

Abstract
Investigation of the effect of disintegrants on the disintegration time and hardness of rapidly disintegrating tablets (RDTs) was carried out using a quality by design (QbD) paradigm. Ascorbic acid, aspirin, and ibuprofen, which have different water solubilities, were chosen as the drug models. Disintegration time and hardness of RDTs were determined and modeled by executing combined optimal design. The generated models were validated and used for further analysis. Sodium starch glycolate, croscarmellose sodium, and crospovidone were found to lengthen disintegration time when utilized at high concentrations. Sodium starch glycolate and crospovidone worked synergistically in aspirin RDTs to decrease disintegration time. Sodium starch glycolate-crospovidone mixtures, as well as croscarmellose sodium-crospovidone mixtures, also decreased disintegration time in ibuprofen RDTs at high compression pressures as compared to the disintegrants used alone. The use of sodium starch glycolate in RDTs with highly water soluble active ingredients like ascorbic acid slowed disintegration, while microcrystalline cellulose and crospovidone drew water into the tablet rapidly and quickened disintegration. Graphical optimization analysis demonstrated that the RDTs with desired disintegration times and hardness can be formulated with a larger area of design space by combining disintegrants at difference compression pressures. QbD was an efficient and effective paradigm in understanding formulation and process parameters and building quality in to RDT formulated systems.
KEY WORDS: disintegrants, quality by design, rapidly disintegrating tablets
INTRODUCTION
Rapidly disintegrating tablets (RDTs), also known as orally disintegrating, orodispersible, fast melting, or mouth dissolving tablets, are tablets that disperse readily in the mouth, usually within seconds. The United States Food and Drug Administration (US FDA) defined an RDT as “a solid dosage form containing medicinal substance or active ingredient which disintegrates rapidly usually within a matter of seconds when placed upon the tongue” (1). Such tablets are advantageous to patients with dysphagia or swallowing difficulties such as pediatric, geriatric, stroke, Parkinson’s disease, or cerebral palsy patients as well as those who have undergone head/neck radiation therapy. Travelers who do not have access to water can also benefit from RDTs (2–5). Furthermore, RDTs provide a line extension opportunity for companies facing the end of patent protection for their products (6–8).
RDTs have been formulated using several techniques, lyophilization, direct compression, molding, spray drying, and effervescence (7,9–13). Lyophilization and direct compression have become the most commonly used techniques. Lyophilized tablets have very porous structures and hence allow quick penetration of saliva resulting in faster disintegration. However, the high tablet porosity leads to poor physical strength, and the lyophilization process itself is less economical and not productive. On the other hand, direct compression tablets can be produced by existing tabletting machines as relatively hard tablets of low friability. The main disadvantage is that the tablets formed generally have slower disintegration than those produced by lyophilization. Direct compression requires the efficient use of disintegrant combinations in the formulation to enhance the breaking up of tablets in contact with water. First generation disintegrants are starches (e.g., corn starch, potato starch) and cellulose-based disintegrants (e.g., microcrystalline cellulose, low substituted hydroxypropyl cellulose). The need for shorter disintegration times or more efficient disintegrants led to the development of superdisintegrants such as modified cellulose (croscarmellose sodium), cross-linked polyvinylpolypyrrolidone (crospovidone) and modified starches (sodium starch glycolate) (14).
Croscarmellose sodium is reported to aid disintegration by rapid swelling and wicking upon contact with water (2,15,16). Wicking is a “whipping” action where material-air or material-material interface is spontaneously replaced by material-water interface and thus helps in maintaining capillary flow. The effectiveness of crospovidone has been claimed to be due to its high capillary activity through wicking with little swelling action (2,17,18), mainly by swelling (19), or by strain recovery or latent viscoelastic recovery (20). Sodium starch glycolate is reported to work via rapid uptake of water and swelling (16,18). Microcrystalline cellulose, commonly used as filler in tablet formulations, is not considered to be a superdisintegrant but reported to possess good wicking properties and hydrogen bonds between adjacent matchstick-like bundles that break when exposed to water (21).
The effectiveness of superdisintegrants has been well studied. Haware et al. reported the effectiveness of croscarmellose sodium, crospovidone, and sodium starch glycolate at different concentrations (3). Douroumis et al. reported the effects of compression force on cetirizine RDTs formulated with several superdisintegrants (22). Martino et al. have reported the effects of compression pressure on disintegration with different disintegrants (23). Some reports examined the effects of superdisintegrants but only using a single drug compound (22,24,25). While many studies have examined the use of individual disintegrants, little emphasis has been placed on studying the potential synergistic effects between mixtures of disintegrants, as well as the possibility of improved disintegration by mixing functionally different disintegrants. The interactions of the disintegrants mixtures and their effects on RDT formulations have not been well studied. Hence, it is worthwhile to acquire an in-depth understanding of the effects of mixing disintegrants with different kinds of active ingredients to prepare RDTs.
Quality by design (QbD) paradigm can be useful to understand the effects of disintegrant blends and select disintegrants at particular concentrations for optimal RDT formulation. QbD emphasizes that product and process understanding should be enhanced on the basis of sound scientific principles and design efforts to achieve predefined objectives (26–29). Design of experiments (DOE) and response surface methodology (RSM) are useful components of this paradigm to produce a design space for the input variables of the formulation (30). DOE and RSM allow the collection of data with much fewer runs than would be possible with a traditional experimental design, where one variable is changed at each time. DOE in combination with RSM can fit linear or quadratic functions while assessing response surfaces and thus provide mathematical models of output responses as a function of input variables (30). Optimal settings of these input variables will further lead to achieve desirable performance of the formulation.
In this study, the design space will be the multidimensional region of disintegrant attributes and compression pressure (input variables) within which the disintegration times and hardness of RDTs (output responses) will remain within specifications. RDTs with mixtures of the disintegrants, croscarmellose sodium, crospovidone, sodium starch glycolate, and microcrystalline cellulose, were prepared via direct compression at various compression pressures. Three actives with different water solubilities, ascorbic acid (freely soluble in water), aspirin (slightly soluble), and ibuprofen (practically insoluble), were used with the disintegrant mixtures. DOE and RSM were employed to generate the experimental matrix for the experiment, in which several components such as disintegrant mixture composition and compression pressure were concurrently varied. Combined optimal design was implemented for eliciting the effect of disintegrants mixtures (mixture component) and compression force (numeric-process factor). The objective of this study was to investigate possible synergistic behavior of disintegrants in ascorbic acid, aspirin, and ibuprofen RDTs. The relationships between compression pressure, disintegrant mixture, disintegration time, and hardness were also evaluated to explore design space for RDTs.
