mouse model of pressure overload, but the CHIP-AMPK signaling axis may also be important in other metabolically active tissues, such as the brain, where both CHIP and AMPK are expressed. Recently, 5 independent reports (70–74) identified numerous coding mutations in CHIP in patients with various forms of cerebellar ataxia. Altered AMPK activity is known to affect various neurological pathologies (75). Therefore, it is easy to envision a model of how loss-of-function mutations in CHIP can lead to derangements of the CHIP-AMPK signaling axis in the brain and directly impact various neuropathophysiologies. As described below, other E3 ligases appear to regulate AMPK activation and activity through indirect mechanisms rather than directly affecting AMPK protein stability.
Indirect Regulation of AMPK Via E3 Ubiquitin Ligases
An additional example of a genetic mouse model providing insight into the link between the UPS and metabolic homeostasis involved the molecular scaffold protein casitas B-lineage lymphoma protooncogene (c-Cbl), a characterized E3 ubiquitin ligase that regulates tyrosine kinase receptor signaling (76) and insulin signaling in adipocytes (77). Mice that lack the expression of c-Cbl (c-Cbl−/−) exhibited increased energy expenditure, higher resting metabolic rates, and decreased adiposity compared with wild-type control mice (78). Given the known function of c-Cbl, the authors hypothesized that c-Cbl regulates AMPK via ubiquitination. Consistent with a low-energy phenotype, AMPK phosphorylation was increased in muscle homogenates derived from c-Cbl−/− mice compared with wild-type mice. However, total AMPKα levels were not affected by c-Cbl deletion, suggesting that c-Cbl itself does not directly mediate ubiquitin-dependent proteasomal degradation of AMPK (78). To determine whether the ubiquitin ligase activity of c-Cbl is involved in metabolic homeostasis, a mouse was engineered expressing only a single copy of c-Cbl that was modified with a point mutation in the ubiquitin ligase domain that abolishes ubiquitin ligase activity (c-Cbl−/A) (79). Interestingly, c-Cbl−/A mice metabolically phenocopied c-Cbl−/− mice, including increased AMPK phosphorylation in skeletal muscle. These data suggest that the ubiquitin ligase activity of c-Cbl, although not directly regulating the protein stability of AMPK, plays an important role in metabolic homeostasis, resulting in increased AMPK activity.
In addition to c-Cbl, 2 other ubiquitin ligases, cereblon (CRBN) and breast cancer-associated gene 2 (BCA2), have been demonstrated to affect AMPK phosphorylation without directly affecting AMPK protein stability. CRBN is a thalidomide-binding protein that exists in a ubiquitin ligase complex with cullin proteins (80). CRBN interacts with AMPKα1 and inhibits Thr172 phosphorylation. Additionally, depletion of CRBN in a cell culture model increases AMPK activity without affecting total AMPK levels (81). BCA2, a RING (really interesting new gene family) E3 ubiquitin ligase overexpressed in many breast tumors (82), similarly decreases Thr172 phosphorylation independent of any change in AMPKα1 protein levels, effects that are dependent on an intact RING domain (83). The precise manner in which CRBN and BCA2 inhibit AMPK phosphorylation is unknown. However, both rely on indirect mechanisms that likely involve the degradation of an unknown intermediate. Identifying the substrates of CRBN and BCA2 is an important research goal to identify direct regulators of AMPK kinase activity.
Noncanonical Polyubiquitination Regulation of AMPK-Related Kinases
Although canonical polyubiquitination and proteasomal-mediated degradation is perhaps the most studied form of ubiquitination, there are also noncanonical forms of polyubiquitination that can alter protein function vs protein stability. For example, members of the AMPK-related kinase family of proteins, including nua (novel) kinase family 1 (NUAK1) and microtubule affinity-regulating kinase 4 (MARK4), are noncanonically ubiquitinated with lysine 29/33-linked chains. This noncanonical ubiquitin modification appears to decrease the activity of the AMPK-related kinases, as well as inhibit activation via LKB1 phosphorylation (84). This is in contrast to the another AMPK-related kinase, brain-selective kinase 2 (BRSK2), which is canonically ubiquitinated by the ubiquitin ligases Jun activation domain-binding protein 1 and anaphase-promoting complex, leading to BRSK2 degradation (85, 86). It is unclear at this time whether these ubiquitin ligases also modify AMPK, although in similar studies, it was clear that AMPKα1-containing complexes are also polyubiquitinated (84).
Nutrient-Dependent Regulation of AMPK Via the UPS
The availability and metabolism of glucose is essential to maintaining nearly all physiological processes. Glucose acts as a signaling molecule by affecting AMPK activity, and recent studies suggest that glucose also regulates AMPK protein levels. In a cell culture model, AMPKα2 protein levels decreased dramatically due to increased ubiquitination and UPS-mediated degradation after long-term glucose exposure (59). In a screen for AMPK-binding proteins, WW domain-containing protein 1 (WWP1), a neural precursor cell expressed, developmentally down-regulated protein 4 (Nedd4)-like E3 ubiquitin ligase (87), was found to associate with AMPKα2 in high glucose conditions. Consistent with the hypothesis that WWP1 ubiquitinates and promotes the degradation of AMPK, WWP1 expression was required for the glucose-dependent decrease in AMPKα2 expression (59). However, additional experiments including in vitro ubiquitination assays are necessary to determine whether AMPKα2 is a substrate of WWP1. Furthermore, although hyperglycemic conditions appear to promote AMPK degradation, AMPKα2 ubiquitination decreased in cells cultured in serum from caloric-restricted mice (60), suggesting that caloric restriction stabilizes AMPK in combination with other endocrine factors (60). From a cellular energetics perspective, UPS activity is energetically expensive (88). Therefore, it is reasonable for AMPK to be spared from degradation during conditions of nutrient stress. It is not surprising that the E3 ligases that regulate AMPK stability, as well as other metabolically sensitive substrates, are regulated by glucose levels in an effort to prevent aberrant degradation during nutrient-depleted conditions.
Regulation of AMPK Via DUBs
In addition to enzymes that covalently decorate a substrate with ubiquitin, deubiquitinating enzymes (DUBs) also play an important role in regulating protein stability and degradation (89), and AMPK is no exception. The yeast homolog of AMPK, sucrose nonfermenting protein kinase 1 (Snf1), mediates the adaptation to energy stress (90). Snf1 is deubiquitinated by ubiquitin carboxy-terminal hydrolase 8 (Ubp8), a component of the Spt-Ada-Gcn5-acetyltransferase (SAGA) complex, a histone-modifying complex (91). This deubiquitination of Snf1 by Ubp8 increases Snf1 protein stability, phosphorylation of the catalytic Snf1 subunit, and Snf1-mediated phosphorylation of glucose repression protein Gal83, the homolog of AMPKβ (91). Deubiquitination may also be an important aspect of AMPK regulation. As detailed above, the AMPK-related kinases NUAK1 and MARK4 are polyubiquitinated with noncanonical ubiquitin chains. NUAK1 and MARK4 directly interact with the DUB USP9X (84, 92). Consistent with a role for ubiquitin in the regulation of AMPK, depletion of USP9X in cells increases the stability of AMPK (60), suggesting a model of regulation in which AMPK is stabilized by noncanonical ubiquitin chains that, depending on upstream signals, are removed by USP9X to allow destabilizing canonical chain modifications to AMPK, decreasing overall AMPK stability. These preliminary data on USP9X and AMPK warrant in-depth studies of the effects of USP9X and other DUBs on AMPK regulation.
