

Abstract
1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methane (C-DIM) compounds exhibit antineoplastic activity in multiple cancer cell lines and the p-hydroxyphenyl analog (DIM-C-pPhOH) inactivates nuclear receptor 4A1 (NR4A1) in lung and pancreatic cancer cell lines. Using a series of 14 different p-substituted phenyl C-DIMs, we show that several compounds including DIM-C-pPhOH directly interacted with the ligand binding domain of NR4A1. Computational-based molecular modeling studies showed high-affinity interactions of DIM-C-pPhOH and related compounds within the ligand binding pocket of NR4A1, and these same compounds decreased NR4A1-dependent transactivation in colon cancer cells transfected with a construct containing 3 tandem Nur77 binding response elements linked to a luciferase reporter gene. Moreover, we also show that knockdown of NR4A1 by RNA interference (small interfering NR4A1) or treatment with DIM-C-pPhOH and related compounds decreased colon cancer cell growth, induced apoptosis, decreased expression of survivin and other Sp-regulated genes, and inhibited mammalian target of rapamycin signaling. Thus, C-DIMs such as DIM-C-pPhOH directly bind NR4A1 and are NR4A1 antagonists in colon cancer cells, and their antineoplastic activity is due, in part, to their interactions with nuclear NR4A1.
The nuclear receptor (NR) superfamily are critical regulators of homeostasis at all stages of development, and many of these receptors such as steroid hormone and retinoid receptors are important drug targets for treating multiple diseases including cancer (1, 2). The 48 human NRs have been divided into 3 main groups, including the endocrine receptors, adopted orphan receptors, and orphan receptors, and endogenous ligands have been characterized only for the former 2 groups of receptors. The 3 members of the NR4A orphan receptor subfamily (3, 4) include NR4A1 (Nur77, TR3, NGFI-B), NR4A2 (Nurr1), and NR4A3 (Nor1), which were initially identified as immediate-early response genes induced by nerve growth factor in PC12 cells (5). The well-conserved DNA binding and C-terminal ligand binding domains (LBDs) of the NR4A receptors exhibit ∼91% to 95% and ∼60% homology, respectively, whereas their N-terminal A/B domains are highly divergent (6–8). NR4A receptors are induced by multiple stimuli/stressors and play essential roles in metabolic processes, inflammation, vascular function, steroidogenesis, and the central nervous system (3, 4). NR4A1 is also overexpressed in multiple tumors and cancer cell lines (9–11). In colon cancer patients, our initial studies showed that high NR4A1 expression was observed in 60% of colon tumors, whereas only 10% of normal colon tissue exhibited high staining (12), and overexpression of NR4A1 in colon tumors has been confirmed in other studies (13, 14). RNA interference (RNAi) and antisense oligonucleotides have been used to investigate the role of NR4A1 in cancer cells, and in solid tumors, NR4A1 exhibits pro-oncogenic activity and enhances both cancer cell proliferation and survival (13, 15–21), whereas results from NR4A1/NR4A3-knockout mouse models suggest that NR4A is a tumor suppressor for acute myelocytic leukemia (22).
The anticancer activity of several apoptosis-inducing agents is due to induced expression and nuclear export of NR4A1, and NR4A1 interactions with bcl-2 results in a proapoptotic complex that decreases mitochondrial membrane potential and activates stress responses (9, 23–25). Research in this laboratory demonstrated that among a series of synthetic 1,1-bis(3′-indolyl)-1-(substituted phenyl)methane analogs (C-DIMs), a select group of these compounds activated or inactivated nuclear NR4A1-dependent transactivation in cancer cell lines and did not induce nuclear export of NR4A1 (15, 16, 26, 27). For example, the p-hydroxyphenyl analog [1,1-bis(3′-indolyl)-1-(p-hydroxyphenyl)methane (DIM-C-pPhOH)] inhibits NR4A1-dependent transactivation and exhibits antineoplastic activities and effects on gene expression similar to that observed in pancreatic cells after knockdown of NR4A1 (by RNAi) (15, 16).
Cytosporone B (Csn-B) and structural analogs were first identified as NR4A1 ligands, and these compounds induce NR4A1-dependent transactivation but also induce expression of NR4A1 and nuclear export of the receptor to mitochondria (28, 29). In addition, ethyl 2-[2,3,4-trimethoxy-6-(1-octanoyl)phenyl]acetate (30) and 1-(3,4,5-trihydroxypenyl)nonan-1-one (31) also bind NR4A1 and modulate functions of the receptor that results in therapeutic applications. In this study, we show structure-dependent binding of C-DIMs to the LBD of NR4A1 and demonstrate that selected compounds exhibit NR4A1 antagonist activity and inactivate nuclear NR4A1 in colon cancer cell lines and exhibit antineoplastic activities similar to that observed after receptor knockdown.
Materials and Methods
Fluorescence quenching and circular dichroism assays
Fluorescence spectra were obtained as previously described (30). Briefly, His-LBD and glutathione S-transferase (GST) were expressed in Escherichia coli strain BL21, purified, and dialyzed against PBS (pH 7.4). For analyzing the interactions between the protein and compounds, 5μM proteins were incubated with various concentrations of compounds, and the fluorescence quenching was monitored at 25°C with a slit width of 5 nm for excitation and a slit width of 2.5 nm for emission. The excitation wavelength was 280 nm, and the emission spectra were recorded from 285 to 410 nm. To estimate the binding affinity, the fluorescence intensities at 334 nm with increasing concentrations of quencher were measured, GST was used as the inner filter controls, and the Kd values were calculated. The circular dichroism (CD) spectroscopy assay was used to determine the DIM-C-pPhOH–induced conformational changes in the His-LBD and was carried out essentially as previously described (28–31). Mutation of the NR4A1-LBD (H516W) was also carried out (28–31) and used in the fluorescence binding assay.