MATERIALS AND METHODS
Materials
The active pharmaceutical ingredients (APIs), ascorbic acid, and aspirin were obtained from Chempure Pte. Ltd., Singapore, while ibuprofen was from CFS (S) Pte. Ltd., Singapore. Mannitol (Pearlitol 400 DC) was the main filler and was obtained from Roquette, Lestrem, France. The disintegrants, microcrystalline cellulose (Avicel PH-102) and croscarmellose sodium (Ac-Di-Sol) were sourced from FMC Biopolymer, PA, USA. Sodium starch glycolate (Primojel) was obtained from DFE Pharma, Goch, Germany. Crospovidone (Kollidon CL-SF) was obtained from BASF, Ludwigshafen, Germany. Magnesium stearate was purchased from Productos Metalest, Spain. APIs were passed through a 500-μm aperture size sieve before use. All other materials were used as received and used percentage concentrations are based on w/w.
Experimental Design
A combined optimal design was crafted using Design-Expert 8.0.7.1 (Minneapolis, MN, USA), comprising five mixture (variable fraction) components (croscarmellose sodium (CCS), crospovidone (CP), sodium starch glycolate (SSG), microcrystalline cellulose (MCC), and mannitol) and one numeric factor (compression pressure). Our aim was to use disintegrant mixtures ≤ 10%, which represent the variable fraction (four disintegrants and one filler) of the tablet composition. Thus, five variable fraction components were each set to range from 0–10% of the tablet composition, with the total sum of the five components being 10% of the tablet composition. The API used comprised 50% of the composition and magnesium stearate (Mg St) was used at 0.5%. Mannitol was also used as filler in the remaining fraction of the total composition. Compression pressure was set as a discrete variable with four levels (88.7, 177.3, 266, and 354.7 MPa). An IV-optimal, reduced quadratic x quadratic model was selected. IV optimality is desirable for RSM where prediction is important. The algorithm picks points that minimize the integral of the prediction variance across the design space. The experiment was designed as 1 block. A total of 55 runs was generated, and the runs were randomized to avoid systematic error. Each set of 55 runs was prepared for an API. Tablets prepared were tested for disintegration time and hardness.
Preparation of Tablets
Mannitol, active ingredient, and disintegrants were weighed out and manually mixed in a polyethylene bag for 8 min. Magnesium stearate was added to the mixture and further blended for 2 min. The blend was compressed using 8-mm flat-faced punches in a manual press (Natoli Engineering Company, MO, USA). Compression pressures used were as determined by the RSM. A total of twenty tablets (each weighing 200 mg) were compressed per run, and tablets were stored for at least a week before testing.
Disintegration Test
US FDA described RDT as a solid oral preparation that disintegrates rapidly in mouth. Based upon the USP disintegration test method and apparatus (for conventional tablets), the disintegration time of RDT should be less than 30 s. No specific disintegration test method is identified for RDT by the US FDA (1). European Pharmacopeia (EP) has defined RDT as tablets which disperses or disintegrates in less than 3 min using the conventional disintegration test method and apparatus (31). Japanese Pharmacopeia (JP) has not suggested any particular disintegration test method and apparatus for RDT (32). Since no special disintegration apparatus and method has been identified by US FDA, EP, or JP, disintegration tests for RDTs were performed using the USP disintegration apparatus, without disks, in water at 34°C (to simulate temperature of the tongue). Disintegration times of six tablets per batch were determined, and the mean value was entered into Design-Expert.
Hardness Test
Hardness was determined using a hardness tester (Copley TBF 1000, Nottingham, UK). The hardness values for six tablets per batch were determined, and the mean value was entered into Design-Expert.
Statistical Analysis
Generation of Disintegration Time and Hardness Models for Active Ingredients
Models were generated by Design-Expert to describe possible relationships between mixture components and compression pressures. Analysis of variance (ANOVA) was carried out for each model. R2, p value, lack of fit, and Adeq precision values were checked and the best model was selected. “Adeq precision” measures signal to noise ratio. The value should be greater than 4 to indicate an adequate signal and imply that the model can be used to navigate the design space. Backward elimination regression was used to remove insignificant terms (p > 0.05).
Validation of Selected Models
Validation of the selected model and optimization was determined by experimental confirmation of predicted disintegration time and hardness of numerically optimized solutions. Three numerically optimized formulations for each API were chosen for validation (Table I). Numerical optimization criteria were the minimization of disintegrant composition and minimization of predicted disintegration time. The target hardness for tablets was at least 20 N. New batches of RDTs with predicted values of input variables were prepared and output responses measured to confirm the validity of generated models.
Table I.
Experimental Matrix for Validation Runs
| Run | Mannitol | MCC | SSG | CP | CCS | API | Mg St | Pressurea |
|---|---|---|---|---|---|---|---|---|
| (%) | (%) | (%) | (%) | (%) | (%) | (%) | (MPa) | |
| Aspirin | ||||||||
| 1 | 44.07 | 0 | 0 | 0.33 | 5.10 | 50 | 0.5 | 354.7 |
| 2 | 44.20 | 0 | 0 | 0 | 5.30 | 50 | 0.5 | 354.7 |
| 3 | 41.20 | 2.30 | 0 | 0 | 6.00 | 50 | 0.5 | 266 |
| Ascorbic acid | ||||||||
| 1 | 44.64 | 0 | 0 | 4.04 | 0.82 | 50 | 0.5 | 354.7 |
| 2 | 42.52 | 1.40 | 2.89 | 2.69 | 0 | 50 | 0.5 | 177.3 |
| 3 | 43.94 | 1.34 | 0 | 3.38 | 0.83 | 50 | 0.5 | 266 |
| Ibuprofen | ||||||||
| 1 | 43.72 | 0 | 1.67 | 0 | 4.12 | 50 | 0.5 | 88.7 |
| 2 | 42.89 | 0 | 2.15 | 1.15 | 3.31 | 50 | 0.5 | 88.7 |
| 3 | 39.50 | 2.50 | 2.51 | 2.49 | 2.50 | 50 | 0.5 | 88.7 |
MCC microcrystalline cellulose, SSG sodium starch glycolate, CCS croscarmellose sodium, CP crospovidone, API active pharmaceutical ingredients, Mg St magnesium stearate
aCompression pressure
Graphical Optimization Analysis Using Validated Models
The optimized regions within the design space were further identified based on the desired criteria of output responses. Graphical optimization option was used for this purpose, and an overlay plot was generated to depict the effect of input variables on output responses. Output response limits were required to specify for overlay plot generation. Limits specified in the software were as follows: (a) disintegration time of RDT ≤ 15 s and (b) hardness ≥ 20 N.