Regulation of AMPK Through Small Ubiquitin-Like Modifier (SUMO)ylation
Like ubiquitin, SUMO is a small protein that “tags” a substrate protein through reversible covalent attachment to affect substrate activity or localization. Although SUMOylation does not directly lead to proteasome-mediated degradation, there is also evidence that modification by SUMO regulates AMPK activity and stability. For example, in yeast cultured under high glucose, the catalytic subunit of Snf1 was SUMOylated by methyl methanesulfonate-sensitivity protein 21, and this modification decreased Snf1 activity and promoted ubiquitination mediated by the synthetic lethal of unknown function 8 SUMO-targeted ubiquitin ligase (93, 94). In mammalian cells, AMPKβ2 (but not AMPKβ1) was modified with SUMO2 by protein inhibitor of activated STAT protein 4, and this modification increased activity of the AMPK α2β2γ1 complex (62). SUMOylation does not affect the protein stability of AMPKβ2 but does increase AMPKβ2 aggregation, and SUMOylation may compete with ubiquitination of AMPKβ2 (62).
It is clear that there are several UPS proteins that either directly or indirectly regulate AMPK. Given the central role of AMPK activation and activity in both metabolic homeostasis and numerous pathologies, understanding the precise mechanisms of AMPK protein regulation by ubiquitinating, deubiquitinating, and SUMOylating enzymes represents both a unique research niche and an opportunity to develop therapeutic strategies targeting AMPK.
Regulation of the UPS by AMPK
UPS-dependent protein degradation is an energy-consuming process, requiring upwards of 150 molecules of ATP per protein molecule (88). With the central role of AMPK in regulating ATP generation and the high ATP requirements of the UPS, it is easy to envision a regulatory link between the 2 systems. Consistent with this concept, experimental evidence suggests that AMPK does regulate components of the UPS.
AMPK Effects on Proteasome Activity
In addition to the effects of extracellular high glucose on AMPK stability (59), glucose also increases 26S proteasome activity. The glucose-dependent increase in proteasome activity appears to be dependent on a decrease AMPK activity (which occurs in high glucose conditions, discussed above), because either the addition of AICAR or of an exogenously expressed constitutively activated AMPK construct to human umbilical vein endothelial cells completely blocked glucose-induced increases in 26S activity (95). AICAR- or metformin-mediated proteasome inhibition appears to be dependent on AMPK catalytic activity; supportive of the AMPK-UPS link, blocking AMPK activity increased proteasome activity (95). Interestingly, cell culture models using isolated aortas, fibroblasts, and endothelial cells suggest that the exogenous expression of constitutively active AMPK had a relatively small effect on proteasome activity inhibition under basal conditions. However, loss of AMPK activity through genetic deletion or exogenous expression of a dominant negative AMPK construct led to a substantial increase in proteasome activity (95–97). Therefore, it should be taken in consideration that pharmacological manipulation of AMPK activity may affect 26S proteasome activity and proteolytic homoeostasis.
How does AMPK directly regulate proteasome function? A recent study proposed a mechanism in which increased AMPK activity inhibited the physical association between 19S and the catalytic 20S proteasome subunit, preventing aberrant proteasome activity, through AMPK-dependent association of the O-GlcNAcylating enzyme O-linked N-acetylglucosamine transferase (OGT) with the regulatory 19S subunit of the proteasome (98). When AMPK is inhibited or depleted, OGT-mediated O-GlyNAcylation of 19S decreases, a posttranslational modification of 19S that facilitates 26S proteasome assembly and activity (98). The specific mechanism by which AMPK alters the affinity of OGT for substrates is presently unclear. A second potential mechanism of AMPK-dependent proteasome regulation is the interaction between AMPK and proteasome 26S subunit non-ATPase regulatory subunit 11 (PSMD11), a component of the 19S subunit. PSMD11 is a substrate of AMPK. However, whether PSMD11 phosphorylation or other outcomes of the AMPK-PSMD11 interaction mediate proteasome activity remains unknown (99). These studies suggest that the negative correlation between AMPK and 26S proteasome activity is mediated by more than an indirect link of cellular energetic status, and uncovering the molecular underpinnings of the AMPK-proteasome relationship warrants further investigation.
Regulation of E3 Ubiquitin Ligases by AMPK
In addition to the effect of AMPK on 26S proteasome activity, AMPK appears to regulate the UPS through the regulation of specific E3 ubiquitin ligases.
S-phase kinase associated protein 2 (Skp2)
AMPK regulates the activity of the E3 ubiquitin ligase Skp2 through the nuclear factor-κB signaling pathway (100, 101). Vascular smooth muscle cells (VSMCs) from AMPKα2−/− mice, but not AMPKα1−/− mice, had higher proliferation rates compared with control cells, and the increase in proliferation in AMPKα2−/− VSMCs was associated with decreased levels of the cell cycle inhibitor p27Kip1 (100–102), a known substrate for the ubiquitin ligase activity of Skp2 (103). The authors determined that the decrease in p27Kip1 was due to increased nuclear factor-κB signaling in AMPKα2−/− cells, in part due to increased expression of Skp2, an effect that was recapitulated using the AMPK inhibitor compound C (100, 101). Skp2 up-regulation also coincided with decreased protein levels of E-cadherin (101). Given the potent effect of AMPKα2 deletion on VSMC proliferation and migration, the authors suggested that AMPKα2 prevents neointima formation (100, 101). Interestingly, AMPK purified from liver phosphorylated p27Kip1 and increased p27Kip1 stability (104), providing multiple nodes where AMPK can act on the regulation of the cell cycle.