Cell lines and plasmids
RKO and SW480 human colon cancer cell lines were obtained from the American Type Culture Collection and maintained as previously described (27). The Flag-tagged full-length FLAG-NR4A1 and mutant FLAG-NR4A1(A–D) and FLAG-NR4A1(C–F) expression plasmids were constructed by inserting PCR-amplified full-length NR4A1 (amino acids 1–599) into the EcoRI/BamHI site and C-terminal NR4A1 (amino acids 67–599) and N-terminal NR4A1 (amino acids 1–354) into the EcoRI/KpmI site of p3XFLAG-CMV-10 expression vector (Sigma-Aldrich). NBREx3-luc was generously provided by Dr Jacques Drouin (University of Montreal, Quebec, Canada). All other plasmids used in this study were previously described (15, 16).
Reagents, antibodies, and Western blot analysis
NR4A1 antibody was purchased from Abcam, and all other antibodies were purchased from Cell Signaling Technology. The p-substituted phenyl C-DIMs were synthesized and purified in this laboratory as previously described (32). The N-methyl C-DIM analogs were prepared by condensing N-methylindole (Sigma-Aldrich) with the corresponding p-substituted benzaldehyde and the ortho- and meta-substituted isomers were prepared from condensing indole plus the corresponding substituted benzaldehydes as described (32). Purities were >95% to 98% as determined by gas chromatography mass spectometry. Reporter lysis buffer, luciferase reagent, and β-galactosidase reagent were supplied by Promega. Western blot analysis was carried out as previously described (15, 16).
Transfection, small interfering RNA oligonucleotides, cell proliferation assay, and reporter gene assay
Cells were transfected with 100nM of each small interfering RNA (siRNA) duplex using LipofectAMINE 2000 reagent (Invitrogen) following the manufacturer's protocol. The sequences of siNR4A1 oligonucleotides used were 5′-CAG UCC AGC CAU GCU CCU C dTdT-3′. As a negative control, a nonspecific scrambled siRNA (siScr) oligonucleotide was used (QIAGEN), and all other siRNAs were purchased from Santa Cruz Biotechnology. Cell proliferation and reporter gene assays were performed as previously described (15, 16).
Computation-based molecular modeling
All molecular modeling studies were conducted using Accelrys Discovery Studio version 3.5 (Accelrys Software, Inc; http://accelrys.com). The crystal structure coordinates for rat orphan nuclear receptor NR4A1 (33) was downloaded from the Protein Data Bank (http://www.pdb.org; PDB ID 1YJE). The crystal structure was subjected to implicit solvent-based energy minimization using the conjugate gradient minimization protocol (34) with a CHARMm force field (35) and the generalized Born implicit solvent model with simple switching (36) to an root mean square convergence of <0.001 kcal/mol before use in the modeling studies. The flexible docking algorithm (37) was used to predict the binding orientation of the 14 compounds within the ligand binding site of NR4A1. Protein-ligand complexes underwent energy minimization in situ (10 000 iterations of the conjugate gradient protocol) before calculating the predicted binding energies using the generalized Born implicit solvent model with simple switching (36).
Statistical analysis
Statistical significance of differences were determined by ANOVA with Dunnett's post hoc test. The results are expressed as means with error bars representing 95% confidence intervals for 3 experiments for each group unless otherwise indicated, and a P value < .05 was considered statistically significant. Correlation between predicted binding energy and in vitro Kd was determined by calculating the Pearson's correlation coefficient.
Results
C-DIM binding and interactions with NR4A1
A panel of 14 p-substituted phenyl C-DIM analogs (Figure 1A and Table 1) were used to investigate their binding to the LBD of NR4A1 using a fluorescence assay as previously described (28–31). Figure 1, B–D, illustrates the binding curves for the 8 different C-DIMs that bound the LBD, including the para trifluoromethyl (CF3), bromo (Br), unsubstituted (H), hydroxyl (OH), cyano (CN), chloro (Cl), iodo (I), and carboxymethyl (CO2Me) analogs. KD values for these compounds ranged from 0.1μM to 0.74μM (Table 1). Binding was not observed for the fluoro (F), t-butyl (tBu), methoxy (OCH3), and methyl (CH3) compounds; the phenyl (C6H5) analog bound to GST, and the dimethylamino (N[CH3]2) compound gave abnormal fluorescent curves that interfered with the assay (data not shown). In addition, one of the most active ligands (DIM-C-pPhOH) also induced conformational changes as determined by CD spectroscopy (Figure 1E). These results demonstrate direct binding of C-DIMs with the LBD of NR4A1, and this was further investigated by molecular modeling.
Figure 1.

C-DIM structure and the receptor binding KD values. A, Composite structure of C-DIMs (DIM-C-pPh-X). B–D, KD values for binding to the LBD of NR4A1 using a fluorescent binding assay as outlined in Materials and Methods. E, CD spectroscopy. CD spectroscopy was used to determine ligand (DIM-C-pPhOH)-induced conformational changes essentially as described previously (28–31).
Table 1.
NR4A1 Receptor Binding Affinities and Binding Energies of C-DIM Compounds
| Kd Value, μM | Binding Energies, kcal/mol | |
|---|---|---|
| DIM-C-pPhCF3 | 0.1 | −54.3 |
| DIM-C-pPhBr | 0.41 | −52.1 |
| DIM-C-pPhFa | NB | −53.8 |
| DIM-C-pPht-Bua | NB | −53 |
| DIM-C-pPhOCH3a | NB | −81.5 |
| DIM-C-pPhN(CH3)2b | −48.7 | |
| DIM-C-pPhH | 0.71 | −55.9 |
| DIM-C-pPhOH | 0.11 | −86.2 |
| DIM-C-pPhC6H5c | −43.5 | |
| DIM-C-pPhCN | 0.26 | −75.3 |
| DIM-C-pPhCH3a | NB | −52.2 |
| DIM-C-pPhCl | 0.48 | −52.8 |
| DIM-C-pPhl | 0.13 | −76.9 |
| DIM-C-pPhCO2Me | 0.25 | −58.0 |
Net binding was not observed (NB).
More than one fluorescent binding site; gave variable data.
Compound also binds GST.