RESULTS
Disintegration Time and Hardness Models for Aspirin RDTs
Disintegration time of aspirin RDTs fitted best to a combined reduced quadratic x linear model (for mixture components and compression pressure, respectively). Results of ANOVA are given in Table II. The generated model reached statistical significance, with non-significant lack of fit, high Adeq precision value, and a predicted R2 value that was in reasonable agreement with the adjusted R2 value. Hardness of aspirin RDTs was also described by a combined reduced quadratic x linear model. The model was significant, lack of fit was non-significant, predicted R2 value was in reasonable agreement with the adjusted R2 value, and Adeq precision value was high.
Table II.
Results of ANOVA for Aspirin, Ascorbic acid, and Ibuprofen RDTs
| Model | SS | df | MS | F | p value | Lack of fit | Adjusted R 2 | Predicted R 2 | Adeq precision |
|---|---|---|---|---|---|---|---|---|---|
| Aspirin | |||||||||
| Disintegration time | 233.19 | 14 | 16.66 | 148.55 | <0.0001 | 0.79 | 0.97 | 0.96 | 38.42 |
| (4.49) | (40) | (0.11) | |||||||
| Hardness | 159.81 | 16 | 9.99 | 59.52 | <0.0001 | 0.48 | 0.95 | 0.91 | 23.07 |
| (6.38) | (38) | (0.17) | |||||||
| Ascorbic acid | |||||||||
| Disintegration time | 3442.21 | 22 | 156.46 | 6.69 | <0.0001 | 0.20 | 0.79 | -3.65 | 12.01 |
| (724.79) | (31) | (23.38) | |||||||
| Hardness | 7093.67 | 32 | 221.68 | 36.78 | <0.0001 | 0.71 | 0.96 | undefined | 21.81 |
| (126.55) | (21) | (6.03) | |||||||
| Ibuprofen | |||||||||
| Disintegration time | 19556.86 | 33 | 592.63 | 222.66 | <0.0001 | 0.44 | 0.99 | −1.58 | 44.48 |
| (55.89) | (21) | (2.66) | |||||||
| Hardness | 8406.72 | 11 | 764.25 | 18.53 | <0.0001 | 0.91 | 0.78 | 0.71 | 18.34 |
| (1773.94) | (43) | (41.25) | |||||||
SS sum of squares, df degrees of freedom, MS mean of squares, F Fischer’s ratio, R 2 regression coefficient. Values in bracket are residuals from regression models
The model that describes the relationship between USP disintegration time, tablet composition, and compression pressure is as follows:
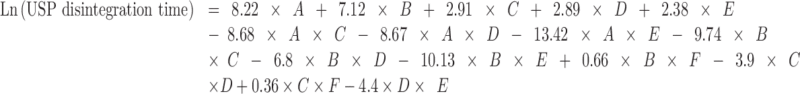 |
The relationship between hardness, tablet composition, and compression pressure is described as follows:
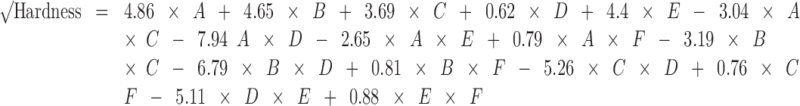 |
where A = mannitol, B = MCC, C = SSG, D = CP, E = CCS, and F = pressure.
Disintegration Time and Hardness Models for Ascorbic Acid RDTs
Disintegration of ascorbic acid RDTs had a combined reduced quadratic x linear model (Table II). The generated model reached statistical significance, with non-significant lack of fit. The predicted R2 value was negative, implying that the overall mean was a better predictor of disintegration time than the generated model. High Adeq precision value suggested that the model can be used to predict the desired design space. Hardness of ascorbic acid RDTs followed a combined reduced quadratic x quadratic model with desirable ANOVA results.
The equation that describes the relationship between USP disintegration time, tablet composition, and compression pressure is as follows:
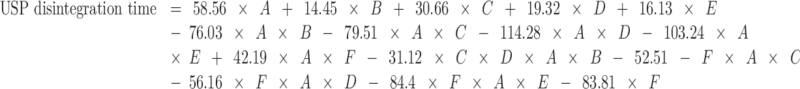 |
Hardness, tablet composition, and compression pressure can be described by:
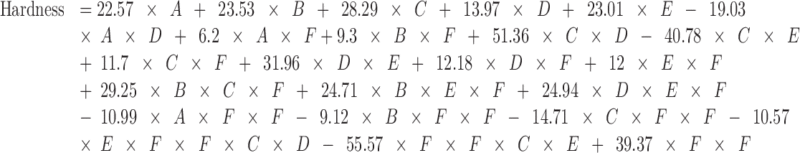 |
where A = mannitol, B = MCC, C = SSG, D = CP, E = CCS, and F = pressure.
Disintegration Time and Hardness Value Models for Ibuprofen RDTs
The disintegration behavior of ibuprofen RDTs was described by a combined reduced quadratic x quadratic model. Results of ANOVA are given in Table II. The model reached statistical significance and had nonsignificant lack of fit. The predicted R2 value was negative. High Adeq value was achieved. Hardness of ibuprofen RDTs had a combined reduced linear x quadratic relationship. The model was statistically significant with high Adeq precision value. Lack of fit was nonsignificant, and the predicted R2 value was in reasonable agreement with the adjusted R2 value.
The model that describes the relationship between USP disintegration time, tablet composition, and compression pressure is as follows:
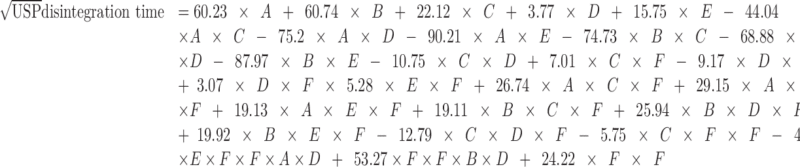 |
The relationship between hardness, tablet composition, and compression pressure is described as follows:
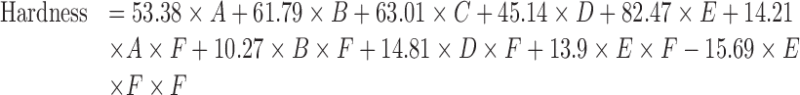 |
where A = mannitol, B = MCC, C = SSG, D = CP, E = CCS, and F = pressure.
