

Abstract
Among the many scientific advances to come from the study of nanoscience, the development of ligand-targeted nanoparticles to eliminate solid tumors is predicted to have a major impact on human health. There are many reports describing novel designs and testing of targeted nanoparticles to treat cancer. While the principles of the technology are well demonstrated in controlled lab experiments, there are still many hurdles to overcome for the science to mature into truly efficacious targeted nanoparticles that join the arsenal of agents currently used to treat cancer in humans. One of these hurdles is overcoming unwanted biodistribution to the liver while maximizing delivery to the tumor. This almost certainly requires advances in both nanoparticle stealth technology and targeting. Currently, it continues to be a challenge to control the loading of ligands onto polyethylene glycol (PEG) to achieve maximal targeting. Nanoparticle cellular uptake and subcellular targeting of genes and siRNA also remain a challenge. This review examines the types of ligands that have been most often used to target nanoparticles to solid tumors. As the science matures over the coming decade, careful control over ligand presentation on nanoparticles of precise size, shape, and charge will likely play a major role in achieving success.
KEY WORDS: cancer, nanoparticle, targeted delivery
INTRODUCTION
Nanoparticles, such as liposomes and gene delivery polyplexes, are designed to carry drug or gene cargos and maximize payload delivery to tumor cells. The size of nanoparticles restricts their ability to passively diffuse into organs that lack fenestrated endothelial cells. The liver, spleen, and bone marrow possess natural fenestrated endothelial cells that allow nanoparticles of <100 nm to extravagate (1). Solid tumors as well as other diseased tissues such as inflamed joints possess an unnatural vasculature through which nanoparticles of micrometer size traverse through leaky tight junctions in the vascular endothelium and accumulate in the extracellular space of the diseased tissue (2).
Successful nanoparticle delivery depends on optimal passive targeting to direct accumulation in the tumor and active targeting to gain intracellular access (3). Passive targeting takes advantage of the propensity for nanoparticles to selectively accumulate approximately 10% of dose in a solid tumor (4). Matsumara and Maeda first described tumors as tightly packed with blood vessels yet also having a leaky vasculature, which results in the “enhanced permeability and retention effect” (EPR). They noted that molecules larger than 30–45 kDa are retained within solid tumors while smaller molecules are able to diffuse back into the blood stream (5,6).
To take advantage of the EPR effect, nanoparticles must be biocompatible with the blood and must also avoid accumulation in the liver and spleen. The charge on nanoparticles is a key factor for achieving these attributes. Negatively and neutrally charged nanoparticles are much more biocompatible compared to positively charged particles as a result of less protein binding in the blood (7). Albumin is the most abundant blood protein at a concentration of 50 mg/ml. Its overall negative charge results in ionic binding to positively charged nanoparticles resulting in aggregation, charge reversal, and entrapment in the lung (8,9). However, blood proteins also bind to all nanoparticles which can lead to accumulation of ligands on negatively and neutrally charged nanoparticles that influence their biodistribution (10).
In addition to blood protein binding, nanoparticle biodistribution is strongly influenced by scavenger receptor binding and internalization into Kupffer cells and fenestrated endothelial cells of the liver (11–14). Scavenger receptors bind negatively charged nanoparticles, accounting for the large percent of the dose (50–80%) that is immediately captured by the liver (12,15). Nearly all liposomes, polyplexes, and nanoparticles are made biocompatible by attaching polyethylene glycol (PEG) (16–20). Covalent attachment of a surface layer of PEG onto the nanoparticle masks the nanoparticle charge. The length and density of the PEG attached to the nanoparticles significantly influence the efficiency of charge masking (21). If the polyplexes are positively charged, a PEG layer that partially blocks positive charge will not completely block albumin binding but will minimize albumin-induced aggregation (22,23). A primary goal of attaching PEG to nanoparticles is to mask charge and minimize protein binding to allow nanoparticles to evade scavenger receptor recognition. Under these conditions, nanoparticle accumulation in the liver over time is approximately 10% of dose (24), which allows the remainder of the dose to freely circulate. Since the known family of scavenger receptors only binds anionic molecules, cationic PEGylated polyplexes are presumed to undergo charge reversal in the blood as a result of albumin binding, leading to their recognition by scavenger receptors in the liver (25).
While PEGylated nanoparticles are more biocompatible, they are also more difficult to functionalize with ligands. Small molecule ligands directly attached to nanoparticles are masked by covalently attached PEG (26). This necessitates the development of a bioconjugation strategy to attach the ligand to the end of a PEG spacer, resulting in tethering of ligands to the nanoparticle surface (26). Due to the flexibility of PEG, this strategy does not guarantee proper ligand presentation, which may be buried in the PEG layer surrounding the nanoparticle (Fig. 1). In addition to the uncertainties of ligand presentation to allow receptor recognition, the attachment of ligand to the ends of PEG is chemically challenging. Most often, this requires the use of heterobifunctionalized PEGs that possess unique terminal groups that react selectively with a functional group on the ligand and the nanoparticle surface. Approaches to achieve bioconjugation of ligands to PEG on liposomes and other nanoparticles has been recently reviewed (26). While it is most convenient to react a heterobifunctionalized PEG with the nanoparticle and then load the ligand onto the PEGylated nanoparticle in a second reaction, it is also more difficult to chemically characterize the assembled nanoparticle system, compared to preparing and characterizing a PEG-ligand conjugate. The lack of careful chemical characterization leads to undefined and heterogeneous ligand density that ultimately causes irreproducible targeting results. However, attempts to directly label PEGs with ligands prior to loading onto nanoparticles are also complicated by the difficulty of chromatographically separating labeled PEGs from unlabeled PEGs and uncertainties in the mass spectral characterization of polydisperse ligand-modified PEGs. These complications multiply as the PEG length is increased, which is often an inevitable consequence of the need to achieve a sufficient PEG layer to avoid scavenger receptor binding. Even when performed with great care, modification of ligands with PEG can disrupt receptor recognition, depending on the location of the functional group on the ligand and the type of reactive group on PEG. Still, most nanoparticle formulations are PEGylated and proper ligand presentation is assumed.
Fig. 1.
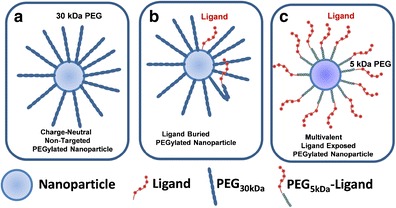
Ligand presentation on PEGylated nanoparticles. a The influence of PEG to mask surface charge. b Ligand presentation is masked by 30 kDa PEG and by PEG folding. c A sufficiently short PEG modified with ligand on the termini can result in exposed ligand. It is essential to find the shortest PEG that masks nanoparticle charge to maximize ligand exposure
Evading detection by scavenger receptors on nonparenchymal cells in the liver is the first major obstacle to achieve tumor targeting (27). However, the liver possesses a second potential trap to sequester nanoparticles. The liver-fenestrated endothelial cells allow extravagation of small nanoparticles (<100 nm) into the space of Disse that surrounds hepatocytes (28). The space of Disse is composed of extracellular matrix proteins including collagen and proteoglycans. Compared to the saturable binding capacity of scavenger receptors on nonparenchymal cells, the space is very large and difficult to saturate (15,29,30). Positively charged PEGylated polyplexes that evade capture by scavenger receptors on Kupffer cells and fenestrated endothelial cells bind and accumulate in the space of Disse (25). Negatively and neutrally charged nanoparticles that are sufficiently small (<100) also cross the liver-fenestrated endothelial cells and sample the space of Disse, but appear not to accumulate in the liver most likely because of their lack of ionic binding to extracellular matrix proteins (24,25). Some evidences suggest a similar charge relationship exists for extravagated nanoparticles binding ionically to the tumor extracellular matrix (31).
LIGAND PRESENTATION
The selection of a targeting ligand is primarily dictated by the receptors present on the target cells. Not all receptors are suitable for targeting nanoparticles. Cell surface receptors that bind their ligand but do not internalize are generally inadequate for the delivery of poorly diffusible cargos such as DNA and siRNA. Since nanoparticles are typically 50–100 nm in diameter, receptors that undergo receptor-mediated endocytosis through coated pits and recycle to the cell surface are considered necessary because they are capable of internalizing larger cargo. The receptor and ligand typically dissociate in endosomes, with the ligand continuing to traffic to lysosomes. Disrupting the intracellular trafficking by effecting endosomal lysis to release the cargo into the cytosol is an important event for successful gene and siRNA delivery (32).