Previous crystallographic analysis of NR4A1 ligand binding has established that there are at least 2 separate and distinct ligand binding sites on different faces of the NR4A1 LBD (30). These 2 sites correspond to the coactivator binding site (Figure 2A, left) and the ligand binding site (Figure 2A, right) of the classical NRs. Initial docking of a subset of the 14 C-DIM compounds with the LBD of NR4A1 returned very few potential binding orientations and scoring analysis, indicating that the compounds had very low affinity for the coactivator site, suggesting a low probability that the C-DIM analogs bind in this region (data not shown). Therefore, we concentrated our efforts on docking each of the 14 compounds into the ligand binding site. Our results indicate that the NR4A1 ligand binding pocket is capable of accommodating all 14 compounds, and the modeling studies predict that the majority bind in an orientation similar to that observed for the p-hydroxyphenyl (DIM-C-pPhOH) compound (Figure 2B). This binding involves hydrogen bond interactions with Glu445 and His516 and π-cation interactions with Arg515 (Figure 2C). The exceptions were the unsubstituted (C-DIM 7) and the alkyl (C-DIMs 4 and 11), dimethylamine (C-DIM 6), and phenyl (C-DIM 9) substituted compounds, which were predicted to adopt a flipped binding orientation with the phenyl substituent pointing up and away from the binding pocket (data not shown). Despite similar predicted binding orientations, there was a range of predicted binding energies among the various compounds (Table 1) that generally correlated with the observed ligand binding in vitro (Pearson's r = 0.6467, P = .0415 (1-tailed), P = .0830 (2-tailed). We also investigated the binding of DIM-C-pPhOH to the NR4A1 LBD mutated in His516 using the fluorescence binding assay and observed no change in fluorescence, thus confirming the importance of this amino acid for binding DIM-C-pPhOH (Figure 2D).
Figure 2.
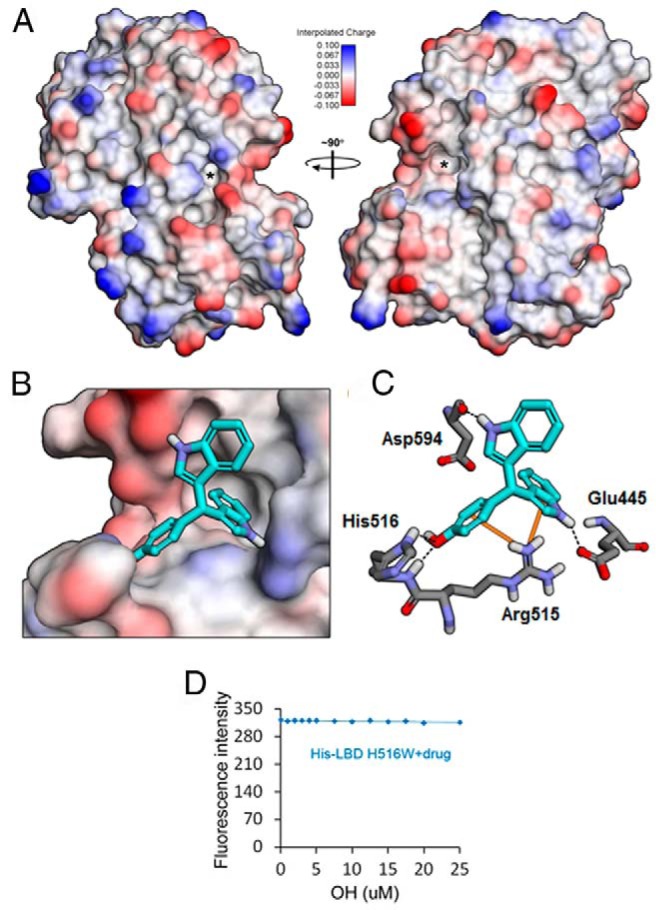
Predicted interactions between NR4A1 and DIM-C-pPhOH. A, Molecular surface representation of the crystal structure of the orphan nuclear receptor NR4A1 (PDB ID 1YJE) colored by interpolated charge from positive (blue) to neutral (white) to negative (red). Asterisks indicate the locations of 2 different potential ligand binding sites equivalent to the coactivator (left panel) and ligand (right panel) binding bites of classical nuclear receptors. B, Predicted binding orientation of DIM-C-pPhOH (C-DIM 8) within the ligand binding site. C, Specific nonbonded interactions between C-DIM 8 (cyan) and the residues of NR4A1 (gray). Dashed lines indicate predicted hydrogen bonds, and solid orange lines indicate predicted π interactions. D, Binding of DIM-C-pPhOH to mutant NR4A1-LD(H516W). The mutant NR4A1-LBD(H516W) protein was incubated with DIM-C-pPhOH, and receptor binding was determined as outlined in Materials and Methods.
C-DIMS inhibit NR4A1-dependent transactivation
The effects of C-DIMs on NR4A1-dependent transactivation were initially investigated in RKO cells transfected with NBRE3-luc and NuRE3-luc constructs containing 3 binding sites for NR4A1 monomer and homodimer, respectively (38). Basal activity was low for both constructs but significantly enhanced by cotransfection with a FLAG-TR3 expression plasmid (Supplemental Figure 1A); Figure 3A summarizes the effects of the p-substituted phenyl C-DIMs on luciferase activity in RKO cells transfected with NBRE3-luc. Results of this assay show that most of the compounds significantly inhibit transactivation, and the effects of the p-substituted C6H5 and CH3 analogs were minimal. Similar results were observed in cells transfected with the NuRE3-luc construct (Supplemental Figure 1B).
Figure 3.

C-DIMs and NR4A1-dependent transactivation. A, Activation of an NBRE3-luc. RKO cells were transfected with FLAG-NR4A1 (38) and NBRE3-luc and treated with DMSO or different concentrations of 14 p-substituted phenyl C-DIMs, and luciferase activity was determined as described in Materials and Methods. B, Cells were transfected as outlined in A and treated with ortho- and meta-substituted phenyl and N-methyl p-substituted phenyl compounds, and luciferase activity was determined as outlined in Materials and Methods. C, RKO cells were transfected with NBRE3-luc or wild-type or truncated FLAG-NR4A1 expression plasmids and treated with selected C-DIMs, and luciferase activity was determined as outlined in Materials and Methods. Results are expressed as means ± SE for at least 3 replicate determinations, and values <0.8 are significantly (P < .05) decreased compared with control (DMSO).