Validation of Selected Models
Models for the disintegration time of aspirin, ascorbic acid, and ibuprofen RDTs obtained were validated. The models for hardness of aspirin and ibuprofen RDTs were validated, and that for ascorbic acid RDTs was partially validated (the actual hardness in one of the three optimized formulations was outside the predicted hardness range so the model was termed partially validated). The predicted and actual values of the validation runs are given in Table III (99% prediction interval, α = 0.01). The results confirmed that the validated models are good predictive tools. Also, output responses for input variables (within the experimental domain) could be computed and analyzed since the models were validated.
Table III.
Predicted and Actual Values for Validation Runs
| Run | Predicted DT | Actual DT | Predicted hardness | Actual hardness |
|---|---|---|---|---|
| (s) | (s) | (N) | (N) | |
| Aspirin | ||||
| 1 | 5.7 (3.2–10.1) | 7.6 | 20.0 (13.5–27.8) | 21.5 |
| 2 | 5.9 (3.2–10.9) | 8.9 | 23.0 (15.7–31.7) | 23.6 |
| 3 | 5.7 (3.5–9.4) | 9.4 | 20.0 (15.2–25.4) | 22.7 |
| Ascorbic acid | ||||
| 1 | 12.7 (1.7–23.7) | 14.9 | 20.0 (14.7–25.4) | 15.5 |
| 2 | 10.3 (2.9–17.7) | 13.9 | 20.0 (16.2–23.8) | 9.1 |
| 3 | 10.1 (2.4–17.8) | 14.1 | 20.0 (15.9–24.1) | 17.2 |
| Ibuprofen | ||||
| 1 | 13.7 (0.0–53.9) | 35.7 | 46.1 (36.5–55.7) | 45.0 |
| 2 | 13.7 (0.4–46.1) | 34.8 | 44.4 (35.5–53.2) | 36.6 |
| 3 | 6.8 (0.1–23.7)a | 19.0 | 45.4 (36.8–54.0) | 37.4 |
DT disintegration time
a95% prediction interval (α = 0.05) as interval could not be generated at 99% level
DISCUSSION
Trends found using the validated models are discussed in the discussion section below. Contour plots of variable fraction components, overlay plots of variable fraction components, and relevant trends are provided to support the discussion wherever possible.
Effects of Individual Disintegrants on RDT Disintegration
SSG and CCS have been known to result in lengthened disintegration times when used at high concentrations. This is likely due to partial gelling that potentially could form a viscous barrier and delay the entry of water into the tablet (3, 15). Similar results were observed for aspirin, ibuprofen, and ascorbic acid RDTs for all compression pressures (88.7–354.7 MPa range). SSG was optimal at 8% in aspirin and ibuprofen RDTs, and at 6% in ascorbic acid RDTs. CCS performed best at about 7% in all three drug models. CP was also observed to demonstrate similar effects especially for aspirin and ascorbic acid RDTs. Disintegration time decreased with an increase in CP concentration, until a minimum at 8% for aspirin and ibuprofen RDTs, and 6% for ascorbic acid RDTs. The typical observed effects are presented in Fig. 1 at compression pressure 177.3 MPa. Decrease in disintegration time could also be caused by a decrease in hardness of the RDTs at that particular composition. For this particular example, the lowest hardness values (or range of values) have been included in the Fig. 1, depicted by closed circles. Aspirin RDTs had the lowest hardness values when single disintegrant, CP was used in the range of 6.5–8.5%, SSG was used in the range of 6–8% or CCS was used in the range of 5.5–6.5%. Thus, for aspirin RDT, lowest disintegration time at a particular disintegrant composition could be due to decreased hardness value. However, for ibuprofen RDT, hardness was not low when the disintegration time decreased. Hardness was increased with increase in SSG and CCS concentrations (discussion in more details under “Effect of Disintegrant Compositions on RDT Hardness” ). Hardness was not at the lowest point when the disintegration time was low. However, hardness decreased with increase in CP and was lowest at highest CP amount. The model for hardness of ascorbic acid RDT was partially validated and therefore hardness and disintegration time relation for this RDT is not discussed.
Fig. 1.
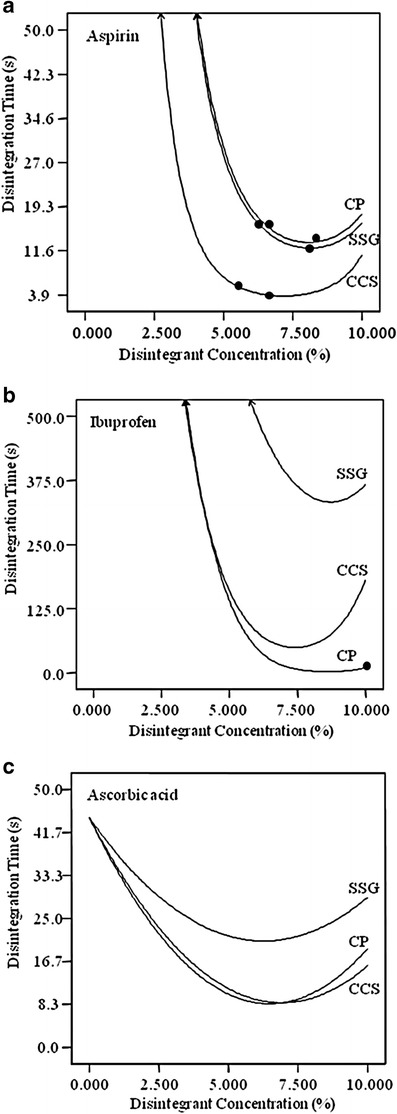
Effects of disintegrant concentration on disintegration time of a aspirin, b ibuprofen, and c ascorbic acid RDTs (compression pressure, 177.3 MPa). Similar effects were observed for other compression pressures (not shown)
Effect of Compression Pressure on RDT Disintegration Time
For the optimal individual disintegrant concentration (mentioned above), the effect of compression pressure on RDT disintegration time was studied. Generally, increase in compression pressure increased the disintegration time of RDT or had no significant effect on the disintegration time of RDT. This typical observed effect is presented in Fig. 2a for aspirin RDT when SSG (8%) was used as disintegrant. However, CP and CCS showed different trends in selected cases. When CP alone was used at its optimal concentration (8%), disintegration time of ibuprofen RDT decreased with increase in compression pressure (up to 177.3 MPa) and then increased again with increase in compression pressure (Fig. 2b). This particular effect could be due to the strain recovery mechanism of CP. For ascorbic acid RDT, increase in compression pressure decreased the disintegration time when CCS and CP were used at their optimal concentrations (7 and 6%, respectively).