AMPK and muscle ring finger 1 (MuRF1)/Atrogin-1
MuRF1 and Atrogin-1 are E3 ubiquitin ligases that are termed “atrogenes” given their role in mediating skeletal muscle atrophy by ubiquitinating myofibrillar proteins (105). Additionally, expression of either MuRF1 or Atrogin-1 is sufficient to inhibit cardiac hypertrophy induced by pressure overload (106–108). MuRF1 contains a RING domain that mediates ubiquitination (107), whereas Atrogin-1 contains an F-box domain that requires the association with the Skp, Cullin, F-box containing complex ubiquitin ligase complex to form a functional E3 ubiquitin ligase (109). In C2C12 myotubes, treatment with AICAR, 2-deoxyglucose, or metformin increased transcription of MuRF1 and Atrogin-1, in addition to the anticipated effect of increased AMPK phosphorylation (110, 111). Furthermore, the transcriptional response of the atrogenes was blocked with addition of compound C (110), suggesting that AMPK activation results in the transcriptional activation of MuRF1 and Atrogin-1. One intriguing candidate proposed to mediate the transcriptional effects of AMPK activation is the Forkhead box O (FoxO) family of transcription factors, including FoxO1 and FoxO3; the protein levels of these factors increased with AICAR stimulation in some models (111) and are known to regulate MuRF1 and Atrogin-1 transcription (112, 113). Interestingly, AICAR and other AMPK-activating compounds have produced contradictory effects on atrogene and FoxO expression in other models (114–116), suggesting that cell-type specificity plays a role in the AMPK-atrogene signaling axis. The effect of AMPK on FoxO transcription factors appears to be posttranslational, because AMPK can directly phosphorylate and activate FoxO3a at sites not targeted by the FoxO3a-inhibiting kinase Akt (117). AICAR also prevented the IGF-dependent accumulation of cytosolic Akt-phosphorylated FoxO3a by increasing AMPK-dependent FoxO3a phosphorylation (118). In C2C12 myotubes treated with combinations of AICAR and IGF, AICAR also increased MurF1 and Atrogin-1 mRNA and protein levels (118). Other work has shown that activation of AMPK activity through AICAR or nutrient deprivation in cardiomyocytes up-regulated MuRF1 and Atrogin-1 transcription through increased activity of the myocyte enhancer factor 2A (MEF-2) transcription factor (119). The authors observed an increase in total protein degradation involving both proteasomal and macroautophagy pathways that was dependent on both AMPK and MuRF-1 expression (119). 26S proteasome activity, however, was not measured in this context, and it is still unclear how AMPK affects MEF-2 transcriptional activity. Although there are many details within the AICAR-AMPK-FoxO/MEF-2-atrogene signaling axis that are currently unknown, the regulation and consequences of atrogene expression by AMPK is a pathway that warrants further examination (Figure 3A).
Figure 3.

Regulation of UPS components by AMPK. A, Activation of AMPK using AICAR in cell culture models leads to increased activity of the FoxO1/3 and MEF-2 transcription factors, which up-regulate the expression of the E3 ubiquitin ligase atrogenes MuRF1 and Atrogin-1 and subsequent degradation of atrogene substrates. B, AMPK affects the activity of the Laforin/Malin E3 ligase complex and regulation/degradation of the glycogen-associated protein PTG. AMPK phosphorylates Ser25 on Laforin, promoting the association between Laforin and Malin. AMPK also phosphorylates PTG, which decreases PTG protein stability by promoting recognition and canonical ubiquitination (red) by Malin. Malin polyubiquitinates Laforin under low glucose conditions, and Malin also polyubiquitinates AMPK with stabilizing, noncanonical K63-linked chains (yellow). C, AMPK phosphorylates and activates the E3 ubiquitin ligase Nedd4-2, which polyubiquitinates a variety of membrane-associated transporters resulting in transporter removal from the plasma membrane and the reduction of membrane transport activity.
AMPK and Laforin/Malin complex
Under high glucose conditions, glucose that is not metabolized is synthesized into long polysaccharide chains of glycogen, which serves as a storage mechanism until low-energy conditions return. AMPK regulates glycogen metabolism by the Laforin/Malin complex (Figure 3B). Laforin is a tyrosine phosphatase (120), and Malin is a RING domain-containing E3 ubiquitin ligase (121). Simultaneous mutations in these 2 proteins cause Lafora's progressive myoclonus epilepsy, a disease characterized by seizures, neurodegeneration, and early mortality (122). Laforin contains a carbohydrate binding domain that mediates the association with, and dephosphorylation of, complex carbohydrates and other glycogen-related substrates, including protein targeting to glycogen (PTG) (123–125). The genetic deletion of the Lafora gene in mice led to the emergence of Lafora bodies, which are insoluble glycogen deposits that accumulate in tissues that normally store little glycogen (124). Low glucose promoted the association of AMPK and Laforin, resulting in the phosphorylation of Laforin Ser25, which then promoted complex formation with Malin (126, 127). The importance of phosphorylation of Laforin at Ser25 and proper Laforin activity was highlighted by the association of mutations in Laforin Ser25 and Lafora disease (126). When Laforin and Malin are in complex, Laforin acts as a scaffold to recruit PTG into proximity with Malin (127); Malin subsequently canonically ubiquitinates PTG, leading to PTG degradation and preventing glycogen synthesis (128). AMPK also phosphorylates Ser8 and Ser268 of PTG, which also promotes canonical ubiquitination by the Laforin/Malin complex and subsequent degradation (129). An additional link between Malin and AMPK was established in a study that demonstrated noncanonical K63-linked polyubiquitination of AMPKβ mediated by Malin that appeared to stabilize AMPK holoenzyme protein levels (130), suggesting a feed-forward mechanism between AMPK and Malin to block glycogen synthesis. Incidentally, AMPK is ubiquitinated with noncanonical ubiquitin chains not linked through K48 or K63 by the ubiquitin ligase tripartite motif-containing protein 32 (131), which has high homology to Malin and targets cytoskeletal components, similar to MuRF1 (132). In addition to regulating proteins involved in glycogen synthesis, the Laforin/Malin complex also limited glucose availability by regulating the translocation of glucose transporters into the plasma membrane (133). Laforin levels increased with high glucose and were degraded by the proteasome under low glucose by Malin in an AMPK-dependent manner (133, 134), a system which allows Laforin to regulate abnormal glycogen phosphorylation during periods of high glucose availability. In summary, AMPK regulates multiple aspects of Laforin/Malin activity and glycogen metabolism by promoting the formation of the Laforin/Malin complex, phosphorylating Laforin to promote activity, phosphorylating PTG to promote ubiquitination and degradation mediated by the Laforin/Malin complex, and promoting degradation of Laforin by Malin during low-energy conditions (Figure 3B). This pathway demonstrates the multiple nodes by which AMPK influences UPS-dependent effects on cellular metabolism.