The structure-dependent effects of ortho-, meta-, and para-substituted phenyl C-DIM analogs and the importance of the free indole group on C-DIM–mediated inhibition of transactivation were also investigated for selected compounds in RKO cells (Figure 3B). Treatment of RKO cells with the CN-, OH-, F-, and Br-phenyl analogs and the N-methyl (indole) derivatives of the para-substituted phenyl compounds showed that the differences in the position of the phenyl ring substituents (ortho/meta/para) had minimal effects on antagonism activity (decreased luciferase), whereas methylation of the indole groups attenuated their NR4A1 antagonist activities in the transactivation assay. This is consistent with results of the modeling studies that indicated hydrogen bond interactions between NR4A1 and the indole ring amine group (Figure 2C). The requirements for different domains of NR4A1 for C-DIM–dependent inhibition of transactivation were investigated in RKO cells transfected with wild-type FLAG-NR4A1 and mutants that contain the A–D (N-terminal plus DNA binding domain) and C–F (C-terminal) domains. Cells were transfected with the NBRE3-luc construct, the wild-type and mutant NR4A1 expression plasmids and treated with the p-Br, p-OH and p-CO2Me analogs. All three compounds decreased transactivation in cells transfected with wild-type NR4A1 and mutant NR4A1 containing the C-terminal LBD. However, we also observed decreased luciferase activity with RKO cells transfected with NR4A1 containing the N-terminal domain of the receptor, and we are currently investigating the underlying mechanisms for antagonism of N-terminal activation function-1 (AF1) by C-DIMs in colon cancer cells.
Functional effects of NR4A1 antagonists
The p-hydroxyphenyl C-DIM analog (DIM-C-pPhOH), which exhibits high NR4A1 binding activity in both the fluorescence binding and molecular modeling assays exhibits activity similar to that observed after NR4A1 knockdown (siNR4A1) in pancreatic and lung cancer cells (15, 16) and was used as a model NR4A1 antagonist in colon cancer cells. Results in Figure 4A demonstrate that DIM-C-pPhOH inhibits growth of RKO and SW480 cells with IC50 values (48 hours) of 21.2μM and 21.4μM, respectively. Moreover, in RKO cells, the growth-inhibitory effects of DIM-C-pPhOH were attenuated by overexpression of NR4A1 (Figure 4B). DIM-C-pPhOH also induced cleavage of caspases 3 and 7 and poly(ADT-ribose) polymerase in RKO and SW480 cells (Figure 4C), indicative of the proapoptotic effects of this NR4A1 antagonist. Supplemental Figure 2, A and B, demonstrates the NR4A1 antagonist activities of the p-bromophenyl (DIM-C-pPhBr) and p-carboxymethyl (DIM-C-pPhCO2Me), which also inhibited RKO cell growth and induced apoptosis. Treatment of RKO and SW480 cells with dimethylsulfoxide (DMSO) or 20μM DIM-C-pPhOH followed by staining with TR3 antibodies and 4′,6-diamidino-2-phenylindole followed by merging shows that NR4A1 is nuclear in both treatment groups (Figure 4D) as previously observed in pancreatic cancer cells (15).
Figure 4.

DIM-C-pPhOH inhibits cell growth and induces apoptosis in colon cancer cells. A, Cell survival. RKO and SW480 cells were treated with various concentrations of DIM-C-pPhOH or DMSO as a control for 3 days, and the number of cells in each well was counted on days 1, 2, and 3. B, RKO cells were transfected with FLAG-NR4A1 and treated with DIM-C-pPhOH for 24 hours, and the number of cells in each well was counted. C, RKO and SW480 cells were treated with either DMSO or various concentrations of DIM-C-pPhOH for 24 hours, and whole-cell lysates were analyzed by Western blot. β-Actin was used as a loading control. D, RKO and SW480 cells were treated with DMSO or 20μM DIM-C-pPhOH for 12 hours, and fluorescent images after staining were determined as described (15). All data points were determined at least 3 times (A–C).
Knockdown of NR4A1 by RNAi also decreased growth of RKO and SW480 cells (Figure 5, A and B), and results in Figure 5, A and B, show that the growth-inhibitory effects of DIM-C-pPhOH were significantly decreased after knockdown of NR4A1, confirming a role for NR4A1 in mediating the antiproliferative activity of DIM-C-pPhOH. It should be noted that although the results are reported as percentage of DMSO control (set at 100%), cells transfected with siNR4A1 exhibited a significantly decreased rate of growth. Knockdown of NR4A1 in RKO and SW480 cells also induced markers of apoptosis including cleaved caspases 3 and 7 and poly(ADP-ribose) polymerase (Figure 5, C and D), and the effects of NR4A1 knockdown was similar to the effects observed after treatment with the C-DIM/NR4A1 antagonists (Figure 4 and Supplemental Figure 2B). Quantitation of protein levels for results in Figure 5, C and D, is shown in Figure 5E.
Figure 5.

Knockdown of NR4A1 inhibits cell growth and induces apoptosis in colon cancer cells. A and B, Cell proliferation. RKO (A) and SW480 (B) cells were transfected with a nonspecific oligonucleotide (siCtl) or siNR4A1 and treated with DMSO or 20μM DIM-C-pPhOH, and percent cell number (relative to DMSO) was determined in both cell lines. Results are expressed as means ± SD for 3 replicated determinations. *, Significant (P < .05) inhibition by DIM-C-pPhOH; **, significant (P < .05) attenuation of this response after siNR4A1. C and D, Induction of apoptosis. RKO (C) and SW480 (D) cells were transfected with siCtl or siNR4A1, and whole-cell lysates were analyzed by Western blots. E, Quantitation of the blots (3 replicates) using β-actin as an internal control standard is shown. *, Significant (P < .05) induction.