In addition to endocytosis, receptors present in abundance on target cells are an advantage toward achieving in vivo efficacy. Some naturally occurring receptors are present at 10 (6) per cell; however, this is rarely experienced on cancer cells that most often upregulate or selectively express a receptor in lower abundance. To achieve greater selectivity over normal cells, some targeting antibodies are directed against cancer cell surface molecules, such as mucines or glycolipids. In addition to these considerations, ligands should have high affinity (dissociation constant (Kd) = nM) for binding the cell surface receptors, although lower binding affinity can be compensated through clustering as discussed below (16,33). Extensive structure activity relationship (SAR) knowledge of the ligand is essential to successfully attach it to PEG while maintaining its receptor binding affinity.
While developing targeted nanoparticle delivery systems, it is advantageous to perform preliminary tests in vitro on receptor expressing cells (34). Often, these may be cancer cell lines that have been shown to express the receptor endogenously. Alternatively, an appropriate cell can be derived from a cancer cell line that lacks the receptor by genetical transformation to achieve stable expression of the receptor. Transformed cell lines offer the advantage of direct comparison with a nontransformed receptor cell line. A major disadvantage of this approach is variability and unpredictability in the number of receptors expressed. Also, expressed receptors may lose their ability to endocytose ligands. In either case, it is important to verify the presence of cell surface receptors which is normally performed by fluorescence assisted cell sorting (FACS) using a fluorescently labeled antibody. This verification is important since subtle experimental parameters, such as cell passage number, can influence the level of expression of the cell surface receptors. When developing targeted nanoparticles for in vitro binding or uptake assays, essential controls must be taken into consideration which include the use of receptor-blocking antibodies and blocking with excess ligand or antagonists. Most in vitro assays also require the use of a radiolabel or fluorophore to monitor nanoparticle binding to receptors (34).
Clustering ligands on nanoparticles has a major benefit of enhancing receptor binding affinity (Fig. 2). This well-known phenomenon can amplify ligand affinity by several orders of magnitude due to the simultaneous occupation of receptor binding sites on the cell surface (35–41). The multivalent effect also opens the possibility of incorporating two different ligands to increase affinity and selectivity for cells expressing multiple target receptors. High binding affinity is essential to achieve in vivo efficacy since circulating nanoparticles are subject to strong shearing pressures in the vasculature (35,42).
Fig. 2.
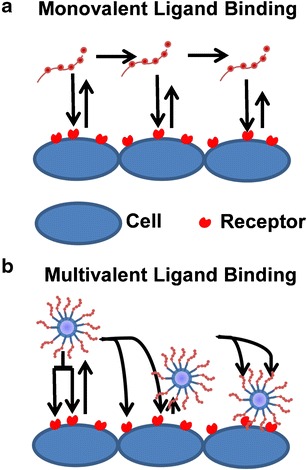
Benefits of multivalent nanoparticles. Nanoparticles are subjected to forces such as rotation and moment in the blood. A single ligand can bind its receptor diminishing these forces slowly with each interaction (a). Multivalent nanoparticles can be bound by multiple receptors simultaneously leading to increased receptor-ligand interactions. The exponential increase in affinity and increased interactions caused by the multivalency of the nanoparticle increase the amount of nanoparticles that remain bound and that consequently can be internalized (b)
The importance of a targeting ligand for nanoparticle accumulation in solid tumor is dependent on the specific application. Most studies conclude that targeting ligands do not increase the percent of nanoparticle dose accumulated in solid tumor, which is dictated by EPR, but targeting ligands do increase nanoparticle binding and internalization into tumor cells (Fig. 3) (43,44). For example, Wu et al. used tumor xenografts to show an increase in tumor localization of (64) Cu-labeled antibody fragments conjugated to DOTA when containing a targeting ligand (45). Likewise, Hussain et al. showed tritium-labeled PEGylated liposomes (110 ± 10 nm) containing an antiepithelial cell adhesion molecule antibody, conjugated with a cysteine-maleimide linkage on the distal end of PEG on stabilized liposomes, had the same biodistribution and pharmacokinetics as nontargeted liposomes (46). Similarly, Kirpotin et al. tracked (67) Ga- or gold-loaded lipid nanoparticles (80–100 nm) targeted with anti-HER2 antibodies, conjugated with terminal cysteine-maleimide linkage on the end of PEG, and found a similar biodistribution and pharmacokinetics between targeted and controls (47). However, the nontargeted particles remained in the tumor stroma while targeted particles were internalized by the cells. In addition, Davis used positron emission tomography (PET) and bioluminescent imaging to quantify the biodistribution of cyclodextrin nanoparticles (90–125 nm, ~+10 mV) containing (64) Cu-labeled siRNA with or without the transferrin ligand. Moreover, they determined that targeted nanoparticles, where transferrin-carbohydrate was attached to the distal end of PEG, showed equivalent uptake in tumors as untargeted molecules due to the EPR effect, but the targeted molecules were capable of internalization leading to superior gene delivery and siRNA knockdown (44).
Fig. 3.
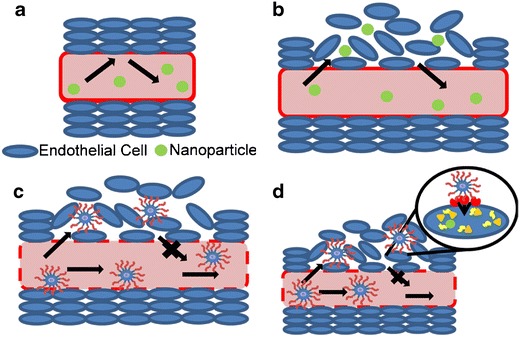
The EPR effect and targeting nanoparticles. Normal endothelial cells line the vasculature forming a barrier that prevents nanoparticle entrance (a). However, tumors form a leaky vasculature that allows small nanoparticles entrance and exit from the vasculature to the extracellular space (b). Larger PEGylated nanoparticles gain access to the extracellular space of the tumor and their larger size cause them to become trapped (c). Targeted nanoparticles also gain entrance into the tumor through the EPR effect, but the multivalent ligand receptor interactions result in cellular uptake of the nanoparticle where the therapeutic cargo can be released
ANTIBODIES
Antibodies and FABs are some of the most selective and potent targeting agents to deliver nanoparticles to tumor cells. The vast subject of immunoliposomes targeting encapsulated anticancer agents, DNA and siRNA, has been recently reviewed and will only be touched on briefly for completeness (2,34). As discussed earlier, the development of humanized monoclonal antibodies and single-chain FABs to target nanoparticles is largely dependent on the type of cancer and the antigenic epitope being targeted. The recent review by Pazko and Senge is comprehensive in its coverage of this subject (34). However, as described by Nobs et al., a major difficulty still lies in developing reliable chemistry to attach antibodies to liposomes and nanoparticles. Despite the commercial availability of an array of heterobifunctionalized PEGs of different lengths and chemistry that allow a diversity of chemical conjugation approaches, the low efficiency of attaching antibodies to the surface of PEGylated nanoparticles and liposomes continues to be a major challenge to this field (34).
APTAMERS
Aptamers are DNA or RNA sequences that fold into a secondary structure that allow the evolution of high-affinity targeting agents for cell surface receptors (4,48). The development and use of aptamers has been extensively reviewed by Cho et al. in 2011 and Song et al. in 2012 (49,50). Aptamers have advantages over antibodies in that they can be produced rapidly and are nonimmunogenic (51). However, aptamers are rapidly cleared by renal filtration and are degraded by nucleases if not modified with protecting groups including: 2′-fluorine-substituted pyrimidines, polyethylene glycol (PEG), 3′capping, and locking nucleic acids (52). The first use of aptamers (AS1411, Aptamera) in humans was a guanosine-rich oligonucleotide that inhibits growth of tumors (53). Wilner et al. targeted siRNA containing stable nucleic acid lipid particles (SNALPS) to the transferrin receptor where the aptamer-guided SNALPS bound to Jurkat T cells and were rapidly internalized as shown via flow cytometry even when incubated with endogenous levels of serum transferrin (54). Other aptamer targets include the HER2 receptor in breast cancer tissue, the tyrosine kinase receptor Axl and the Muc1 receptor on cancer cells (55–57). Polyethylenimine (58) polyplexes targeted with Muc-1 aptamers resulted in a 10-fold increase in gene expression over nontargeted polyplexes and a twofold increase over PEI polyplexes alone. In vivo results showed aptamer-targeted PEI polyplexes achieved approximately threefold higher expression in mice tumors compared to nontargeted polyplexes (59).