AMPK and Transporter Regulation
A third example of AMPK's direct effect on ubiquitin ligase activity is derived from numerous examples of AMPK-dependent degradation of sodium, potassium, calcium, and glutamate transporters by the ubiquitin ligase Nedd4-2. AMPK expression decreased the membrane expression and conductivity of the epithelial sodium transporter (135–137), neuronal glutamate transporters (138), potassium channels (139–143), and calcium channels (144). The degradation of these various transporters involved the AMPK-dependent phosphorylation of Nedd4-2, which in turn increased the affinity of Nedd4-2 for plasma membrane-inserted transporters; Nedd4-2 then canonically polyubiquitinated the transporters, promoting their removal from the plasma membrane in a mechanism first described in Ref. 136 and later found to be relevant to other channels regulated by Nedd4-2 (Figure 3C). The AMPK-Nedd4-2 relationship represents a negative feedback cycle, because ion or amino acid entry activates energy-consuming sodium-potassium ATP-coupled transporters or ATP-consuming contractile proteins. Incidentally, AMPK also inhibited the activity of some ion channels directly via phosphorylation, such as the cystic fibrosis transmembrane receptor (145, 146) and a variety of potassium channels (147, 148), whereas AMPK increased the channel activity of sodium-coupled glucose transporter to increase glucose availability and ATP generation (149). This discussion of channels regulated by AMPK is by no means exhaustive, and additional information can be found in excellent in-depth reviews (150, 151). However, it is clear that some of the AMPK-dependent effects on plasma membrane transporters clearly involve the UPS.
Conclusion, Future Directions, and Clinical Implications
AMPK is a therapeutic target in a wide variety of common diseases, including those associated with aging and/or imbalances in metabolism. Significant progress has been made in understanding the kinase cascades that promote AMPK activity, but the regulation of AMPK by proteins within the UPS is less understood. Furthermore, the regulation of AMPK by the UPS and counterregulation of various components of the UPS by AMPK forms the basis of an integrated system that links cellular metabolism and proteasome-dependent protein degradation. Glucose levels appear to impact the ubiquitination of both AMPK as well as other metabolically sensitive substrates in what seems to be a coordinated system to prevent aberrant degradation during nutrient-depleted conditions. Conversely, under nutrient-rich conditions, AMPK activity decreases along with the AMPK-dependent inhibition of the 26S proteasome, creating a permissive environment for proteasome activity.
The observations detailed throughout this review support a model whereby a key regulator of metabolism, AMPK, and multiple components of the UPS, including E3 ubiquitin ligases and the proteasome itself, coordinate to balance cellular energetics and protein degradation. However, there is still a lack of direct and targeted experiments that interrogate this relationship. The intention of this review is to promote awareness and collaboration between basic researchers studying metabolism and the UPS, so that the discrete mechanisms by which AMPK and UPS proteins interact can be elucidated. It is essential that claims made about ubiquitin ligase activities towards AMPK are verified through direct assay measurements, such as in vitro ubiquitination assays coupled with mass spectrometry to validate ubiquitination, identification of the residues targeted for ubiquitination, and determination of the type of ubiquitin chain added. In addition, measuring UPS activity in situations in which AMPK activity is experimentally modified will be essential for future studies that link metabolic and proteasome systems.
Targeting AMPK activity either through biguanidine drugs or salicylate is a validated clinical approach to treat insulin resistance and type 2 diabetes. Furthermore, retrospective analysis of clinical data support a relationship between these same drugs and a reduction in cancer risk, as recently reviewed (5). Proteasome inhibitors are approved to treat multiple myeloma, a subject of a well-written review and perspective (152). Likewise, studies on curcumin, a known proteasome inhibitor, improves insulin resistance in several rodent models (153–155) and reduced type 2 diabetes development in prediabetics (156), suggesting that selective proteasome inhibition could be used to treat type 2 diabetes. It is enticing to link the efficacy of some of these therapies to the emerging relationship between AMPK and the UPS, although a causal relationship in a clinical setting remains to be seen. Future studies both in the lab and in the clinic will hopefully further our understanding of the relationship between metabolic and proteolytic homeostasis and lead to new therapeutic options for cancer, neurological, and metabolic disorders.
Acknowledgments
This work was supported by the National Institutes of Health Grant R01-GM061728 and by Foundation Leducq.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AICAR
- 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- AMPK
- 5′-AMP-activated protein kinase
- BCA2
- breast cancer-associated gene 2
- BRSK2
- brain-selective kinase 2
- c-Cbl
- casitas B-lineage lymphoma protooncogene
- CHIP
- C terminus of Hsp70-interacting protein
- Cidea
- cell death-inducing DFF45-like effector A
- CoA
- coenzyme A
- CRBN
- cereblon
- DUB
- deubiquitinase
- FoxO
- Forkhead box O
- LKB1
- liver kinase B1
- MARK4
- microtubule affinity-regulating kinase 4
- MEF-2
- myocyte enhancer factor 2A
- MuRF1
- muscle ring finger 1
- Nedd4
- neural precursor cell expressed, developmentally down-regulated protein 4
- NUAK1
- nua (novel) kinase family 1
- OGT
- O-linked N-acetylglucosamine transferase
- PP2
- protein phosphatase 2
- PSMD11
- proteasome 26S subunit non-ATPase regulatory subunit 11
- PTG
- protein targeting to glycogen
- RING
- really interesting new gene family
- SAGA
- Spt-Ada-Gcn5-acetyltransferase
- SBD
- subunit binding domain
- Snf1
- sucrose nonfermenting protein kinase 1
- Skp2
- S-phase kinase associated protein 2
- SUMO
- small ubiquitin-like modifier
- Ubp8
- ubiquitin carboxy-terminal hydrolase 8
- UPS
- ubiquitin-proteasome system
- VSMC
- vascular smooth muscle cell
- WWP1
- WW domain-containing protein 1.