Fig. 2.
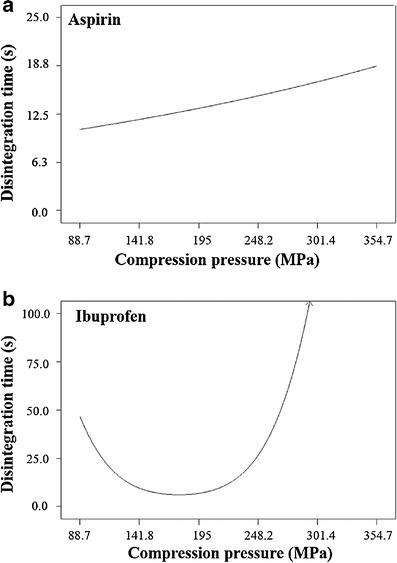
Effects of compression pressure on disintegration time of a aspirin (8% SSG) and b ibuprofen (8% CP) RDTs
Synergism Between Disintegrants
It is possible for synergism to occur between disintegrants with different mechanisms of action. For example, a swelling disintegrant may be complemented by another that draws in water. Since each disintegrant has been reported to have various mechanisms of action, it is conceivable that synergism behavior would occur with concurrence of reinforcing disintegration mechanisms or improvements in the physicochemical microenvironment for rapid disintegrant action.
Synergism was observed between SSG and CP in aspirin RDTs at all modeled compression pressures from 4 to 10% of total disintegrant concentration. A typical graph depicting the synergism was presented in Fig. 3a at a compression pressure of 354.7 MPa. The swelling and wicking abilities of SSG appeared to contribute positively to the strain recovery effect of CP, decreasing total disintegration time.
Fig. 3.
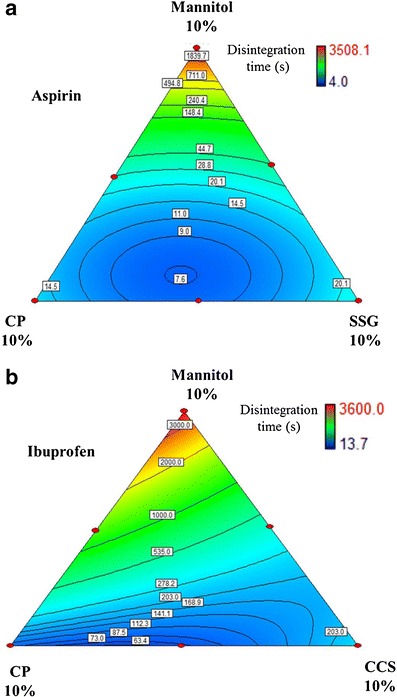
Effects of a SSG-CP composite concentration on aspirin RDTs disintegration time and b CP-CCS composite concentration on ibuprofen RDTs disintegration time (compression pressure, 354.7 MPa)
A similar behavior was observed for ibuprofen RDTs between CCS and CP at 354.7 MPa compression pressure (Fig. 3b). However, the effect was not seen at other modeled compression pressures. The effect became significant from 8 to 10% of total disintegrant concentration. Again, the viscoelastic recovery of CP under high strain might be important in synergism with CCS in this case.
Disintegrant Optimization
All tested RDTs had shorter disintegration times if CCS was used instead of SSG. Figure 4 demonstrates this effect for RDTs pressed at 177.3 MPa compression pressure. The phenomenon was more prominent at higher compression pressures. The observation showed that CCS performed as a disintegrant better than SSG under the experimental conditions used. No synergism was seen between CCS and SSG.
Fig. 4.
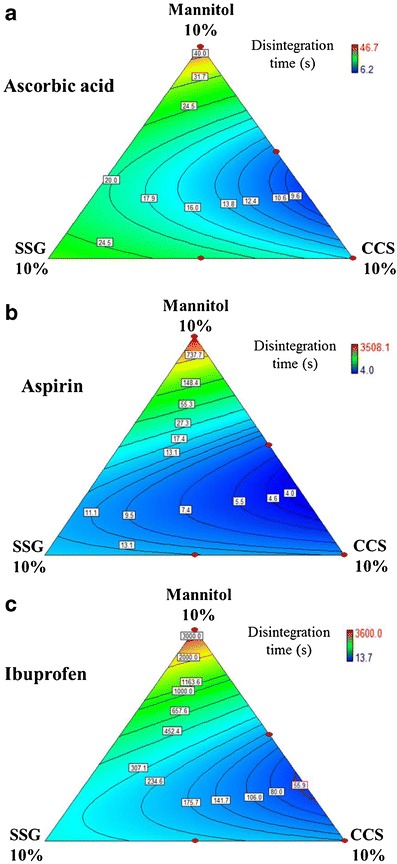
Effects of SSG-CCS composite concentration on disintegration time of a ascorbic acid, b aspirin, and c ibuprofen RDTs (compression pressure, 177.3 MPa)
Ascorbic acid RDTs had improved disintegration times when MCC was used instead of SSG at all compression pressures. Figure 5a depicts the effects of SSG–MCC composite concentration on the disintegration time of ascorbic acid RDTs prepared at 354.7 MPa compression pressure. MCC provided the capillary network for water intake into the tablet. The combined effect of extensive water entry with rapid dissolution of highly soluble ascorbic acid enhanced the swelling effect of SSG. This trade-off between SSG and MCC combination was similar to that between SSG and CP. Disintegration was improved by using CP in comparison with SSG. Figure 5b depicts the effects of SSG–CP composite concentration on the disintegration time of ascorbic acid RDTs prepared at 354.7 MPa compression pressure.
Fig. 5.