TRANSFERRIN
Transferrin is an iron-binding beta-globulin found in plasma that binds to transferrin receptors on the plasma membrane of cells and transfers ferric ions (4,60). The receptor is expressed in most human tissues, but is upregulated in tumor tissue (4,60). Upon binding transferrin, transferrin receptors are internalized and the ferric ions are released in the late endosome at pH 6 and transported to the cytoplasm through an unknown mechanism. The surface receptor is then recycled back to the cell membrane. This mechanism and its use in drug delivery were extensively reviewed in 2011 by Daniels et al. (60). Weissleder’s lab designed a system utilizing cells engineered to overexpress transferrin receptors in which uptake of mono-crystalline iron oxide nanocompounds could be imaged (61,62). Also, Davis et al. performed the first phase I clinical trial by systemically administering siRNA directed against the M2 subunit of ribonucleotide reductase to patients with solid tumors using a transferrin-targeted PEGylated cyclodextrin-based polymer. Preclinical data showed decreased RRM2 messenger RNA (mRNA) levels in tumor samples (clinical trial NCT00689065) (63). Another phase I clinical trial has begun in which transferrin receptors are targeted with an antibody coupled to liposomes containing plasmid encoding the tumor suppressor gene p53. Results thus far confirm the safety of the liposomes and dose-dependent transgene expression (64). Other uses of transferrin for targeted gene transfer include coating herpes virus with transferrin-DOTAP, cholesterol liposomes, and other studies with phage λ particles coupled to holotransferrin (diferric transferrin) which are capable of targeting green fluorescent protein (GFP) to human 293T cells (65).
FOLATE TARGETING
Folate (vitamin B9) is transported into the cells via a reduced folate carrier that functions as a bidirectional anion transporter, which delivers folate across the plasma membrane of cells in all tissues (66,67). Following ligand binding, folate receptors (FRs) internalize via receptor-mediated endocytosis and constitutively recycled back to the cell surface. Since folate is an essential nutrient building block for the synthesis of purines and pyrimidines, the FR is often upregulated in malignant cells and each of the four receptor isoforms (alpha, beta, gamma, and delta) can be differentially upregulated in different cancer subsets (66–69).
Shiokawa et al. demonstrated attachment of folate-PEG of varying PEG molecular weight to lipid emulsions resulted in a ligand-dependent decrease in tumor volume in xenografted mice (69). Lee et al. probed multivalent presentation of folate by assembling oligonucleotide particles (ONPs) that consist of six double-stranded DNA molecules and six double-stranded siRNA molecules which form nanoparticles using the overhang hybridization method. SiRNA gene knockdown required the proper display of a minimum of three folate molecules. Biodistribution analysis indicated an increase in fluorescently labeled ONPs when targeted with folate (70). Polyamidoamine dendrimer conjugates of cyclodextrin and folate-PEG were used to target siRNA to demonstrate dose- and receptor-dependent knockdown in FR-expressing cells versus non-FR-expressing cells (71). Adenoviral vectors coated with folate-PEI or folate-PEG demonstrated enhanced gene delivery. Similarly, folate-targeted cationic liposomes or nanolipoplexes have been recently published (72–75).
There is strong evidence that inflammation is a contributing factor in the progression of cancer (20,76–80). Folate receptor β (FRβ) has been shown to be upregulated in activated macrophages, present in inflammatory disease, but absent on other blood cells or quiescent macrophages. The activation of macrophages to selectively internalize folate-modified nanoparticles makes folate an attractive ligand for targeting nanoparticles to treat inflammation (67). The link between inflammation and the FR dates back to 1988 when Furst et al. found that methotrexate was a FR antagonist with anti-inflammatory effects (81–83). Folate has also been incorporated into nanoparticles encapsulating fluorescent dye IR750 to image cites of inflammation (84). The capability for folate modified nanoparticles to target malignant cells pose an opportunity for a novel two-front treatment approach for chemotherapeutics (85,86).
ANISAMIDE
Anisamide is a small molecule ligand that binds membrane-bound sigma receptors. Sigma receptors are overexpressed in different malignancies including melanoma, non-small lung carcinoma, some breast tumors, and prostate cancer (87,88). Huang’s group has extensively studied the incorporation of anisamide into cancer targeting nanoparticles. Liposome-protamine-heparin (LPH) PEG anisamide nanoparticles were used to deliver the peptide EEEEpYFELV (EV) to block downstream phosphorylation of EGFR in human H60 lung cancer cells (89). In another example, anisamide was used to target lipid-calcium-phosphate nanoparticles to deliver siRNA to HDM2, c-myc, and VEGF (90). Moreover, VEGF was used to co-dose siRNA and gemcitabine (91). Anisamide liposome protamine mRNA nanoparticles were also used to co-deliver mRNA for the herpes simplex virus 1-thymidine kinase (HSV-tk) and ganciclovir (92). In all cases, in vitro data suggested improved targeted uptake and expression in cancer cell lines as well as increased cell toxicity. These results are confirmed in vivo where anisamide-targeted nanoparticles demonstrated improved biodistribution of nanoparticles to the tumor and decreased tumor volumes over nontargeted controls. Experiments with co-dosed treatments of siRNA and anticancer agents led to synergistic effects over nontargeted or individual treatments. Anisamide targeting was also used to target chitosan-PEG-anisamide nanoparticles to A549 lung epithelial cancer cells in order to deliver gemcitabine (87). The results demonstrated a ligand-dependent increase in cell toxicity and increased tumor uptake and decreased tumor volume in mice xenograft models. Similarly, cyclodextrin-PEG-anisamide nanoparticles targeting PC3 prostate tumor cells were used to deliver anti-VEGF siRNA and exhibited 80% silencing of luciferase and decreased tumor volume over nontargeted controls (88).
PEPTIDE LIGANDS
Peptide-mediated targeting is an attractive approach due to the diversity of target receptors that are upregulated on cancer cells (4,93,94). The most common peptide used for targeting is arginine-glycine-aspartic acid (RGD) (95). RGD peptides bind integrins on the cell surface that affect cell migration, growth, differentiation and apoptosis, in addition to cell-cell interactions (18,96). Integrin αvβ3 that bind to RGD peptides with high affinity is involved in intracellular signaling and direct roles in tumor angiogenesis (97–100). Cyclizing RGD peptides increases receptor binding affinity; however, scrambling the peptide sequence or modifying the amino acids removes all binding capability (96). Recent uses of RGD peptides for treating cancer include conjugation of PEGylated RGD peptides to gold nanoshells. These nanoparticles targeted U87 glioblastoma cells with a 267-nM IC50. This simultaneously allowed imaging of (64) Cu-labeled nanoparticles in a rat tumor model and increased thermal necrosis of tumors in mice (93). Most recently, cyclic RGD-targeted polymeric micelles loaded with the oxaliplatin parent drug (1,2-diaminocyclohexane)platinum(II) (DACHPt) effectively showed in vitro drug accumulation in a glioblastoma cell line and produced a fivefold decrease in luminescent intensity in a luciferase-expressing mouse brain tumor model (101). Other recent uses of RGD peptides for nanoparticle targeting to cancer are summarized in a 2013 review by Marelli et al. (102).