Previous studies show that NR4A1 expression is required for activation of specificity protein 1 (Sp1)-regulated genes such as survivin (15, 16). Figure 6A shows that treatment of RKO and SW480 cells with DIM-C-pPhOH or transfection with siNR4A1 decreased expression of 3 Sp-regulated genes (survivin, Id1, and PTTG) (39–41) without affecting Sp protein expression, and the knockdown results are quantitated (Supplemental Figure 2C). DIM-C-pPhBr and DIM-C-pPhCO2Me also decreased expression of these Sp-regulated genes (Supplemental Figure 2D); however, this was also due, in part, to downregulation of Sp1. In RKO cells transfected with GC-rich survivin promoter-reporter constructs (pGL3-SVV[−269] or pGL3-SVV[−150]) (Figure 6B) or a GC-rich construct containing 3 tandem consensus GC-rich binding sites (pGL3[GC]3-TK) (Figure 6C), DIM-C-pPhOH or siNR4A1 significantly decreased transactivation, and these effects were also observed in SW480 cells (data not shown), demonstrating that NR4A1 coregulated Sp-dependent promoters as previously observed in lung and pancreatic cancer cells (15). siNR4A1 and DIM-C-pPhOH decreased mammalian target of rapamycin (mTOR) activation in p53-positive lung cancer cell lines, reversing NR4A1-dependent inhibition of p53; this resulted in activation of sestrin 2 and activation of AMP-activated protein kinase (AMPKα) resulting in inhibition of mTOR signaling (decreased phosphorylation of 70S6K and S6RP) (16). Figure 7, A and B, demonstrates that DIM-C-pPhOH and siNR4A1 induced sestrin 2 and phosphorylation of AMPKα and decreased phosphorylation of mTOR-regulated p70S6K and S6RP in p53-positive RKO but not in p53-negative SW480 cells, and the results in Figure 7, A and B, are quantitated (Supplemental Figure 3, A and B). Similar results were also observed for the DIM-C-pPhBr and DIM-C-pPhCO2Me NR4A1 antagonists in RKO cells (Supplemental Figure 3C). DIM-C-pPhOH and siNR4A1 also activated luciferase activity in RKO cells transfected with a p53-responsive construct ([p53]X14-luc) (Figure 7C), and the effects of DIM-C-pPhOH as an mTOR inhibitor in RKO cells were attenuated after knockdown of p53 (sip53). We also showed that the p-methylphenyl analog (DIM-C-pPhCH3) that does not bind NR4A1 (Table 1) did not affect expression of Sp-regulated genes or the mTOR pathway (Supplemental Figure 3D).
Figure 6.

DIM-C-pPhOH and knockdown of NR4A1 inhibit Sp1-regulated gene expression through downregulation of Sp1 transactivation in colon cancer cells. A, Western blot analysis. Cells were treated with DIM-C-pPhOH for 24 hours or transfected with siNR4A1 for 72 hours, and whole-cell lysates were analyzed by Western blot analysis. β-Actin was used as a loading control. B and C, RKO cells were transfected with 0.1 μg of either pGL3-SVV (−269), pGL3-SVV (−150) or pGL3-(GC)3-TK for 4 hours and treated with various concentrations of DIM-C-pPhOH for another 18 hours or transfected with siNR4A1 and siCtl. Luciferase activity (relative to β-galactosidase activity) was determined. Data are presented as means with SD. *, Significantly (P < .05) decreased activity. Results were replicated (3 times).
Figure 7.

DIM-C-pPhOH and knockdown of NR4A1 inhibit mTOR signaling through activation of p53/sestrin2/AMPKα axis in colon cancer cells expressing wild-type p53. A and B, Cells were treated with DIM-C-pPhOH for 24 hours or transfected with indicated siRNA for 72 hours, and whole-cell lysates were analyzed by Western blot. β-Actin was used as a loading control. C, RKO cells were transfected with p53x14-Luc and treated with DIM-C-pPhOH for 18 hours (left panel) or RKO cells were cotransfected with each siRNA and p53x14-Luc (right panel). Luciferase activity (relative to β-galactosidase activity) was determined, and the corresponding empty vector was used as a control. The data are presented as means with SD for at least 3 replicate determinations. **, P < .01 vs DMSO plus p53x14-Luc; #, P < .001 vs siScr. D, RKO cells were transfected with either siScr or sip53 for 48 hours and treated with DIM-C-pPhOH for another 24 hours. Whole-cell lysates were analyzed by Western blot, and β-actin was used as a loading control.
These results confirm that representative NR4A1 ligands DIM-C-pPhOH, DIM-C-pPhBr, and DIM-C-pPhCO2 Me (Table 1) decrease NR4A1-dependent transactivation from NBRE3-luc and also antagonize 2 important NR4A1-regulated pro-oncogenic pathways in colon cancer cells as previously observed in lung and pancreatic cancer cell lines (15, 16). Thus, in colon cancer cells, C-DIMs such as DIM-C-pPhOH, DIM-C-pPhBr, and DIM-C-pPhCO2Me that bind NR4A1 (Table 1) act as NR4A1 antagonists.
Discussion
Endogenous ligands for the NR4A orphan receptors have not yet been identified; however, it is clear from knockout and transgenic mouse models that these receptors play a critical role in cellular homeostasis and disease. NR4A receptors are immediate-early genes induced by multiple stimuli and play essential roles in metabolism in the liver, pancreas, and adipose tissue and neuronal and neurobehavioral functions, inflammation, cardiovascular function, and steroidogenesis (3, 4). NR4A1 is also overexpressed and plays a pro-oncogenic role in multiple solid tumors; however, results from the double-knockout NR4A1−/−/NR4A3−/− mouse, which rapidly develops leukemia, suggest that NR4A1 may have tumor suppressor-like activity for acute myelocytic leukemia (22). Csn-B was the first compound characterized as an NR4A1 agonist that directly bound the LBD of NR4A1; Csn-B also inhibited colon tumor growth in a mouse xenograft study, and this inhibitory effect was NR4A1-dependent (28). Csn-B not only activated NR4A1-dependent genes through a classical ligand-activated nuclear receptor but also induced nuclear export of NR4A1 to the mitochondria to induce apoptosis as previously described for other apoptosis-inducing agents (9, 23–25).