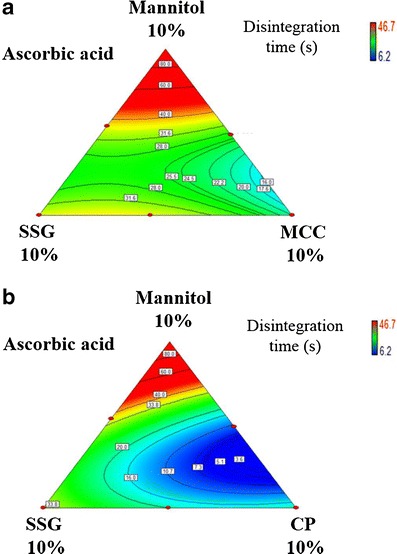
Effects of a SSG-MCC composite concentration and b SSG-CP composite concentration on ascorbic acid RDT disintegration time (compression pressure, 354.7 MPa)
Effect of Disintegrant Compositions on RDT Hardness
Hardness of aspirin RDTs was decreased when the disintegrant proportion increased. However, a minima existed, after which hardness values increased for MCC, CCS, and SSG. Hardness was lowest in aspirin RDTs when MCC or CCS was used at 6% and when SSG was 7% (Fig. 6). Relating the disintegration time (Fig. 1) and hardness value (Fig. 6), the disintegration time range of aspirin RDT was lowest when hardness range was low (also depicted by closed circles in Fig. 6). On the other hand, hardness varied linearly with disintegrant concentration in ibuprofen RDTs. CCS, SSG, and MCC increased hardness when used at higher concentrations while CP lowered hardness as the concentration was increased. For ibuprofen RDTs, disintegration time (Fig. 1) was not reduced at lower hardness value (Fig. 6). The decrease in hardness with CP was possibly due to the relative lack of hydrogen bonding sites, and hence, decreased the overall bond strength between tablet constituents. Cellulose excipients are generally good for hydrogen bonding. Therefore, the increase in CCS, SSG, or MCC concentrations had improved the hardness of ibuprofen RDTs (Fig. 6).
Fig. 6.
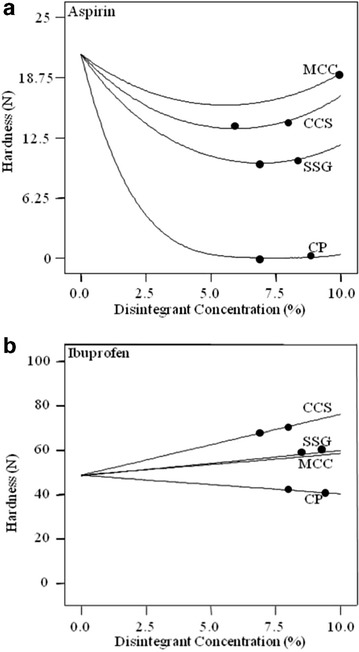
Effects of disintegrant concentration on hardness of a aspirin and b ibuprofen RDTs (compression pressure, 177.3 MPa)
Hardness values of aspirin RDTs were lower when MCC was combined with SSG than when with either disintegrant used alone (Fig. 7a). Hardness was lowest when SSG was used alone as compared to MCC and CCS. The best tablet hardness values with a single component disintegrant were achieved using MCC. Ibuprofen RDTs displayed a different relationship between tablet hardness and the disintegrant concentration used. Tablets formulated with CCS had the highest hardness values, while those containing MCC or SSG had similar hardness values. Figure 7b shows the effects of MCC-CCS disintegrant composites on ibuprofen RDTs at 177.3 MPa compression pressure.
Fig. 7.
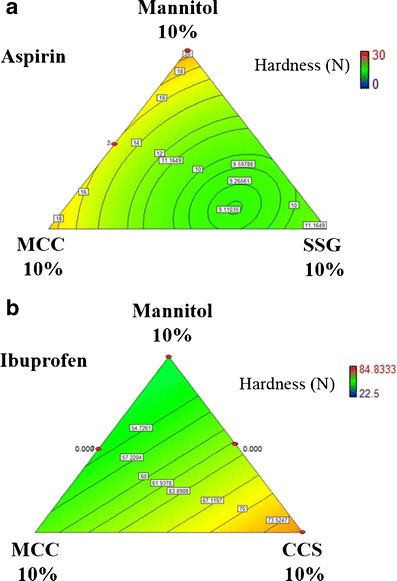
Effects of a MCC-SSG composite concentration on aspirin RDT hardness and b MCC-CCS composite concentration on ibuprofen RDT hardness (compression pressure, 177.3 MPa)
Formulation Optimization
Overlay plots of aspirin, ibuprofen, and ascorbic acid RDTs were studied to determine the disintegrant attributes and compression pressure from which robust fast disintegrating and sufficient hard tablets can be produced. For aspirin RDTs, MCC-CCS, SSG-CCS, and CP-CCS composite combinations at higher compression pressures (221.7–354.7 MPa) provided acceptable output responses. Here, the main disintegrant proposed was CCS at 4% or more. The use of other disintegrants would be optional. Figure 8a shows a sample overlay plot for aspirin RDTs depicting an acceptable region (white region) of CCS-SSG composite combination requirements at 354.7 MPa compression pressure to produce RDTs with required output responses. Areas that did not meet the feasible output response criteria are shaded gray. Overlay plots for other disintegrant composite combinations at different compression pressures can be similarly generated for aspirin RDTs.
Fig. 8.