Bombesin is a 14-amino acid peptide that targets the G protein-coupled receptor (GPCR) family, neuromedin B (NMBR), and gastrin releasing peptide receptor (GRPR). Bombesin receptors are upregulated in human tumor cell membranes including breast, prostate, small-cell lung, and pancreatic cancer and have been used to target imaging agents and cytotoxic drugs to tumors (4,103). Ming et al. attached bombesin to the 5′ end of a splice-shifting oligonucleotide that can repair a splice variant in luciferase-reporter cells. The authors reported a 30-fold increase in luciferase mRNA and a threefold increase in luciferase expression in PC3 prostate cancer cells that could be removed by co-dosing free bombesin (104). Recently, bombesin-conjugated polymersomes were shown to effectively target PC-3 cells by FACS analysis, opening the possibility to create polymersomes that can be dual loaded with iron oxide and camptothecin for MRI imaging and treatment (105). A bombesin analogue was also conjugated to liposomes to increase the targeting of doxorubicin. In vitro analysis resulted in a twofold increase in the percent of bound liposomes compared to nontargeted liposomes, and in vivo tumor growth studies confirmed a 43% inhibition of tumor growth compared to nontargeted liposomes at the 19th day (106).
Somatostatin, also called somatotropin-release-inhibiting factor (SRIF), was first discovered in 1973 by Roger Guillemin’s group as a peptide that inhibited the secretion of growth hormones in the pituitary gland (107). Somatostatin is endogenously expressed in two forms; a 28-amino acid cyclic peptide and a cleaved cyclic 14-amino acid peptide (108). The ligand receptor interaction plays a role in controlling the release of numerous compounds including growth hormone, thyrotropin, prolactin, adrenocorticotropin, glucagon, vascular endothelial grown factor (VEGF; which induces nerve growth factor), regulation of calcium currents, and the adenylyl cyclase pathway via pertussis toxin sensitive G proteins (109–113). Somatostatin’s inhibition of these processes leads to antiangiogenic, apoptotic, and cell growth inhibition effects.
While each of the five somatostatin receptor subtypes are endogenously expressed in normal cells, they are each upregulated in a wide range of tumor and cancer cells (covered in a review by Patel et al.) (114,115). This includes but is not limited to: (a) neuroendocrine tumors and their metastases, (b) hepatocellular cancer, (c) small-cell lung cancer and other pulmonary malignancies, (d) breast, (e) stomach, (f) colon, (g) rectal, (h) gastrointestinal, (i) pancreatic, (j) bone, and (k) prostate cancer and almost all human meningiomas, medulloblastomas, and pheochromocytomas (116–126). Somatostatin receptor (SSTR) upregulation in tumors and its natural apoptotic and growth inhibitory effects make it a good target for nanoparticle targeting cancer therapies. This upregulation led to the treatment of tumors with somatostatin to slow tumor growth and angiogenesis. However, while 73–77% of patients showed a biological response and tumor growth inhibition, it had little effect in extending survival length (117). This was partially due to somatostatin’s short half-life when dosed in vivo necessitating the design of new somatostatin analogs and its use in conjunction with other drugs (127). N-terminally modified somatostatin compounds have proven successful in the imaging and diagnosis of brain tumors by radioscintigraphy leading to numerous clinically approved drugs such as Sandostatin® (Novartis), OctreoTher (Novartis), and OctreoScan® (Mallinkrodt) (128–130). Reviews of radio imaging peptides, including somatostatin analogs, and isotopes used are available by Reubi et al., Ferro-Flores et al., Schottelius et al., and Eberle et al (103,131–133).
Somatostatin analogs have the ability to target a variety of tumor types making these a viable option for combination therapy with existing chemotherapeutics. Recent reports from the literature indicate that somatostatin analogs have continued to be used in combination with paclitaxel and doxorubicin (121,134). Targeted mixed micelles were loaded with doxorubicin or a mixture of salinomycin and an antibiotic resulting in tissue specificity and prolonged circulation leading to improved antitumor effects (135,136). Liposomes conjugated to somatostatin analogs have also been used in combination with doxorubicin and tubulin-binding agents to achieve inhibition of tumor growth (137,138).
While targeted nanoparticles show better toxicity profiles, somatostatin targeted nanoparticles are being developed as gene delivery agents to further decrease the side effects. Xiao et al. capped gold nanorods with octreotide in order to deliver doxorubicin and siRNA to achaete-scute-complex-like 1 (ASCL-1). They reported that the combination of octreotide, doxorubicin, and siRNA showed the largest antiproliferative effect and the largest gene silencing by immunoblot, especially compared to nontargeted controls (139). In addition, adenoviral vectors that were modified to replace the coat protein which binds to the CAR receptor with somatostatin were shown to efficiently target and express their therapeutic gene in glioma cells. The increased targeting efficiency allowed for low doses to achieve reporter gene expression 75 times that of untargeted viruses in vitro (140).
An extensive range of peptides exist to target other receptors as well. Cholecystokinin, a C-terminal (Gly-Trp-Met-Asp-Phe) peptide, targets gastrin receptors (103,141). Neurotensin, a 14-amino acid peptide, and its analogs that are found in the ileum and hypothalamus can target the neurotensin receptor in neural cells. It is also upregulated in pancreatic and prostate cancer along with the melanocortin 1 receptor (MC1R), vasoactive intestinal polypeptide receptor 1 (VPAC-1), glucagon-like peptide-1 (GLP-1), and C-X-C chemokine receptor type 4 (CXCR4) (142–148). Another targeting peptide, Ile-Thr-Asp-Gly-Glu-Ala-Thr-Asp-Ser-Gly (ITDGEATDSG), has been shown to affect both psoriasis experimental autoimmune encephalomyelitis and can target lung carcinoma (149). The peptide Tyr-Ser-Ala-Try-Pro-Asp-Ser-Val-Pro-Met-Met-Ser(YSAYPDSVPMMS) binds erythropoietin producing hepatocellular receptor (150).
In addition to the numerous peptide ligands discovered for which the receptor is already known, technology such as phage display and high-throughput screening (151) of peptide libraries is leading to new peptides at a rapid rate of which little characterization or binding information is known (152–154). One example is the peptide Ser-Phe-Ser-Ile-Ile-His-Thr-Pro-Ile-Leu-Pro-Leu-Gly-Gly-Cys (SFSIIHTPILPLGGC), found using a phage display technique. This peptide targets unknown receptors on HEP3B human carcinoma cell lines (41). These examples suggest the extensive synthetic flexibility of peptides and make them viable targeting ligands that need to be studied further for more advanced cancer therapies.
CONCLUSIONS
Cancer is a very difficult disease to treat due to the diversity of cancer types, progression, and patient variation. Early forms of treatment with cytotoxic agents relied not only upon the propensity for drugs to act upon rapidly dividing cancer cells but also led to normal tissue damage. The discovery of cancer specific receptors has begun to increase the effectiveness of cancer treatment using nanoparticles. Nanoparticles have the benefit of increasing circulatory half-life of chemotherapeutic agents and limiting their toxicity. In the same way, incorporation of ligands on nanoparticles increases the accumulation of chemotherapeutics in cells creating a safer treatment. Cancer-specific ligand-receptor pairings are growing both in diversity as well as availability. Targeted nanoparticles containing safer chemotherapeutic cargo, such as siRNA or DNA, are proving efficacious in animal and disease models. While cytotoxic chemotherapeutics are effective at destroying diseased cells, the future of cancer therapies will rely on nanoparticles which are efficient at delivering a safer cargo. These nanoparticles must combine an optimized size, charge, and ligand presentation and also require the discovery of new ligands that offer the ability not only to target cancer but also to create individual treatments for different types of cancer to improve patient life expectancy. Targeted nanoparticles will lead the way to decrease the toxic side effects of current cancer treatments and to decrease the number of cancer related deaths worldwide.