Research in the laboratory initially identified C-DIMs that activated mouse NR4A1-dependent transactivation in both pancreatic and colon cancer cells (12, 26); however, in pancreatic cancer cells transfected with human NR4A1-derived constructs, C-DIMs exhibited either minimal induction or decreased transactivation (10, 11), and similar results were observed in colon cancer cells (Figure 3). We also previously reported that C-DIMs induced NR4A1- and NR4A2-dependent expression of genes in several different cancer cell lines; however, we also observed that C-DIMs both increased and decreased expression of specific genes (10, 27, 32, 38, 42), and this is a pattern observed for other ligands and their cognate receptors that induce and repress gene expression. We demonstrated that both siNR4A1 and DIM-C-pPhOH affected a common set of genes and pathways (15, 16). For example, in lung and pancreatic cancer cells, nuclear NR4A1 in combination with p300 acts as a coregulator of Sp-regulated genes such as survivin, and transfection with siNR4A1 or treatment with DIM-C-pPhOH decreased expression of survivin but did not affect Sp1 expression (15). Using a similar approach, we observed comparable responses in colon cancer cells (Figure 5), demonstrating the pro-oncogenic functions of NR4A1 and the antineoplastic activity of DIM-C-pPhOH and other C-DIMs that antagonize nuclear NR4A1 but do not induce nuclear export of this receptor. NR4A1 binds and inactivates p53 (43), and in lung cancer cells, both siNR4A1 and DIM-C-pPhOH activate p53, which in turn induces sestrin 2, and activation of AMPKα results in mTOR inhibition (16). We used a similar approach in colon cancer cells, and both siNR4A1 and DIM-C-pPhOH and related compounds inhibited mTOR signaling in p53-positive RKO but not p53-negative SW480 cells (Figure 7 and Supplemental Figure 2D). These data suggest that DIM-C-pPhOH and related analogs function as NR4A1 antagonists or indirect inhibitors of NR4A1. Results of the transactivation studies with NBRE3-luc also show that DIM-C-pPhOH and related C-DIMs decrease luciferase activity that is dependent on the N-terminal (AF1) domain of NR4A1.
Previous studies show that the N-terminal domain of NR4A1 contains a potent AF1 that interacts with multiple coactivators (44–46). Moreover, the AF1 domain physically associates with the C-terminal LBD (44). Results of transactivation studies show that C-DIMs such as DIM-C-pPhOH that bind NR4A1-LBD antagonize transactivation in cells transfected with FLAG-NR4A1 and the N-terminal and C-terminal deletion mutants (Figure 3C). Moreover, C-DIM compounds that do not bind the LBD of NR4A1 (Table 1) also inhibit full-length NR4A1-dependent transactivation (Figure 3, A and B). These results suggest that inhibition of NR4A1-dependent transactivation by members of the C-DIM class of compounds used in this study is also independent of ligand binding and may be due to direct interactions of these compounds with the A/B domain of NR4A1 and/or with coactivators of NR4A1. These observations are not inconsistent with previous studies with 6-mercapturine and C-DIMs that modulated NR4A2- and NR4A3- (6-mercaptorpurine only)-dependent transactivation in cells transfected with constructs containing N-terminal AF1 (38, 47, 48) and not the LBD of these receptors. However, we also observed that C-DIMs inhibited transactivation in cells transfected with the C-terminal ligand binding of NR4A1 and therefore investigated the direct binding of C-DIM analogs to the ligand binding of NR4A1 using the fluorescent assay as previously described for determining ligand binding to NR4A1 (28–31). The results show that C-DIMs directly bind to the LBD of NR4A1 with variable KD values; not all the compounds exhibited binding in the assay, and some binding interferences were also observed (Table 1).
In addition, we also used ligand docking to the ligand binding pocket of NR4A1, and results of computational-based molecular modeling show that the C-DIMs readily fit into the ligand binding pocket. DIM-C-pPhOH exhibited a low KD in the direct binding assay, and based on the results of the modeling studies, this high affinity involves π-cation interactions with Arg515 and hydrogen bond interactions with His516. In addition, activation or inhibition of nuclear receptors also involves a concomitant conformational change in the protein in response to drug binding, and it may be that these interactions with Arg515 and His516 are necessary not only for optimal binding affinity but also for the requisite conformational changes required to inhibit transcriptional activation. This was confirmed by showing that DIM-C-pPhOH did not bind mutant NR4A1-LBD(H516W) (Figure 2D). It was also apparent from the molecular docking studies with C-DIMs that the amino acid side chains and interactions that facilitate C-DIM interactions with NR4A1 are different from those previously reported for Csn-B and related compounds (28, 29) where hydrogen bonding to Tyr453 was an important interaction in the ligand binding pocket. These results are consistent with the significantly different structures of C-DIMs and other NR4A1 ligands and the existence of several potential binding sites within the LBD of NR4A1 (30) with occupancy of either 1 or multiple sites influencing cell context-dependent agonist or antagonist activity of C-DIMs and other NR4A1 ligands. Furthermore, differences in the contacts between the ligands and the receptor may also play a role in determining whether a compound is an NR4A1 agonist or antagonist. Thus, both an NR4A1 agonist and antagonist may bind the same site, but differences in specific interactions in the LBD will result in different conformational changes in the protein that correspond to activation or inhibition of receptor function.
In summary, results of this study demonstrate that C-DIMs that target nuclear NR4A1 also bind the receptor and function as antineoplastic agents in colon cancer cells by acting as NR4A1 antagonists. The results are consistent with previous studies in lung and pancreatic cancer cells where both siNR4A1 and DIM-C-pPhOH gave responses similar to that observed in Figures 4 to 7. The designation of specific C-DIMs such as DIM-C-pPhOH as an NR4A1 antagonist is based on the modeling and receptor binding data and on the parallel functional effects observed for this compound and for siNR4A1. However, C-DIMs also modulate transactivation in cells transfected with NR4A1-AD (Figure 3C) and NR4A2-AD (38) constructs expressing the N-terminal region of the receptor. These effects are independent of ligand binding and have also been previously reported for 6-mercaptopurine (47, 48), and there is strong evidence that AF1 plays an important role in NR4A-dependent regulation of gene expression (44, 46, 49). The mechanisms of C-DIM–dependent modulation of N-terminal AF1 and possible direct interactions with this domain are currently being investigated. The interaction of C-DIM, Csn-B analogs, and other compounds within the NR4A1 ligand binding pocket suggests that NR4A1 may also bind other structural classes of small molecules that can be used as agonists or antagonists for treatment of cancer and other health problems where NR4A1 plays a prominent role (3, 4).
Additional material
Supplementary data supplied by authors.