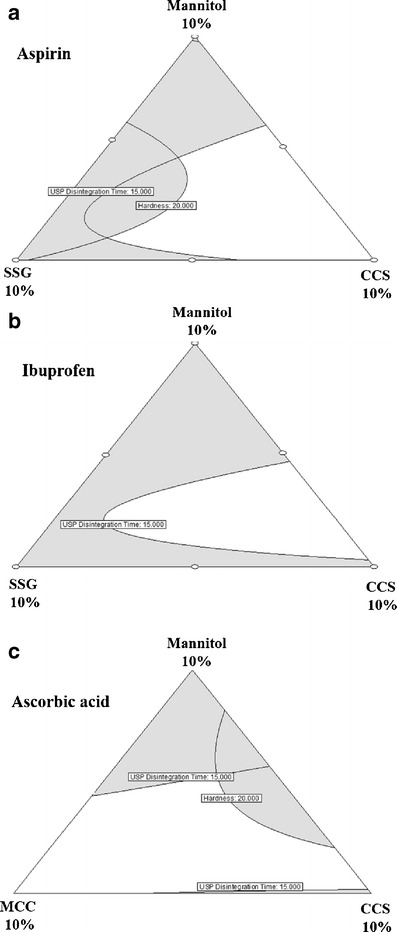
Overlay plots for the effect of disintegrant attributes on disintegration time and hardness of RDTs. a Aspirin (compression pressure—354.7 MPa), b ibuprofen (compression pressure—88.7 MPa), and c ascorbic acid (compression pressure—221.7 MPa)
Ibuprofen RDTs had sufficient hardness even when prepared with 88.7 MPa compression pressure. Low melting point (75–77°C) of ibuprofen could be responsible for particle-particle fusion while compressing ibuprofen RDTs, leading to stronger tablets (33,34). The suggested single disintegrant for ibuprofen RDT was CCS (≥4.5% at compression pressure 88.7 MPa) or disintegrant composite combinations for ibuprofen RDTs were SSG-CP (compression pressure range, 88.7–239.4 MPa), MCC-CP (compression pressure range, 88.7–239.4 MPa), MCC-CCS (compression pressure range, 88.7–133 MPa), SSG-CCS (compression pressure range, 88.7–133 MPa) and CP-CCS (compression pressure range, 88.7–239.4 MPa). Figure 8b shows a sample overlay plot for ibuprofen RDTs depicting SSG-CCS composite combination requirements at 88.7 MPa compression pressure.
For ascorbic acid RDTs, any two disintegrant composite combinations (total 4.5–5%) used in the study provided acceptable output responses when compressed in the range of 177.4–354.7 MPa. Here, the main disintegrants proposed were CCS (≥4.5%) and MCC (≥5%) which can produce acceptable ascorbic acid RDTs even when used alone. Figure 8c shows a sample overlay plot for ascorbic acid RDTs depicting MCC-CCS composite combinations requirements at 221.7 MPa compression pressure.
Based upon all the overlay plot analysis, the suggested optimized combinations of each particular RDT formulations are presented in Table IV.
Table IV.
Optimized Combinations of RDT Formulations
| RDT | Disintegrant(s) | Pressurea (MPa) | Other ingredients | Output responses |
|---|---|---|---|---|
| Aspirin | CCS (4%) | 354.7 | API (50%), Mg St (0.5%), mannitol (45.5%) | DT ≤15 s, hardness ≥20 N |
| Ibuprofen | CCS (4.5%) | 88.7 | API (50%), Mg St (0.5%), mannitol (45%) | DT ≤15 s, hardness ≥20 N |
| Ascorbic acid | CCS (3%) + MCC (1.5%) | 221.7 | API (50%), Mg St (0.5%), mannitol (45%) | DT ≤15 s, hardness ≥20 N |
DT disintegration time, API active pharmaceutical ingredients, CCS croscarmellose sodium, Mg St magnesium stearate, RDT rapidly disintegrating tablet
aCompression pressure
Analysis of any three disintegrant combinations showed that the total disintegrant amount required was similar to single or combination of two disintegrants in providing acceptable outcome responses in any of RDT formulations. Thus, either single disintegrant or combination of two disintegrants is suggested. The final decision may be considered based on relative excipient cost and product stability profiles of formulated products.
CONCLUSIONS
RDTs incorporating active ingredients of different water solubilities were investigated for the effect of different disintegrants on disintegration time and tablet hardness. DOE and RSM proved to be important and useful tools to investigate the effect of disintegrant concentrations, disintegrant combinations, and compression pressure on disintegration time and hardness of RDTs. This study serves as a model for product development based on the QbD framework with specifications of excipients in ranges within the designed acceptance space for optimal product performance.
Acknowledgments
The authors would like to acknowledge funding support from GEA-NUS PPRL (N-148-000-008-001) and SERC Grant No. 102 169 0049 (R-148-000-157-305).
References
- 1.Food and Drug Administration. Guidance for industry. Orally disintegrating tablets. Silver Spring: US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER); 2008.
- 2.Goel H, Vora N, Rana V. A novel approach to optimize and formulate fast disintegrating tablets for nausea and vomiting. AAPS PharmSciTech. 2008;9(3):774–81. doi: 10.1208/s12249-008-9113-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haware RV, Chaudhari PD, Parakh SR, Bauer-Brandl A. Development of a melting tablet containing promethazine HCl against motion sickness. AAPS PharmSciTech. 2008;9(3):1006–15. doi: 10.1208/s12249-008-9133-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu Y, Jeong SH, Park K. Fast-melting tablets based on highly plastic granules. J Control Release. 2005;109(1–3):203–10. doi: 10.1016/j.jconrel.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 5.Douroumis D. Orally disintegrating dosage forms and taste-masking technologies; 2010. Exp Opin Drug Deliv. 2011;8(5):665–75. doi: 10.1517/17425247.2011.566553. [DOI] [PubMed] [Google Scholar]
- 6.Desai PM, Liew CV, Heng PW. Assessment of disintegration of rapidly disintegrating tablets by a visiometric liquid jet-mediated disintegration apparatus. Int J Pharm. 2013;442(1–2):65–73. doi: 10.1016/j.ijpharm.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Saigal N, Baboota S, Ahuja A, Ali J. Fast-dissolving intra-oral drug delivery systems. Exp Opin Ther Pat. 2008;18(7):769–81. doi: 10.1517/13543776.18.7.769. [DOI] [Google Scholar]
- 8.Jeong SH, Fu Y, Park K. Frosta®: a new technology for making fast-melting tablets. Exp Opin Drug Deliv. 2005;2(6):1107–16. doi: 10.1517/17425247.2.6.1107. [DOI] [PubMed] [Google Scholar]
- 9.Bi Y, Sunada H, Yonezawa Y, Danjo K, Otsuka A, Iida K. Preparation and evaluation of a compressed tablet rapidly disintegrating in the oral cavity. Chem Pharm Bull. 1996;44(11):2121–7. doi: 10.1248/cpb.44.2121. [DOI] [PubMed] [Google Scholar]
- 10.Corveleyn S, Remon JP. Formulation and production of rapidly disintegrating tablets by lyophilisation using hydrochlorothiazide as a model drug. Int J Pharm. 1997;152(2):215–25. doi: 10.1016/S0378-5173(97)00092-6. [DOI] [Google Scholar]
- 11.Watanabe Y, Koizumi KI, Zama Y, Kiriyama M, Matsumoto Y, Matsumoto M. New compressed tablet rapidly disintegrating in saliva in the mouth using crystalline cellulose and a disintegrant. Biol Pharm Bull. 1995;18(9):1308–10. doi: 10.1248/bpb.18.1308. [DOI] [PubMed] [Google Scholar]
- 12.Van Scoik KG, inventor; Abbott Laboratories, assignee. Solid pharmaceutical dosage in tablet triturate form and method of producing same. United States patent 5082667. 1992 Jan 21.