References
- 1.Wisse E, De Zanger RB, Charels K, Van Der Smissen P, McCuskey RS. The liver sieve: considerations concerning the structure and function of endothelial fenestrae, the sinusoidal wall and the space of Disse. Hepatology. 1985;5(4):683–692. doi: 10.1002/hep.1840050427. [DOI] [PubMed] [Google Scholar]
- 2.Torchilin VP. Targeted pharmaceutical nanocarriers for cancer therapy and imaging. AAPS J. 2007;9(2):E128–E147. doi: 10.1208/aapsj0902015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rettig GR, Rice KG. Non-viral gene delivery: from the needle to the nucleus. Expert Opin Biol Ther. 2007;7(6):799–808. doi: 10.1517/14712598.7.6.799. [DOI] [PubMed] [Google Scholar]
- 4.Das M, Mohanty C, Sahoo SK. Ligand-based targeted therapy for cancer tissue. Expert Opin Drug Deliv. 2009;6(3):285–304. doi: 10.1517/17425240902780166. [DOI] [PubMed] [Google Scholar]
- 5.Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11(17–18):812–818. doi: 10.1016/j.drudis.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer-chemotherapy—mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12):6387–6392. [PubMed] [Google Scholar]
- 7.Fleischer CC, Payne CK. Nanoparticle surface charge mediates the cellular receptors used by protein-nanoparticle complexes. J Phys Chem B. 2012;116(30):8901–8907. doi: 10.1021/jp304630q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Z, Jiao Y, Wang T, Zhang Y, Xue W. Interactions between solubilized polymer molecules and blood components. J Cont Rel. 2012;160(1):14–24. doi: 10.1016/j.jconrel.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Merdan T, Kunath K, Petersen H, Bakowsky U, Voigt KH, Kopecek J, et al. PEGylation of poly(ethylene imine) affects stability of complexes with plasmid DNA under in vivo conditions in a dose-dependent manner after intravenous injection into mice. Bioconj Chem. 2005;16(4):785–792. doi: 10.1021/bc049743q. [DOI] [PubMed] [Google Scholar]
- 10.Yan X, Kuipers F, Havekes LM, Havinga R, Dontje B, Poelstra K, et al. The role of apolipoprotein E in the elimination of liposomes from blood by hepatocytes in the mouse. Bioch Biophys Res Commun. 2005;328(1):57–62. doi: 10.1016/j.bbrc.2004.12.137. [DOI] [PubMed] [Google Scholar]
- 11.Jansen RW, Molema G, Harms G, Kruijt JK, van Berkel TJC, Hardonk MJ, et al. Formaldehyde treated albumin contains monomeric and polymeric forms that are differently cleared by endothelial and kupffer cells of the liver: evidence for scavenger receptor heterogeneity. Biochem Biophys Res Commun. 1991;180:23–32. doi: 10.1016/s0006-291x(05)81249-5. [DOI] [PubMed] [Google Scholar]
- 12.Kawabata K, Takakura Y, Hashida M. The fate of plasmid DNA after intravenous injection in mice: involvement of scavenger receptors in its hepatic uptake. Pharm Res. 1995;12(6):825–830. doi: 10.1023/a:1016248701505. [DOI] [PubMed] [Google Scholar]
- 13.Kamps JA, Morselt HW, Swart PJ, Meijer DK, Scherphof GL. Massive targeting of liposomes, surface-modified with anionized albumins, to hepatic endothelial cells. Proc Natl Acad Sci U S A. 1997;94(21):11681–11685. doi: 10.1073/pnas.94.21.11681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Z, Tian J, Smith JS, Byrnes AP. Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. J Virol. 2008;82(23):11705–11713. doi: 10.1128/JVI.01320-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamps JA, Scherphof GL. Receptor versus non-receptor mediated clearance of liposomes. Adv Drug Del Rev. 1998;32(1–2):81–97. doi: 10.1016/s0169-409x(97)00133-6. [DOI] [PubMed] [Google Scholar]
- 16.Wong SY, Pelet JM, Putnam D. Polymer systems for gene delivery-past, present, and future. Prog Polym Sci. 2007;32(8–9):799–837. [Google Scholar]
- 17.Tseng Y-C, Mozumdar S, Huang L. Lipid-based systemic delivery of siRNA. Adv Drug Deliv Rev. 2009;61(9):721–731. doi: 10.1016/j.addr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin ME, Rice KG. Peptide-guided gene delivery. AAPS J. 2007;9(1):E18–E29. doi: 10.1208/aapsj0901003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatakeyama H, Akita H, Harashima H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: a strategy for overcoming the PEG dilemma. Adv Drug Deliv Rev. 2011;63(3):152–160. doi: 10.1016/j.addr.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 20.Lozza C, Navarro-Teulon I, Pelegrin A, Pouget J-P, Vives E. Peptides in receptor-mediated radiotherapy: from design to the clinical application in cancers. Front Oncol. 2013;3(247):1–13. doi: 10.3389/fonc.2013.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kizzire K, Khargharia S, Rice KG. High-affinity PEGylated polyacridine peptide polyplexes mediate potent in vivo gene expression. Gene Ther. 2013;20(4):407–416. doi: 10.1038/gt.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oupicky D, Konak C, Dash PR, Seymour LW, Ulbrich K. Effect of albumin and polyanion on the structure of DNA complexes with polycation containing hydrophilic nonionic block. Bioconjug Chem. 1999;10(5):764–772. doi: 10.1021/bc990007+. [DOI] [PubMed] [Google Scholar]
- 23.Dash PR, Read ML, Barrett LB, Wolfert MA, Seymour LW. Factors affecting blood clearance and in vivo distribution of polyelectrolyte complexes for gene delivery. Gene Ther. 1999;6:643–650. doi: 10.1038/sj.gt.3300843. [DOI] [PubMed] [Google Scholar]
- 24.Khargharia S, Kizzire K, Ericson MD, Baumhover NJ, Rice KG. PEG length and chemical linkage controls polyacridine peptide DNA polyplex pharmacokinetics, biodistribution, metabolic stability and in vivo gene expression. J Cont Rel. 2013;170(3):325–333. doi: 10.1016/j.jconrel.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khargharia S, Baumhover NJ, Duskey JT, Crowley ST, Rice KG. Mechanism of PEGyalted DNA polyplex capture by the liver. Molecular therapy. 2014:submitted. [DOI] [PubMed]
- 26.Nobs L, Buchegger F, Gurny R, Allemann E. Current methods for attaching targeting ligands to liposomes and nanoparticles. J Pharm Sci. 2004;93(8):1980–1992. doi: 10.1002/jps.20098. [DOI] [PubMed] [Google Scholar]
- 27.Liu YP, Tong C, Dispenzieri A, Federspiel MJ, Russell SJ, Peng KW. Polyinosinic acid decreases sequestration and improves systemic therapy of measles virus. Cancer Gene Ther. 2012;19(3):202–211. doi: 10.1038/cgt.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burkel WE, Low FN. The fine structure of rat liver sinusoids, space of Disse and associated tissue space. Amer J Anat. 1966;118(3):769–783. doi: 10.1002/aja.1001180307. [DOI] [PubMed] [Google Scholar]
- 29.Scherphof GL, Kamps JA. The role of hepatocytes in the clearance of liposomes from the blood circulation. Prog Lipid Res. 2001;40(3):149–166. doi: 10.1016/s0163-7827(00)00020-5. [DOI] [PubMed] [Google Scholar]
- 30.Kamps JA, Scherphof GL. Biodistribution and uptake of liposomes in vivo. Methods Enzym. 2004;387:257–266. doi: 10.1016/S0076-6879(04)87016-2. [DOI] [PubMed] [Google Scholar]
- 31.Li SD, Huang L. Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. Biochim Biophys Acta. 2009;1788(10):2259–2266. doi: 10.1016/j.bbamem.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. J Cont Rel. 2011;151(3):220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 33.Giacca M, Zacchigna S. Virus-mediated gene delivery for human gene therapy. J Cont Rel. 2012;161(2):377–388. doi: 10.1016/j.jconrel.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 34.Paszko E, Senge MO. Immunoliposomes. Curr Med Chem. 2012;19(31):5239–5277. doi: 10.2174/092986712803833362. [DOI] [PubMed] [Google Scholar]