Acknowledgments
This work was supported by funded by National Institutes of Health (NIH)/National Cancer Institute Grant R01-CA124998 (S.S.), NIH/National Institute of Environmental Health Sciences Grant P30-ES023512 (to S.S.), Texas AgriLife (to S.S.), and NIH/National Center for Advancing Translational Sciences Grant UL1-TR001082 (to D.S.B.).
Disclosure Summary: The authors have nothing to declare.
Footnotes
- AF1
- activation function-1
- AMPK
- AMP-activated protein kinase
- CD
- circular dichroism
- C-DIM
- 1,1-bis(3′-indolyl)-1-(substituted phenyl)methane analog
- Csn-B
- cytosporone B
- DIM-C-pPhOH
- [1,1-bis(3′-indolyl)-1-(p-hydroxyphenyl)methane
- DMSO
- dimethylsulfoxide
- GST
- glutathione S-transferase
- LBD
- ligand binding domain
- mTOR
- mammalian target of rapamycin
- NR
- nuclear receptor
- RNAi
- RNA interference
- siRNA
- small interfering RNA
- Sp1
- specificity protein 1.
References
- 1. Mullican SE, Dispirito JR, Lazar MA. The orphan nuclear receptors at their 25-year reunion. J Mol Endocrinol. 2013;51:T115–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Evans RM. The nuclear receptor superfamily: a rosetta stone for physiology. Mol Endocrinol. 2005;19:1429–1438. [DOI] [PubMed] [Google Scholar]
- 3. Maxwell MA, Muscat GE. The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nucl Recept Signal. 2006;4:e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pearen MA, Muscat GE. Minireview: Nuclear hormone receptor 4A signaling: implications for metabolic disease. Mol Endocrinol. 2010;24:1891–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Milbrandt J. Nerve growth factor induces a gene homologous to the glucocorticoid receptor gene. Neuron. 1988;1:183–188. [DOI] [PubMed] [Google Scholar]
- 6. Murphy EP, Dobson AD, Keller C, Conneely OM. Differential regulation of transcription by the NURR1/NUR77 subfamily of nuclear transcription factors. Gene Expr. 1996;5:169–179. [PMC free article] [PubMed] [Google Scholar]
- 7. Paulsen RF, Granas K, Johnsen H, Rolseth V, Sterri S. Three related brain nuclear receptors, NGFI-B, Nurr1, and NOR-1, as transcriptional activators. J Mol Neurosci. 1995;6:249–255. [DOI] [PubMed] [Google Scholar]
- 8. Saucedo-Cardenas O, Kardon R, Ediger TR, Lydon JP, Conneely OM. Cloning and structural organization of the gene encoding the murine nuclear receptor transcription factor, NURR1. Gene. 1997;187:135–139. [DOI] [PubMed] [Google Scholar]
- 9. Zhang XK. Targeting Nur77 translocation. Expert Opin Ther Targets. 2007;11:69–79. [DOI] [PubMed] [Google Scholar]
- 10. Lee SO, Li X, Khan S, Safe S. Targeting NR4A1 (TR3) in cancer cells and tumors. Expert Opin Ther Targets. 2011;15:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Safe S, Kim K, Li X, Lee SO. NR4A orphan receptors and cancer. Nucl Recept Signal Atlas. 2011;9:e002. [Google Scholar]
- 12. Cho SD, Yoon K, Chintharlapalli S, et al. Nur77 agonists induce proapoptotic genes and responses in colon cancer cells through nuclear receptor-dependent and nuclear receptor-independent pathways. Cancer Res. 2007;67:674–683. [DOI] [PubMed] [Google Scholar]
- 13. Wu H, Lin Y, Li W, et al. Regulation of Nur77 expression by β-catenin and its mitogenic effect in colon cancer cells. FASEB J. 2011;25:192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen HZ, Liu QF, Li L, et al. The orphan receptor TR3 suppresses intestinal tumorigenesis in mice by downregulating Wnt signalling. Gut. 2012;61:714–724. [DOI] [PubMed] [Google Scholar]
- 15. Lee SO, Abdelrahim M, Yoon K, et al. Inactivation of the orphan nuclear receptor TR3/Nur77 inhibits pancreatic cancer cell and tumor growth. Cancer Res. 2010;70:6824–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee SO, Andey T, Jin UH, et al. The nuclear receptor TR3 regulates mTORC1 signaling in lung cancer cells expressing wild-type p53. Oncogene. 2012;31:3265–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith AG, Lim W, Pearen M, Muscat GE, Sturm RA. Regulation of NR4A nuclear receptor expression by oncogenic BRAF in melanoma cells. Pigment Cell Melanoma Res. 2011;24:551–563. [DOI] [PubMed] [Google Scholar]
- 18. Zhan YY, He JP, Chen HZ, Wang WJ, Cai JC. Orphan receptor TR3 is essential for the maintenance of stem-like properties in gastric cancer cells. Cancer Lett. 2013;329:37–44. [DOI] [PubMed] [Google Scholar]
- 19. Brás A, Albar JP, Leonardo E, de Buitrago GG, Martínez-A C. Ceramide-induced cell death is independent of the Fas/Fas ligand pathway and is prevented by Nur77 overexpression in A20 B cells. Cell Death Differ. 2000;7:262–271. [DOI] [PubMed] [Google Scholar]
- 20. Li QX, Ke N, Sundaram R, Wong-Staal F. NR4A1, 2, 3–an orphan nuclear hormone receptor family involved in cell apoptosis and carcinogenesis. Histol Histopathol. 2006;21:533–540. [DOI] [PubMed] [Google Scholar]
- 21. Kolluri SK, Bruey-Sedano N, Cao X, et al. Mitogenic effect of orphan receptor TR3 and its regulation by MEKK1 in lung cancer cells. Mol Cell Biol. 2003;23:8651–8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mullican SE, Zhang S, Konopleva M, et al. Abrogation of nuclear receptors Nr4a3 and Nr4a1 leads to development of acute myeloid leukemia. Nat Med. 2007;13:730–735. [DOI] [PubMed] [Google Scholar]
- 23. Dawson MI, Hobbs PD, Peterson VJ, et al. Apoptosis induction in cancer cells by a novel analogue of 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalenecarboxylic acid lacking retinoid receptor transcriptional activation activity. Cancer Res. 2001;61:4723–4730. [PubMed] [Google Scholar]