References
- 1. Beg ZH, Allmann DW, Gibson DM. Modulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity with cAMP and wth protein fractions of rat liver cytosol. Biochem Biophys Res Commun. 1973;54(4):1362–1369 [DOI] [PubMed] [Google Scholar]
- 2. Carlson CA, Kim KH. Regulation of hepatic acetyl coenzyme A carboxylase by phosphorylation and dephosphorylation. J Biol Chem. 1973;248(1):378–380 [PubMed] [Google Scholar]
- 3. Carling D, Clarke PR, Zammit VA, Hardie DG. Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur J Biochem. 1989;186(1–2):129–136 [DOI] [PubMed] [Google Scholar]
- 4. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hardie DG. AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes. 2013;62(7):2164–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res. 2012;111(6):800–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S. AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem. 2009;109(suppl 1):17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crute BE, Seefeld K, Gamble J, Kemp BE, Witters LA. Functional domains of the α1 catalytic subunit of the AMP-activated protein kinase. J Biol Chem. 1998;273(52):35347–35354 [DOI] [PubMed] [Google Scholar]
- 9. Salt I, Celler JW, Hawley SA, et al. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the α2 isoform. Biochem J. 1998;334(pt 1):177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hudson ER, Pan DA, James J, et al. A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr Biol. 2003;13(10):861–866 [DOI] [PubMed] [Google Scholar]
- 11. Thornton C, Snowden MA, Carling D. Identification of a novel AMP-activated protein kinase β subunit isoform that is highly expressed in skeletal muscle. J Biol Chem. 1998;273(20):12443–12450 [DOI] [PubMed] [Google Scholar]
- 12. Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci. 1997;22(1):12–13 [DOI] [PubMed] [Google Scholar]
- 13. Scott JW, Hawley SA, Green KA, et al. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113(2):274–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase γ-subunit isoforms and their role in AMP binding. Biochem J. 2000;346(pt 3):659–669 [PMC free article] [PubMed] [Google Scholar]
- 15. Li J, Coven DL, Miller EJ, Hu X, et al. Activation of AMPK α- and γ-isoform complexes in the intact ischemic rat heart. Am J Physiol Heart Circ Physiol. 2006;291(4):H1927–H1934 [DOI] [PubMed] [Google Scholar]
- 16. Mahlapuu M, Johansson C, Lindgren K, et al. Expression profiling of the γ-subunit isoforms of AMP-activated protein kinase suggests a major role for γ3 in white skeletal muscle. Am J Physiol Endocrinol Metab. 2004;286(2):E194–E200 [DOI] [PubMed] [Google Scholar]
- 17. Ferrer A, Caelles C, Massot N, Hegardt FG. Activation of rat liver cytosolic 3-hydroxy-3-methylglutaryl coenzyme A reductase kinase by adenosine 5′-monophosphate. Biochem Biophys Res Commun. 1985;132(2):497–504 [DOI] [PubMed] [Google Scholar]
- 18. Hawley SA, Davison M, Woods A, et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271(44):27879–27887 [DOI] [PubMed] [Google Scholar]
- 19. Shaw RJ, Kosmatka M, Bardeesy N, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101(10):3329–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2(1):9–19 [DOI] [PubMed] [Google Scholar]
- 21. Hawley SA, Selbert MA, Goldstein EG, Edelman AM, Carling D, Hardie DG. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem. 1995;270(45):27186–27191 [DOI] [PubMed] [Google Scholar]
- 22. Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281(35):25336–25343 [DOI] [PubMed] [Google Scholar]
- 23. Xie M, Zhang D, Dyck JR, et al. A pivotal role for endogenous TGF-β-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci USA. 2006;103(46):17378–17383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davies SP, Helps NR, Cohen PT, Hardie DG. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C α and native bovine protein phosphatase-2AC. FEBS Lett. 1995;377(3):421–425 [DOI] [PubMed] [Google Scholar]
- 25. Gowans GJ, Hawley SA, Ross FA, Hardie DG. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013;18(4):556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drew BG, Fidge NH, Gallon-Beaumier G, Kemp BE, Kingwell BA. High-density lipoprotein and apolipoprotein AI increase endothelial NO synthase activity by protein association and multisite phosphorylation. Proc Natl Acad Sci USA. 2004;101(18):6999–7004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Minokoshi Y, Kim YB, Peroni OD, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415(6869):339–343 [DOI] [PubMed] [Google Scholar]
- 28. Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8(11):1288–1295 [DOI] [PubMed] [Google Scholar]
- 29. Horman S, Vertommen D, Heath R, et al. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase α-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem. 2006;281(9):5335–5340 [DOI] [PubMed] [Google Scholar]
- 30. Gleason CE, Lu D, Witters LA, Newgard CB, Birnbaum MJ. The role of AMPK and mTOR in nutrient sensing in pancreatic β-cells. J Biol Chem. 2007;282(14):10341–10351 [DOI] [PubMed] [Google Scholar]
- 31. Steinberg GR, Michell BJ, van Denderen BJ, et al. Tumor necrosis factor α-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006;4(6):465–474 [DOI] [PubMed] [Google Scholar]
- 32. Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994;353(1):33–36 [DOI] [PubMed] [Google Scholar]
- 33. Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51(8):2420–2425 [DOI] [PubMed] [Google Scholar]
- 35. Foretz M, Hébrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120(7):2355–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494(7436):256–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fryer LG, Parbu-Patel A, Carling D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277(28):25226–25232 [DOI] [PubMed] [Google Scholar]
- 38. Nawrocki AR, Rajala MW, Tomas E, et al. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor γ agonists. J Biol Chem. 2006;281(5):2654–2660 [DOI] [PubMed] [Google Scholar]
- 39. Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89(3):1025–1078 [DOI] [PubMed] [Google Scholar]
- 40. Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408(3):297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Russell RR, 3rd, Bergeron R, Shulman GI, Young LH. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol. 1999;277(2 pt 2):H643–H649 [DOI] [PubMed] [Google Scholar]
- 42. Luiken JJ, Coort SL, Willems J, et al. Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes. 2003;52(7):1627–1634 [DOI] [PubMed] [Google Scholar]
- 43. Marsin AS, Bertrand L, Rider MH, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10(20):1247–1255 [DOI] [PubMed] [Google Scholar]
- 44. Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem. 1995;270(29):17513–17520 [DOI] [PubMed] [Google Scholar]
- 45. Samari HR, Seglen PO. Inhibition of hepatocytic autophagy by adenosine, aminoimidazole-4-carboxamide riboside, and N6-mercaptopurine riboside. Evidence for involvement of amp-activated protein kinase. J Biol Chem. 1998;273(37):23758–23763 [DOI] [PubMed] [Google Scholar]