- 35.Josan JS, Handl HL, Sankaranarayanan R, Xu L, Lynch RM, Vagner J, et al. Cell-specific targeting by heterobivalent ligands. Bioconjug Chem. 2011;22(7):1270–1278. doi: 10.1021/bc1004284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munoz EM, Correa J, Riguera R, Fernandez-Megia E. Real-time evaluation of binding mechanisms in multivalent interactions: a surface plasmon resonance kinetic approach. JACS.135(16):5966–5969. [DOI] [PubMed]
- 37.Cecioni S, Faure S, Darbost U, Bonnamour I, Parrot-Lopez H, Roy O, et al. Selectivity among two lectins: probing the effect of topology, multivalency and flexibility of “clicked” multivalent glycoclusters. Chem-a Eur J. 17(7):2146–2159. [DOI] [PubMed]
- 38.Lundquist JJ, Toone EJ. The cluster glycoside effect. Chem Rev. 2002;102(2):555–578. doi: 10.1021/cr000418f. [DOI] [PubMed] [Google Scholar]
- 39.Kane RS. Thermodynamics of multivalent interactions: influence of the linker. Langmuir. 2010;26(11):8636–8640. doi: 10.1021/la9047193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem-Int Ed. 1998;37(20):2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 41.Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers (vol 10, pg 389, 2011). Nat Mater. 2011;10(5):389–397. doi:10.1038/nmat2992. [DOI] [PMC free article] [PubMed]
- 42.Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. 2011;118(26):6743–6751. doi: 10.1182/blood-2011-07-343566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pirollo KF, Chang EH. Does a targeting ligand influence nanoparticle tumor localization or uptake? Trends Biotechnol. 2008;26(10):552–558. doi: 10.1016/j.tibtech.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 44.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci U S A. 2007;104(39):15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu AM, Yazaki PJ, Tsai SW, Nguyen K, Anderson AL, McCarthy DW, et al. High-resolution microPET imaging of carcino-embryonic antigen-positive xenografts by using a copper-64-labeled engineered antibody fragment. Proc Natl Acad Sci U S A. 2000;97(15):8495–8500. doi: 10.1073/pnas.150228297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hussain S, Plueckthun A, Allen TM, Zangemeister-Wittke U. Antitumor activity of an epithelial cell adhesion molecule-targeted nanovesicular drug delivery system. Mol Cancer Ther. 2007;6(11):3019–3027. doi: 10.1158/1535-7163.MCT-07-0615. [DOI] [PubMed] [Google Scholar]
- 47.Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66(13):6732–6740. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- 48.Neves MAD, Reinstein O, Saad M, Johnson PE. Defining the secondary structural requirements of a cocaine-binding aptamer by a thermodynamic and mutation study. Biophys Chem. 2010;153(1):9–16. doi: 10.1016/j.bpc.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 49.Song K-M, Lee S, Ban C. Aptamers and their biological applications. Sensors. 2012;12(1):612–631. doi: 10.3390/s120100612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho EJ, Lee J-W, Ellington AD. Applications of aptamers as sensors. Annu Rev Anal Chem. 2009;2:241–264. doi: 10.1146/annurev.anchem.1.031207.112851. [DOI] [PubMed] [Google Scholar]
- 51.Wang H-Q, Wu Z, Tang L-J, Yu R-Q, Jiang J-H. Fluorescence protection assay: a novel homogeneous assay platform toward development of aptamer sensors for protein detection. Nucleic Acids Research. 2011;39(18). [DOI] [PMC free article] [PubMed]
- 52.Cerchia L, de Franciscis V. Nucleic acid-based aptamers as promising therapeutics in neoplastic diseases. Methods Mol Biol. 2007;361:187–200. doi: 10.1385/1-59745-208-4:187. [DOI] [PubMed] [Google Scholar]
- 53.Mongelard F, Bouvet P. AS-1411, a guanosine-rich oligonucleotide aptamer targeting nucleolin for the potential treatment of cancer, including acute myeloid leukemia. Curr Opin Mol Ther. 2010;12(1):107–114. [PubMed] [Google Scholar]
- 54.Wilner SE, Wengerter B, Maier K, de Lourdes Borba Magalhaes M, Del Amo DS, Pai S, et al. An RNA alternative to human transferrin: a new tool for targeting human cells. Mol Ther Nucleic Acids. 2012;1:e21–1. [DOI] [PMC free article] [PubMed]
- 55.Thiel KW, Hernandez LI, Dassie JP, Thiel WH, Liu X, Stockdale KR, et al. Delivery of chemo-sensitizing siRNAs to HER2(+)-breast cancer cells using RNA aptamers. Nucleic Acids Res. 2012;40(13):6319–6337. doi: 10.1093/nar/gks294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gupta S, Thirstrup D, Jarvis TC, Schneider DJ, Wilcox SK, Carter J, et al. Rapid histochemistry using slow off-rate modified aptamers with anionic competition. Appl Immunohistochem Mol Morphol. 2011;19(3):273–278. doi: 10.1097/PAI.0b013e3182008c29. [DOI] [PubMed] [Google Scholar]
- 57.Cerchia L, Esposito CL, Camorani S, Rienzo A, Stasio L, Insabato L, et al. Targeting axl with an high-affinity inhibitory aptamer. Mol Ther. 2012;20(12):2291–2303. doi: 10.1038/mt.2012.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishimoto T, Yamamoto Y, Yoshida K, Goto N, Ohnami S, Aoki K. Development of peritoneal tumor-targeting vector by in vivo screening with a random peptidedisplaying adenovirus library. Plos One. 2012;7(9):e45550–e45555. doi: 10.1371/journal.pone.0045550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurosaki T, Higuchi N, Kawakami S, Higuchi Y, Nakamura T, Kitahara T, et al. Self-assemble gene delivery system for molecular targeting using nucleic acid aptamer. Gene. 2012;491(2):205–209. doi: 10.1016/j.gene.2011.09.021. [DOI] [PubMed] [Google Scholar]
- 60.Daniels TR, Bernabeu E, Rodriguez JA, Patel S, Kozman M, Chiappetta DA, et al. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim Et Biophys Acta-Gen Subj. 2012;1820(3):291–317. doi: 10.1016/j.bbagen.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hogemann D, Josephson L, Weissleder R, Basilion JP. Improvement of MRI probes to allow efficient detection of gene expression. Bioconjug Chem. 2000;11(6):941–946. doi: 10.1021/bc000079x. [DOI] [PubMed] [Google Scholar]
- 62.Hogemann-Savellano D, Bos E, Blondet C, Sato F, Abe T, Josephson L, et al. The transferrin receptor: a potential molecular Imaging marker for human cancer. Neoplasia. 2003;5(6):495–506. doi: 10.1016/s1476-5586(03)80034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6(3):659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 64.Senzer N, Nemunaitis J, Nemunaitis D, Bedell C, Edelman G, Barve M, et al. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol Ther. 2013;21(5):1096–1103. doi: 10.1038/mt.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khalaj-Kondori M, Sadeghizadeh M, Behmanesh M, Saggio I, Monaci P. Chemical coupling as a potent strategy for preparation of targeted bacteriophage-derived gene nanocarriers into eukaryotic cells. J Gene Med. 2011;13(11):622–631. doi: 10.1002/jgm.1617. [DOI] [PubMed] [Google Scholar]
- 66.Hilgenbrink AR, Low PS. Folate receptor-mediated drug targeting: from therapeutics to diagnostics. J Pharm Sci. 2005;94(10):2135–2146. doi: 10.1002/jps.20457. [DOI] [PubMed] [Google Scholar]
- 67.Yang J, Vlashi E, Low P. Folate-linked drugs for the treatment of cancer and inflammatory diseases. Sub-Cell Biochem. 2012;56:163–179. doi: 10.1007/978-94-007-2199-9_9. [DOI] [PubMed] [Google Scholar]