- 24. Li H, Kolluri SK, Gu J, et al. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science. 2000;289:1159–1164. [DOI] [PubMed] [Google Scholar]
- 25. Lin B, Kolluri SK, Lin F, et al. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell. 2004;116:527–540. [DOI] [PubMed] [Google Scholar]
- 26. Chintharlapalli S, Burghardt R, Papineni S, Ramaiah S, Yoon K, Safe S. Activation of Nur77 by selected 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes induces apoptosis through nuclear pathways. J Biol Chem. 2005;280:24903–24914. [DOI] [PubMed] [Google Scholar]
- 27. Cho SD, Lee SO, Chintharlapalli S, et al. Activation of nerve growth factor-induced B alpha by methylene-substituted diindolylmethanes in bladder cancer cells induces apoptosis and inhibits tumor growth. Mol Pharmacol. 2010;77:396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhan Y, Du X, Chen H, et al. Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat Chem Biol. 2008;4:548–556. [DOI] [PubMed] [Google Scholar]
- 29. Liu JJ, Zeng HN, Zhang LR, et al. A unique pharmacophore for activation of the nuclear orphan receptor Nur77 in vivo and in vitro. Cancer Res. 2010;70:3628–3637. [DOI] [PubMed] [Google Scholar]
- 30. Zhan YY, Chen Y, Zhang Q, et al. The orphan nuclear receptor Nur77 regulates LKB1 localization and activates AMPK. Nat Chem Biol. 2012;8:897–904. [DOI] [PubMed] [Google Scholar]
- 31. Wang WJ, Wang Y, Chen HZ, et al. Orphan nuclear receptor TR3 acts in autophagic cell death via mitochondrial signaling pathway. Nat Chem Biol. 2014;10:133–140. [DOI] [PubMed] [Google Scholar]
- 32. Lee SO, Jin UH, Kang JH, et al. The orphan nuclear receptor NR4A1 (Nur77) regulates oxidative and endoplasmic reticulum stress in pancreatic cancer cells. Mol Cancer Res. 2014;12:527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Flaig R, Greschik H, Peluso-Iltis C, Moras D. Structural basis for the cell-specific activities of the NGFI-B and the Nurr1 ligand-binding domain. J Biol Chem. 2005;280:19250–19258. [DOI] [PubMed] [Google Scholar]
- 34. Fletcher R, Reeves CM. Function minimization by conjugate gradients. Comput J. 1964;7:149–154. [Google Scholar]
- 35. Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Feig M, Brooks CL., 3rd Recent advances in the development and application of implicit solvent models in biomolecule simulations. Curr Opin Struct Biol. 2004;14:217–224. [DOI] [PubMed] [Google Scholar]
- 37. Koska J, Spassov VZ, Maynard AJ, et al. Fully automated molecular mechanics based induced fit protein-ligand docking method. J Chem Inf Model. 2008;48:1965–1973. [DOI] [PubMed] [Google Scholar]
- 38. Li X, Lee SO, Safe S. Structure-dependent activation of NR4A2 (Nurr1) by 1,1-bis(3′-indolyl)-1-(aromatic)methane analogs in pancreatic cancer cells. Biochem Pharmacol. 2012;83:1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li Y, Xie M, Yang J, et al. The expression of antiapoptotic protein survivin is transcriptionally upregulated by DEC1 primarily through multiple sp1 binding sites in the proximal promoter. Oncogene. 2006;25:3296–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takeda T, Sakata M, Isobe A, et al. Involvement of Sp-1 in the regulation of the Id-1 gene during trophoblast cell differentiation. Placenta. 2007;28:192–198. [DOI] [PubMed] [Google Scholar]
- 41. Chintharlapalli S, Papineni S, Lee SO, et al. Inhibition of pituitary tumor-transforming gene-1 in thyroid cancer cells by drugs that decrease specificity proteins. Mol Carcinog. 2011;50:655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yoon K, Lee SO, Cho SD, Kim K, Khan S, Safe S. Activation of nuclear TR3 (NR4A1) by a diindolylmethane analog induces apoptosis and proapoptotic genes in pancreatic cancer cells and tumors. Carcinogenesis. 2011;32:836–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao BX, Chen HZ, Lei NZ, et al. p53 mediates the negative regulation of MDM2 by orphan receptor TR3. EMBO J. 2006;25:5703–5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wansa KD, Harris JM, Muscat GE. The activation function-1 domain of Nur77/NR4A1 mediates trans-activation, cell specificity, and coactivator recruitment. J Biol Chem. 2002;277:33001–33011. [DOI] [PubMed] [Google Scholar]
- 45. Maira M, Martens C, Batsché E, Gauthier Y, Drouin J. Dimer-specific potentiation of NGFI-B (Nur77) transcriptional activity by the protein kinase A pathway and AF-1-dependent coactivator recruitment. Mol Cell Biol. 2003;23:763–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rambaud J, Desroches J, Balsalobre A, Drouin J. TIF1beta/KAP-1 is a coactivator of the orphan nuclear receptor NGFI-B/Nur77. J Biol Chem. 2009;284:14147–14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ordentlich P, Yan Y, Zhou S, Heyman RA. Identification of the antineoplastic agent 6-mercaptopurine as an activator of the orphan nuclear hormone receptor Nurr1. J Biol Chem. 2003;278:24791–24799. [DOI] [PubMed] [Google Scholar]
- 48. Wansa KD, Harris JM, Yan G, Ordentlich P, Muscat GE. The AF-1 domain of the orphan nuclear receptor NOR-1 mediates trans-activation, coactivator recruitment, and activation by the purine anti-metabolite 6-mercaptopurine. J Biol Chem. 2003;278:24776–24790. [DOI] [PubMed] [Google Scholar]
- 49. Sohn YC, Kwak E, Na Y, Lee JW, Lee SK. Silencing mediator of retinoid and thyroid hormone receptors and activating signal cointegrator-2 as transcriptional coregulators of the orphan nuclear receptor Nur77. J Biol Chem. 2001;276:43734–43739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.