- 46. Carling D, Hardie DG. The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim Biophys Acta. 1989;1012(1):81–86 [DOI] [PubMed] [Google Scholar]
- 47. Henin N, Vincent MF, Gruber HE, Van den Berghe G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J. 1995;9(7):541–546 [DOI] [PubMed] [Google Scholar]
- 48. Sim AT, Hardie DG. The low activity of acetyl-CoA carboxylase in basal and glucagon-stimulated hepatocytes is due to phosphorylation by the AMP-activated protein kinase and not cyclic AMP-dependent protein kinase. FEBS Lett. 1988;233(2):294–298 [DOI] [PubMed] [Google Scholar]
- 49. Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277(27):23977–23980 [DOI] [PubMed] [Google Scholar]
- 50. Horman S, Browne G, Krause U, et al. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol. 2002;12(16):1419–1423 [DOI] [PubMed] [Google Scholar]
- 51. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590 [DOI] [PubMed] [Google Scholar]
- 52. Banko MR, Allen JJ, Schaffer BE, et al. Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol Cell. 2011;44(6):878–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25(18):1895–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hardie DG, Ashford ML. AMPK: regulating energy balance at the cellular and whole body levels. Physiology (Bethesda). 2014;29(2):99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase–development of the energy sensor concept. J Physiol. 2006;574(pt 1):7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feng Z, Hu W, de Stanchina E, et al. The regulation of AMPK β1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67(7):3043–3053 [DOI] [PubMed] [Google Scholar]
- 57. Rock KL, Gramm C, Rothstein L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78(5):761–771 [DOI] [PubMed] [Google Scholar]
- 58. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229 [DOI] [PubMed] [Google Scholar]
- 59. Lee JO, Lee SK, Kim N, et al. E3 ubiquitin ligase, WWP1, interacts with AMPKα2 and down-regulates its expression in skeletal muscle C2C12 cells. J Biol Chem. 2013;288(7):4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang P, Zhang RY, Song J, et al. Loss of AMP-activated protein kinase-α2 impairs the insulin-sensitizing effect of calorie restriction in skeletal muscle. Diabetes. 2012;61(5):1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Qi J, Gong J, Zhao T, et al. Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. EMBO J. 2008;27(11):1537–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rubio T, Vernia S, Sanz P. Sumoylation of AMPKβ2 subunit enhances AMP-activated protein kinase activity. Mol Biol Cell. 2013;24(11):1801–1811, S1801–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang W, Lv N, Zhang S, et al. Cidea is an essential transcriptional coactivator regulating mammary gland secretion of milk lipids. Nat Med. 2012;18(2):235–243 [DOI] [PubMed] [Google Scholar]
- 64. Xu L, Zhou L, Li P. CIDE proteins and lipid metabolism. Arterioscler Thromb Vasc Biol. 2012;32(5):1094–1098 [DOI] [PubMed] [Google Scholar]
- 65. Zhou Z, Yon Toh S, Chen Z, et al. Cidea-deficient mice have lean phenotype and are resistant to obesity. Nat Genet. 2003;35(1):49–56 [DOI] [PubMed] [Google Scholar]
- 66. Dai Q, Zhang C, Wu Y, et al. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003;22(20):5446–5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Min JN, Whaley RA, Sharpless NE, Lockyer P, Portbury AL, Patterson C. CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol Cell Biol. 2008;28(12):4018–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schisler JC, Rubel CE, Zhang C, Lockyer P, Cyr DM, Patterson C. CHIP protects against cardiac pressure overload through regulation of AMPK. J Clin Invest. 2013;123(8):3588–3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gaude H, Aznar N, Delay A, et al. Molecular chaperone complexes with antagonizing activities regulate stability and activity of the tumor suppressor LKB1. Oncogene. 2012;31(12):1582–1591 [DOI] [PubMed] [Google Scholar]
- 70. Cordoba M, Rodriguez-Quiroga S, Gatto EM, Alurralde A, Kauffman MA. Ataxia plus myoclonus in a 23-year-old patient due to STUB1 mutations. Neurology. 2014 [DOI] [PubMed] [Google Scholar]
- 71. Depondt C, Donatello S, Simonis N, et al. Autosomal recessive cerebellar ataxia of adult onset due to STUB1 mutations. Neurology. 2014;82(19):1749–1750 [DOI] [PubMed] [Google Scholar]
- 72. Shi CH, Schisler JC, Rubel CE, et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet. 2014;23(4):1013–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shi Y, Wang J, Li JD, et al. Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS One. 2013;8(12):e81884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Synofzik M, Gonzalez MA, Lourenco CM, et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain. 2014;137(pt 1):69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Spasic MR, Callaerts P, Norga KK. AMP-activated protein kinase (AMPK) molecular crossroad for metabolic control and survival of neurons. Neuroscientist. 2009;15(4):309–316 [DOI] [PubMed] [Google Scholar]
- 76. Lupher ML, Jr, Andoniou CE, Bonita D, Miyake S, Band H. The c-Cbl oncoprotein. Int J Biochem Cell Biol. 1998;30(4):439–444 [DOI] [PubMed] [Google Scholar]
- 77. Saltiel AR, Pessin JE. Insulin signaling in microdomains of the plasma membrane. Traffic. 2003;4(11):711–716 [DOI] [PubMed] [Google Scholar]
- 78. Molero JC, Jensen TE, Withers PC, et al. c-Cbl-deficient mice have reduced adiposity, higher energy expenditure, and improved peripheral insulin action. J Clin Invest. 2004;114(9):1326–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Molero JC, Turner N, Thien CB, Langdon WY, James DE, Cooney GJ. Genetic ablation of the c-Cbl ubiquitin ligase domain results in increased energy expenditure and improved insulin action. Diabetes. 2006;55(12):3411–3417 [DOI] [PubMed] [Google Scholar]
- 80. Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345–1350 [DOI] [PubMed] [Google Scholar]
- 81. Lee KM, Jo S, Kim H, Lee J, Park CS. Functional modulation of AMP-activated protein kinase by cereblon. Biochim Biophys Acta. 2011;1813(3):448–455 [DOI] [PubMed] [Google Scholar]
- 82. Burger A, Amemiya Y, Kitching R, Seth AK. Novel RING E3 ubiquitin ligases in breast cancer. Neoplasia. 2006;8(8):689–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Buac D, Kona FR, Seth AK, Dou QP. Regulation of metformin response by breast cancer associated gene 2. Neoplasia. 2013;15(12):1379–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Al-Hakim AK, Zagorska A, Chapman L, Deak M, Peggie M, Alessi DR. Control of AMPK-related kinases by USP9X and atypical Lys(29)/Lys(33)-linked polyubiquitin chains. Biochem J. 2008;411(2):249–260 [DOI] [PubMed] [Google Scholar]
- 85. Li R, Wan B, Zhou J, et al. APC/C(Cdh1) targets brain-specific kinase 2 (BRSK2) for degradation via the ubiquitin-proteasome pathway. PLoS One. 2012;7(9):e45932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhou J, Wan B, Li R, et al. Jab1 interacts with brain-specific kinase 2 (BRSK2) and promotes its degradation in the ubiquitin-proteasome pathway. Biochem Biophys Res Commun. 2012;422(4):647–652 [DOI] [PubMed] [Google Scholar]