- 68.Muller C, Schibli R. Prospects in folate receptor-targeted radionuclide therapy. Front Oncol. 2013;3:249. doi: 10.3389/fonc.2013.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shiokawa T, Hattori Y, Kawano K, Ohguchi Y, Kawakami H, Toma K, et al. Effect of polyethylene glycol linker chain length of folate-linked microemulsions loading aclacinomycin a on targeting ability and antitumor effect in vitro and in vivo. Clin Cancer Res. 2005;11(5):2018–2025. doi: 10.1158/1078-0432.CCR-04-1129. [DOI] [PubMed] [Google Scholar]
- 70.Lee H, Lytton-Jean AKR, Chen Y, Love KT, Park AI, Karagiannis ED, et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat Nanotechnol. 2012;7(6):389–393. doi: 10.1038/nnano.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arima H, Yoshimatsu A, Ikeda H, Ohyama A, Motoyama K, Higashi T, et al. Folate-PEG-appended dendrimer conjugate with alpha-cyclodextrin as a novel cancer cell selective siRNA delivery carrier. Mol Pharm. 2012;9(9):2591–2604. doi: 10.1021/mp300188f. [DOI] [PubMed] [Google Scholar]
- 72.Kwon O-J, Kang E, Choi J-W, Kim SW, Yun C-O. Therapeutic targeting of chitosan-PEG-folate-complexed oncolytic adenovirus for active and systemic cancer gene therapy. J Cont Rel. 2013;169(3):257–265. doi: 10.1016/j.jconrel.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 73.Yao H, Chen S-C, Shen Z, Huang Y-C, Zhu X, Wang X-M, et al. Functional characterization of a PEI-CyD-FA-coated adenovirus as delivery vector for gene therapy. Curr Med Chem. 2013;20(20):2601–2608. doi: 10.2174/0929867311320200008. [DOI] [PubMed] [Google Scholar]
- 74.Zhao F, Yin H, Zhang Z, Li J. Folic acid modified cationic gamma-Cyclodextrin-oligoethylenimine star polymer with bioreducible disulfide linker for efficient targeted gene delivery. Biomacromolecules. 2013;14(2):476–484. doi: 10.1021/bm301718f. [DOI] [PubMed] [Google Scholar]
- 75.Mornet E, Carmoy N, Laine C, Lemiegre L, Le Gall T, Laurent I, et al. Folate-equipped nanolipoplexes mediated efficient gene transfer into human epithelial cells. Int J Mol Sci. 2013;14(1):1477–1501. doi: 10.3390/ijms14011477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meirovitz A, Goldberg R, Binder A, Rubinstein AM, Hermano E, Elkin M. Heparanase in inflammation and inflammation-associated cancer. Febs J. 2013;280(10):2307. doi: 10.1111/febs.12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fan Y, Mao R, Yang J. NF-kappa B and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell. 2013;4(3):176–185. doi: 10.1007/s13238-013-2084-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kisielewski R, Tolwinska A, Mazurek A, Laudanski P. Inflammation and ovarian cancer—current views. Ginekol Pol. 2013;84(4):293–297. doi: 10.17772/gp/1579. [DOI] [PubMed] [Google Scholar]
- 79.Inagaki-ohara K, Kondo T, Ito M, Yoshimuura A. SOCS, inflammation, and cancer. JAK-STAT. 2013;2(3):e24053–e24063. doi: 10.4161/jkst.24053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rimessi A, Patergnani S, Loannidi E, Pinton P. Chemoresistance and cancer-related inflammation: two hallmarks of cancer connected by an atypical link, PKC(gamma) Front Oncol. 2013;3(232):1–7. doi: 10.3389/fonc.2013.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cronstein BN, Naime D, Ostad E. The antiinflammatory mechanism of methotrexate—increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in-vivo model of inflammation. J Clin Investig. 1993;92(6):2675–2682. doi: 10.1172/JCI116884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Furst DE, Kremer JM. Methotrexate in rheumatoid-arthritis. Arthritis Rheum. 1988;31(3):305–314. doi: 10.1002/art.1780310301. [DOI] [PubMed] [Google Scholar]
- 83.Thomas TP, Goonewardena SN, Majoros IJ, Kotlyar A, Cao Z, Leroueil PR, et al. Folate-targeted nanoparticles show efficacy in the treatment of inflammatory arthritis. Arthritis Rheum. 2011;63(9):2671–2680. doi: 10.1002/art.30459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou J, Tsai Y-T, Weng H, Baker DW, Tang L. Real time monitoring of biomaterial-mediated inflammatory responses via macrophage-targeting NIR nanoprobes. Biomaterials. 2011;32(35):9383–9390. doi: 10.1016/j.biomaterials.2011.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duchatelle V, Kritikou EA, Tardif J-C. Clinical value of drugs targeting inflammation for the management of coronary artery disease. Can J Cardiol. 2012;28(6):678–686. doi: 10.1016/j.cjca.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 86.Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29(11):1005–1010. doi: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Garg NK, Dwivedi P, Campbell C, Tyagi RK. Site specific/targeted delivery of gemcitabine through anisamide anchored chitosan/poly ethylene glycol nanoparticles: an improved understanding of lung cancer therapeutic intervention. Eur J Pharm Sci. 2012;47(5):1006–1014. doi: 10.1016/j.ejps.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 88.Guo J, Ogier JR, Desgranges S, Darcy R, O’Driscoll C. Anisamide-targeted cyclodextrin nanoparticles for siRNA delivery to prostate tumours in mice. Biomaterials. 2012;33(31):7775–7784. doi: 10.1016/j.biomaterials.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 89.Kim SK, Huang L. Nanoparticle delivery of a peptide targeting EGFR signaling. J Control Release. 2012;157(2):279–286. doi: 10.1016/j.jconrel.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang Y, Hu Y, Wang Y, Li J, Liu F, Huang L. Nanoparticle delivery of pooled siRNA for effective treatment of non-small cell lung caner. Mol Pharm. 2012;9(8):2280–2289. doi: 10.1021/mp300152v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Y, Schwerbrock NMJ, Rogers AB, Kim WY, Huang L. Codelivery of VEGF siRNA and gemcitabine monophosphate in a single nanoparticle formulation for effective treatment of NSCLC. Mol Ther. 2013;21(8):1559–1569. doi: 10.1038/mt.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Y, Su H-H, Yang Y, Hu Y, Zhang L, Blancafort P, et al. Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol Ther. 2013;21(2):358–367. doi: 10.1038/mt.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xie H, Diagaradjane P, Deorukhkar AA, Goins B, Bao A, Phillips WT, et al. Integrin alpha(v)beta(3)-targeted gold nanoshells augment tumor vasculature-specific imaging and therapy. Int J Nanomedicine. 2011;6:259–269. doi: 10.2147/IJN.S15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mokhtarieh AA, Kim S, Lee Y, Chung BH, Lee MK. Novel cell penetrating peptides with multiple motifs composed of RGD and its analogs. Biochem Biophys Res Commun. 2013;432(2):359–364. doi: 10.1016/j.bbrc.2013.01.096. [DOI] [PubMed] [Google Scholar]
- 95.Chen J-X, Xu X-D, Yang S, Yang J, Zhuo R-X, Zhang X-Z. Self-assembled BolA-like amphiphilic peptides as viral-mimetic gene vectors for cancer cell targeted gene delivery. Macromol Biosci. 2013;13(1):84–92. doi: 10.1002/mabi.201200283. [DOI] [PubMed] [Google Scholar]
- 96.Park J, Singha K, Son S, Kim J, Namgung R, Yun CO, et al. A review of RGD-functionalized nonviral gene delivery vectors for cancer therapy. Cancer Gene Ther. 2012;19(11):741–748. doi: 10.1038/cgt.2012.64. [DOI] [PubMed] [Google Scholar]
- 97.Martin I, Dohmen C, Mas-Moruno C, Troiber C, Kos P, Schaffert D, et al. Solid-phase-assisted synthesis of targeting peptide-PEG-oligo(ethane amino)amides for receptormediated gene delivery. Org Biomol Chem. 2012;10(16):3258–3268. doi: 10.1039/c2ob06907e. [DOI] [PubMed] [Google Scholar]
- 98.Liu S, Guo Y, Huang R, Li J, Huang S, Kuang Y, et al. Gene and doxorubicin co-delivery system for targeting therapy of glioma. Biomaterials. 2012;33(19):4907–4916. doi: 10.1016/j.biomaterials.2012.03.031. [DOI] [PubMed] [Google Scholar]