- 13.Allen Jr LV, Wang B, Davies JD, inventors; The Board of Regents of the University of Oklahoma, Janseen Pharmaceutica, Inc., assignees. Method of making a rapidly dissolving tablet. United States patent 5635210. 1997 Jun 3.
- 14.Shah U, Augsburger L. Evaluation of the functional equivalence of crospovidone NF from different sources. I. Physical characterization. Pharm Dev Technol. 2001;6(1):39–51. doi: 10.1081/PDT-100000012. [DOI] [PubMed] [Google Scholar]
- 15.Bi YX, Sunada H, Yonezawa Y, Danjo K. Evaluation of rapidly disintegrating tablets prepared by a direct compression method. Drug Dev Ind Pharm. 1999;25(5):571–81. doi: 10.1081/DDC-100102211. [DOI] [PubMed] [Google Scholar]
- 16.Gissinger D, Stamm A. A comparative evaluation of the properties of some tablet disintegrants. Drug Dev Ind Pharm. 1980;6(5):511–36. doi: 10.3109/03639048009068720. [DOI] [Google Scholar]
- 17.Kornblum SS, Stoopak SB. A new tablet disintegrating agent: cross-linked polyvinylpyrrolidone. J Pharm Sci. 1973;62(1):43–9. doi: 10.1002/jps.2600620107. [DOI] [PubMed] [Google Scholar]
- 18.Gould PL, Tan SB. The effect of recompression on the swelling kinetics of wet massed tablets, containing ‘super’ disintegrants. Drug Dev Ind Pharm. 1985;11(9–10):1819–36. doi: 10.3109/03639048509057701. [DOI] [Google Scholar]
- 19.Shu T, Suzuki H, Hironaka K, Ito K. Studies of rapidly disintegrating tablets in the oral cavity using co-ground mixtures of mannitol with crospovidone. Chem Pharm Bull. 2002;50(2):193–8. doi: 10.1248/cpb.50.193. [DOI] [PubMed] [Google Scholar]
- 20.Desai PM, Liew CV, Heng PW. Understanding disintegrant action by visualization. J Pharm Sci. 2012;101(6):2155–64. doi: 10.1002/jps.23119. [DOI] [PubMed] [Google Scholar]
- 21.Moreton RC. Disintegrants in tableting. In: Augsburger LL, Hoag SW, editors. Pharmaceutical dosage forms: tablets. 3. New York: Informa Healthcare; 2008. pp. 217–50. [Google Scholar]
- 22.Douroumis DD, Gryczke A, Schminke S. Development and evaluation of cetirizine HCl taste-masked oral disintegrating tablets. AAPS PharmSciTech. 2011;12(1):141–51. doi: 10.1208/s12249-010-9569-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martino PD, Martelli S, Wehrlé P. Evaluation of different fast melting disintegrants by means of a central composite design. Drug Dev Ind Pharm. 2005;31(1):109–21. doi: 10.1081/DDC-44233. [DOI] [PubMed] [Google Scholar]
- 24.Kasliwal N, Negi JS, Jugran V, Jain R. Formulation, development, and performance evaluation of metoclopramide HCl oro-dispersible sustained release tablet. Arch Pharm Res. 2011;34(10):1691–700. doi: 10.1007/s12272-011-1013-3. [DOI] [PubMed] [Google Scholar]
- 25.Zhao N, Augsburger LL. Functionality comparison of 3 classes of superdisintegrants in promoting aspirin tablet disintegration and dissolution. AAPS PharmSciTech. 2005;6(4):E634–40. doi: 10.1208/pt060479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Awotwe-Otoo D, Agarabi C, Wu GK, Casey E, Read E, Lute S, et al. Quality by design: impact of formulation variables and their interactions on quality attributes of a lyophilized monoclonal antibody. Int J Pharm. 2012;438(1–2):167–75. doi: 10.1016/j.ijpharm.2012.08.033. [DOI] [PubMed] [Google Scholar]
- 27.Charoo NA, Shamsher AAA, Zidan AS, Rahman Z. Quality by design approach for formulation development: a case study of dispersible tablets. Int J Pharm. 2012;423(2):167–78. doi: 10.1016/j.ijpharm.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 28.Park SJ, Choo GH, Hwang SJ, Kim MS. Quality by design: screening of critical variables and formulation optimization of Eudragit e nanoparticles containing dutasteride. Arch Pharm Res. 2013;36(5):593–601. doi: 10.1007/s12272-013-0064-z. [DOI] [PubMed] [Google Scholar]
- 29.Verma S, Lan Y, Gokhale R, Burgess DJ. Quality by design approach to understand the process of nanosuspension preparation. Int J Pharm. 2009;377(1–2):185–98. doi: 10.1016/j.ijpharm.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, Cho BR, Melloy BJ. Quality by design studies on multi-response pharmaceutical formulation modeling and optimization. J Pharm Innov. 2013;8(1):28–44. doi: 10.1007/s12247-012-9145-7. [DOI] [Google Scholar]
- 31.European pharmacopeia, dosage forms, tablets. 67075 Strasbourg, France: Council of Europe; 2008. [Google Scholar]
- 32.Kakutani R, Muro H, Makino T. Development of a new disintegration method for orally disintegrating tablets. Chem Pharm Bull. 2010;58(7):885–90. doi: 10.1248/cpb.58.885. [DOI] [PubMed] [Google Scholar]
- 33.Di Martino P, Beccerica M, Joiris E, Palmieri GF, Gayot A, Martelli S. Influence of crystal habit on the compression and densification mechanism of ibuprofen. J Cryst Growth. 2002;243(2):345–55. doi: 10.1016/S0022-0248(02)01523-3. [DOI] [Google Scholar]
- 34.Le VN, Leterme P, Gayot A, Flament MP. Influence of granulation and compaction on the particle size of ibuprofen—development of a size analysis method. Int J Pharm. 2006;321(1–2):72–7. doi: 10.1016/j.ijpharm.2006.05.010. [DOI] [PubMed] [Google Scholar]