- 87. Wood JD, Yuan J, Margolis RL, et al. Atrophin-1, the DRPLA gene product, interacts with two families of WW domain-containing proteins. Mol Cell Neurosci. 1998;11(3):149–160 [DOI] [PubMed] [Google Scholar]
- 88. Peth A, Nathan JA, Goldberg AL. The ATP costs and time required to degrade ubiquitinated proteins by the 26 S proteasome. J Biol Chem. 2013;288(40):29215–29222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Clague MJ, Urbé S. Ubiquitin: same molecule, different degradation pathways. Cell. 2010;143(5):682–685 [DOI] [PubMed] [Google Scholar]
- 90. Hedbacker K, Carlson M. SNF1/AMPK pathways in yeast. Front Biosci. 2008;13:2408–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wilson MA, Koutelou E, Hirsch C, et al. Ubp8 and SAGA regulate Snf1 AMP kinase activity. Mol Cell Biol. 2011;31(15):3126–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Al-Hakim AK, Goransson O, Deak M, et al. 14-3-3 cooperates with LKB1 to regulate the activity and localization of QSK and SIK. J Cell Sci. 2005;118(pt 23):5661–5673 [DOI] [PubMed] [Google Scholar]
- 93. Simpson-Lavy KJ, Johnston M. SUMOylation regulates the SNF1 protein kinase. Proc Natl Acad Sci USA. 2013;110(43):17432–17437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Uzunova K, Göttsche K, Miteva M, et al. Ubiquitin-dependent proteolytic control of SUMO conjugates. J Biol Chem. 2007;282(47):34167–34175 [DOI] [PubMed] [Google Scholar]
- 95. Wang S, Xu J, Song P, Viollet B, Zou MH. In vivo activation of AMP-activated protein kinase attenuates diabetes-enhanced degradation of GTP cyclohydrolase I. Diabetes. 2009;58(8):1893–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Viana R, Aguado C, Esteban I, et al. Role of AMP-activated protein kinase in autophagy and proteasome function. Biochem Biophys Res Commun. 2008;369(3):964–968 [DOI] [PubMed] [Google Scholar]
- 97. Wang S, Zhang M, Liang B, et al. AMPKα2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes. Circ Res. 2010;106(6):1117–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Xu J, Wang S, Viollet B, Zou MH. Regulation of the proteasome by AMPK in endothelial cells: the role of O-GlcNAc transferase (OGT). PLoS One. 2012;7(5):e36717. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 99. Moreno D, Viana R, Sanz P. Two-hybrid analysis identifies PSMD11, a non-ATPase subunit of the proteasome, as a novel interaction partner of AMP-activated protein kinase. Int J Biochem Cell Biol. 2009;41(12):2431–2439 [DOI] [PubMed] [Google Scholar]
- 100. Song P, Wang S, He C, Liang B, Viollet B, Zou MH. AMPKα2 deletion exacerbates neointima formation by upregulating Skp2 in vascular smooth muscle cells. Circ Res. 2011;109(11):1230–1239 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 101. Song P, Zhou Y, Coughlan KA, et al. Adenosine monophosphate-activated protein kinase-α2 deficiency promotes vascular smooth muscle cell migration via S-phase kinase-associated protein 2 upregulation and E-cadherin downregulation. Arterioscler Thromb Vasc Biol. 2013;33(12):2800–2809 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 102. Polyak K, Kato JY, Solomon MJ, et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-β and contact inhibition to cell cycle arrest. Genes Dev. 1994;8(1):9–22 [DOI] [PubMed] [Google Scholar]
- 103. Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1(4):193–199 [DOI] [PubMed] [Google Scholar]
- 104. Short JD, Dere R, Houston KD, et al. AMPK-mediated phosphorylation of murine p27 at T197 promotes binding of 14-3-3 proteins and increases p27 stability. Mol Carcinog. 2010;49(5):429–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bodine SC, Latres E, Baumhueter S, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294(5547):1704–1708 [DOI] [PubMed] [Google Scholar]
- 106. Arya R, Kedar V, Hwang JR, et al. Muscle ring finger protein-1 inhibits PKC[ϵ] activation and prevents cardiomyocyte hypertrophy. J Cell Biol. 2004;167(6):1147–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Li HH, Kedar V, Zhang C, et al. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114(8):1058–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res. 2007;100(4):456–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA. 2001;98(25):14440–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Krawiec BJ, Nystrom GJ, Frost RA, Jefferson LS, Lang CH. AMP-activated protein kinase agonists increase mRNA content of the muscle-specific ubiquitin ligases MAFbx and MuRF1 in C2C12 cells. Am J Physiol Endocrinol Metab. 2007;292(6):E1555–E1567 [DOI] [PubMed] [Google Scholar]
- 111. Nakashima K, Yakabe Y. AMPK activation stimulates myofibrillar protein degradation and expression of atrophy-related ubiquitin ligases by increasing FOXO transcription factors in C2C12 myotubes. Biosci Biotechnol Biochem. 2007;71(7):1650–1656 [DOI] [PubMed] [Google Scholar]
- 112. Sandri M, Sandri C, Gilbert A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117(3):399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Stitt TN, Drujan D, Clarke BA, et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14(3):395–403 [DOI] [PubMed] [Google Scholar]
- 114. Festuccia WT, Laplante M, Brûlé S, et al. Rosiglitazone-induced heart remodelling is associated with enhanced turnover of myofibrillar protein and mTOR activation. J Mol Cell Cardiol. 2009;47(1):85–95 [DOI] [PubMed] [Google Scholar]
- 115. Nystrom GJ, Lang CH. Sepsis and AMPK activation by AICAR differentially regulate FoxO-1, -3 and -4 mRNA in striated muscle. Int J Clin Exp Med. 2008;1(1):50–63 [PMC free article] [PubMed] [Google Scholar]
- 116. Tabony AM, Yoshida T, Galvez S, et al. Angiotensin II upregulates protein phosphatase 2Cα and inhibits AMP-activated protein kinase signaling and energy balance leading to skeletal muscle wasting. Hypertension. 2011;58(4):643–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Greer EL, Oskoui PR, Banko MR, et al. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282(41):30107–30119 [DOI] [PubMed] [Google Scholar]
- 118. Tong JF, Yan X, Zhu MJ, Du M. AMP-activated protein kinase enhances the expression of muscle-specific ubiquitin ligases despite its activation of IGF-1/Akt signaling in C2C12 myotubes. J Cell Biochem. 2009;108(2):458–468 [DOI] [PubMed] [Google Scholar]
- 119. Baskin KK, Taegtmeyer H. AMP-activated protein kinase regulates E3 ligases in rodent heart. Circ Res. 2011;109(10):1153–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Minassian BA, Lee JR, Herbrick JA, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20(2):171–174 [DOI] [PubMed] [Google Scholar]
- 121. Chan EM, Young EJ, Ianzano L, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35(2):125–127 [DOI] [PubMed] [Google Scholar]
- 122. Girard JM, Turnbull J, Ramachandran N, Minassian BA. Progressive myoclonus epilepsy. Handb Clin Neurol. 2013;113:1731–1736 [DOI] [PubMed] [Google Scholar]
- 123. Fernández-Sánchez ME, Criado-García O, Heath KE, et al. Laforin, the dual-phosphatase responsible for Lafora disease, interacts with R5 (PTG), a regulatory subunit of protein phosphatase-1 that enhances glycogen accumulation. Hum Mol Genet. 2003;12(23):3161–3171 [DOI] [PubMed] [Google Scholar]
- 124. Tagliabracci VS, Turnbull J, Wang W, et al. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci USA. 2007;104(49):19262–19266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wang J, Stuckey JA, Wishart MJ, Dixon JE. A unique carbohydrate binding domain targets the lafora disease phosphatase to glycogen. J Biol Chem. 2002;277(4):2377–2380 [DOI] [PubMed] [Google Scholar]
- 126. Romá-Mateo C, Solaz-Fuster Mdel C, Gimeno-Alcañiz JV, et al. Laforin, a dual-specificity phosphatase involved in Lafora disease, is phosphorylated at Ser25 by AMP-activated protein kinase. Biochem J. 2011;439(2):265–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Solaz-Fuster MC, Gimeno-Alcañiz JV, Ros S, et al. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum Mol Genet. 2008;17(5):667–678 [DOI] [PubMed] [Google Scholar]
- 128. Worby CA, Gentry MS, Dixon JE. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J Biol Chem. 2008;283(7):4069–4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Vernia S, Solaz-Fuster MC, Gimeno-Alcañiz JV, et al. AMP-activated protein kinase phosphorylates R5/PTG, the glycogen targeting subunit of the R5/PTG-protein phosphatase 1 holoenzyme, and accelerates its down-regulation by the laforin-malin complex. J Biol Chem. 2009;284(13):8247–8255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Moreno D, Towler MC, Hardie DG, Knecht E, Sanz P. The laforin-malin complex, involved in Lafora disease, promotes the incorporation of K63-linked ubiquitin chains into AMP-activated protein kinase β subunits. Mol Biol Cell. 2010;21(15):2578–2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Roma-Mateo C, Moreno D, Vernia S, et al. Lafora disease E3-ubiquitin ligase malin is related to TRIM32 at both the phylogenetic and functional level. BMC Evol Biol. 2011;11:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Cohen S, Zhai B, Gygi SP, Goldberg AL. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J Cell Biol. 2012;198(4):575–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Gentry MS, Worby CA, Dixon JE. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci USA. 2005;102(24):8501–8506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Singh PK, Singh S, Ganesh S. The laforin-malin complex negatively regulates glycogen synthesis by modulating cellular glucose uptake via glucose transporters. Mol Cell Biol. 2012;32(3):652–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Almaça J, Kongsuphol P, Hieke B, et al. AMPK controls epithelial Na(+) channels through Nedd4-2 and causes an epithelial phenotype when mutated. Pflugers Arch. 2009;458(4):713–721 [DOI] [PubMed] [Google Scholar]
- 136. Bhalla V, Oyster NM, Fitch AC, et al. AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J Biol Chem. 2006;281(36):26159–26169 [DOI] [PubMed] [Google Scholar]
- 137. Carattino MD, Edinger RS, Grieser HJ, et al. Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J Biol Chem. 2005;280(18):17608–17616 [DOI] [PubMed] [Google Scholar]
- 138. Sopjani M, Alesutan I, Dërmaku-Sopjani M, et al. Down-regulation of Na+-coupled glutamate transporter EAAT3 and EAAT4 by AMP-activated protein kinase. J Neurochem. 2010;113(6):1426–1435 [DOI] [PubMed] [Google Scholar]
- 139. Alesutan I, Föller M, Sopjani M, et al. Inhibition of the heterotetrameric K+ channel KCNQ1/KCNE1 by the AMP-activated protein kinase. Mol Membr Biol. 2011;28(2):79–89 [DOI] [PubMed] [Google Scholar]
- 140. Alesutan I, Munoz C, Sopjani M, et al. Inhibition of Kir2.1 (KCNJ2) by the AMP-activated protein kinase. Biochem Biophys Res Commun. 2011;408(4):505–510 [DOI] [PubMed] [Google Scholar]
- 141. Alzamora R, Gong F, Rondanino C, et al. AMP-activated protein kinase inhibits KCNQ1 channels through regulation of the ubiquitin ligase Nedd4-2 in renal epithelial cells. Am J Physiol Renal Physiol. 2010;299(6):F1308–F1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Andersen MN, Krzystanek K, Jespersen T, Olesen SP, Rasmussen HB. AMP-activated protein kinase downregulates Kv7.1 cell surface expression. Traffic. 2012;13(1):143–156 [DOI] [PubMed] [Google Scholar]
- 143. Mia S, Munoz C, Pakladok T, et al. Downregulation of Kv1.5 K channels by the AMP-activated protein kinase. Cell Physiol Biochem. 2012;30(4):1039–1050 [DOI] [PubMed] [Google Scholar]
- 144. Lang F, Eylenstein A, Shumilina E. Regulation of Orai1/STIM1 by the kinases SGK1 and AMPK. Cell Calcium. 2012;52(5):347–354 [DOI] [PubMed] [Google Scholar]
- 145. King JD, Jr, Fitch AC, Lee JK, et al. AMP-activated protein kinase phosphorylation of the R domain inhibits PKA stimulation of CFTR. Am J Physiol Cell Physiol. 2009;297(1):C94–C101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Kongsuphol P, Cassidy D, Hieke B, et al. Mechanistic insight into control of CFTR by AMPK. J Biol Chem. 2009;284(9):5645–5653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Klein H, Garneau L, Trinh NT, et al. Inhibition of the KCa3.1 channels by AMP-activated protein kinase in human airway epithelial cells. Am J Physiol Cell Physiol. 2009;296(2):C285–C295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Wyatt CN, Mustard KJ, Pearson SA, et al. AMP-activated protein kinase mediates carotid body excitation by hypoxia. J Biol Chem. 2007;282(11):8092–8098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Sopjani M, Bhavsar SK, Fraser S, Kemp BE, Foller M, Lang F. Regulation of Na+-coupled glucose carrier SGLT1 by AMP-activated protein kinase. Mol Membr Biol. 2010;27(2–3):137–144 [DOI] [PubMed] [Google Scholar]
- 150. Andersen MN, Rasmussen HB. AMPK: a regulator of ion channels. Commun Integr Biol. 2012;5(5):480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Lang F, Foller M. Regulation of ion channels and transporters by AMP-activated kinase (AMPK). Channels (Austin). 2014;8(1):20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011;11(3):239–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Na LX, Zhang YL, Li Y, et al. Curcumin improves insulin resistance in skeletal muscle of rats. Nutr Metab Cardiovasc Dis. 2011;21(7):526–533 [DOI] [PubMed] [Google Scholar]
- 154. Shao W, Yu Z, Chiang Y, et al. Curcumin prevents high fat diet induced insulin resistance and obesity via attenuating lipogenesis in liver and inflammatory pathway in adipocytes. PLoS One. 2012;7(1):e28784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Weisberg SP, Leibel R, Tortoriello DV. Dietary curcumin significantly improves obesity-associated inflammation and diabetes in mouse models of diabesity. Endocrinology. 2008;149(7):3549–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Chuengsamarn S, Rattanamongkolgul S, Luechapudiporn R, Phisalaphong C, Jirawatnotai S. Curcumin extract for prevention of type 2 diabetes. Diabetes Care. 2012;35(11):2121–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Stapleton D, Mitchelhill KI, Gao G, et al. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996;271(2):611–614 [DOI] [PubMed] [Google Scholar]