- 99.Wang X-L, Xu R, Wu X, Gillespie D, Jensen R, Lu Z-R. Targeted systemic delivery of a therapeutic siRNA with a multifunctional carrier controls tumor proliferation in mice. Mol Pharm. 2009;6(3):738–746. doi: 10.1021/mp800192d. [DOI] [PubMed] [Google Scholar]
- 100.Iwasaki T, Yamakawa M, Asaoka A, Kawano T, Ishibashi J. Anti-angiogenesis activities of novel peptide complexes: mitochondria-disruptive 9mer peptides conjugated with the integrin alpha V beta 3-homing cyclic RGD motif. Biosci Biotechnol Biochem. 2012;76(11):2044–2048. doi: 10.1271/bbb.120397. [DOI] [PubMed] [Google Scholar]
- 101.Miura Y, Takenaka T, Toh K, Wu S, Nishihara H, Kano MR, et al. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood–brain tumor barrier. ACS Nano. 2013;7(10):8583–8592. doi: 10.1021/nn402662d. [DOI] [PubMed] [Google Scholar]
- 102.Marelli UK, Rechenmacher F, Sobahi TRA, Mas-Moruno C, Kessler H. Tumor targeting via integrin ligands. Front Oncol. 2013;3(222):1–12. doi: 10.3389/fonc.2013.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ferro-Flores G, Ramirez FM, Melendez-Alafort L, Santos-Cuevas CL. Peptides for in vivo target-specific cancer imaging. Mini-Rev Med Chem. 2010;10(1):87–97. doi: 10.2174/138955710791112596. [DOI] [PubMed] [Google Scholar]
- 104.Ming X, Alam MR, Fisher M, Yan Y, Chen X, Juliano RL. Intracellular delivery of an antisense oligonucleotide via endocytosis of a G protein-coupled receptor. Nucleic Acids Res. 2010;38(19):6567–6576. doi: 10.1093/nar/gkq534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bleul R, Thiermann R, Marten GU, House MJ, St Pierre TG, Hafeli UO, et al. Continuously manufactured magnetic polymersomes—a versatile tool (not only) for targeted cancer therapy. Nanoscale. 2013;5(23):11385–11393. doi: 10.1039/c3nr02190d. [DOI] [PubMed] [Google Scholar]
- 106.Accardo A, Mansi R, Salzano G, Morisco A, Aurilio M, Parisi A, et al. Bombesin peptide antagonist for target-selective delivery of liposomal doxorubicin on cancer cells. J Drug Target. 2013;21(3):240–249. doi: 10.3109/1061186X.2012.741138. [DOI] [PubMed] [Google Scholar]
- 107.Burgus R, Ling N, Butcher M, Guillemi R. Primary structure of somatostatin—hypothalamic peptide that inhibits secretion of pituitary growth-hormone. Proc Natl Acad Sci U S A. 1973;70(3):684–688. doi: 10.1073/pnas.70.3.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Reubi JC. New specific radioligand for one subpopulation of brain somatostatin receptors. Life Sci. 1985;36(19):1829–1836. doi: 10.1016/0024-3205(85)90155-9. [DOI] [PubMed] [Google Scholar]
- 109.Lattuada D, Casnici C, Crotta K, Mastrotto C, Franco P, Schmid HA, et al. Inhibitory effect of pasireotide and octreotide on lymphocyte activation. J Neuroimmunol. 2007;182(1–2):153–159. doi: 10.1016/j.jneuroim.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 110.Hershberger RE, Newman BL, Florio T, Bunzow J, Civelli O, Li XJ, et al. The somatostatin receptors Sstr1 and Sstr2 are coupled to inhibition of adenylyl-cyclase in Chinese-hamster ovary cells via pertussis-toxin-sensitive pathways. Endocrinology. 1994;134(3):1277–1285. doi: 10.1210/endo.134.3.7907016. [DOI] [PubMed] [Google Scholar]
- 111.Nurhidayat, Tsukamoto Y, Sigit K, Sasaki F. Sex differentiation of growth hormone-releasing hormone and somatostatin neurons in the mouse hypothalamus: an immunohistochemical and morphological study. Brain Res. 1999;821(2):309–321. doi: 10.1016/s0006-8993(99)01081-1. [DOI] [PubMed] [Google Scholar]
- 112.Kvols LK, Woltering EA. Role of somatostatin analogs in the clinical management of non-neuroendocrine solid tumors. Anti-Cancer Drugs. 2006;17(6):601–608. doi: 10.1097/01.cad.0000210335.95828.ed. [DOI] [PubMed] [Google Scholar]
- 113.Chen F, Odorisio MS, Hermann G, Hayes J, Malarkey WB, Odorisio TM. Mechanisms of action of long-acting analogs of somatostatin. Regul Pept. 1993;44(3):285–295. doi: 10.1016/0167-0115(93)90138-x. [DOI] [PubMed] [Google Scholar]
- 114.Kaupmann K, Bruns C, Hoyer D, Seuwen K, Lubbert H. Distribution and 2nd messenger coupling of 4 somatostatin receptor subtypes expressed in brain. Febs Lett. 1993;331(1–2):53–59. doi: 10.1016/0014-5793(93)80296-7. [DOI] [PubMed] [Google Scholar]
- 115.Patel YC, Greenwood MT, Panetta R, Demchyshyn L, Niznik H, Srikant CB. Mini review—the somatostatin receptor family. Life Sci. 1995;57(13):1249–1265. doi: 10.1016/0024-3205(95)02082-t. [DOI] [PubMed] [Google Scholar]
- 116.Surujpaul PP, Gutierrez-Wing C, Ocampo-Garcia B, Ramirez FM, de Murphy CA, Pedraza-Lopez M, et al. Gold nanoparticles conjugated to [Tyr(3)]Octreotide peptide. Biophys Chem. 2008;138(3):83–90. doi: 10.1016/j.bpc.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 117.Oyen WJG, Bodei L, Giammarile F, Maecke HR, Tennvall J, Luster M, et al. Targeted therapy in nuclear medicine-current status and future prospects. Ann Oncol. 2007;18(11):1782–1792. doi: 10.1093/annonc/mdm111. [DOI] [PubMed] [Google Scholar]
- 118.Imam SK. Molecular nuclear imaging: the radiopharmaceuticals (review) Cancer Biother Radiopharm. 2005;20(2):163–172. doi: 10.1089/cbr.2005.20.163. [DOI] [PubMed] [Google Scholar]
- 119.Susini C, Buscail L. Rationale for the use of somatostatin analogs as antitumor agents. Ann Oncol. 2006;17(12):1733–1742. doi: 10.1093/annonc/mdl105. [DOI] [PubMed] [Google Scholar]
- 120.Pan X, Thompson R, Meng X, Wu D, Xu L. Tumor-targeted RNA-interference: functional non-viral nanovectors. Am J Cancer Res. 2011;1(1):25–42. [PMC free article] [PubMed] [Google Scholar]
- 121.Shen H, Hu D, Du J, Wang X, Liu Y, Wang Y, et al. Paclitaxel-octreotide conjugates in tumor growth inhibition of A549 human non-small cell lung cancer xenografted into nude mice. Eur J Pharmacol. 2008;601(1–3):23–29. doi: 10.1016/j.ejphar.2008.10.035. [DOI] [PubMed] [Google Scholar]
- 122.Parry JJ, Eiblmaier M, Andrews R, Meyer LA, Higashikubo R, Anderson CJ, et al. Characterization of somatostatin receptor subtype 2 expression in stably transfected A-427 human cancer cells. Mol Imaging. 2007;6(1):56–67. [PubMed] [Google Scholar]
- 123.Parry JJ, Chen R, Andrews R, Lears KA, Rogers BE. Identification of critical residues involved in ligand binding and g protein signaling in human somatostatin receptor subtype 2. Endocrinology. 2012;153(6):2747–2755. doi: 10.1210/en.2011-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Koper JW, Markstein R, Kohler C, Kwekkeboom DJ, Avezaat CJJ, Lamberts SWJ, et al. Somatostatin inhibits the activity of adenylate-cyclase in cultured human meningioma cells and stimulates their growth. J Clin Endocrinol Metab. 1992;74(3):543–547. doi: 10.1210/jcem.74.3.1346787. [DOI] [PubMed] [Google Scholar]
- 125.Fruhwald MC, O’Dorisio MS, Pietsch T, Reubi JC. High expression of somatostatin receptor subtype 2 (sst(2)) in medulloblastoma: Implications for diagnosis and therapy. Pediatr Res. 1999;45(5):697–708. doi: 10.1203/00006450-199905010-00016. [DOI] [PubMed] [Google Scholar]
- 126.Hofland LJ, Vankoetsveld PM, Waaijers M, Zuyderwijk J, Breeman WAP, Lamberts SWJ. Internalization of the radioiodinated somatostatin analog [I-125-Tyr(3)]octreotide by mouse and human pituitary-tumor cells—increase by unlabeled octreotide. Endocrinology. 1995;136(9):3698–3706. doi: 10.1210/endo.136.9.7649075. [DOI] [PubMed] [Google Scholar]
- 127.Poell F, Lehmann D, Illing S, Ginj M, Jacobs S, Lupp A, et al. Pasireotide and octreotide stimulate distinct patterns of sst(2A) somatostatin receptor phosphorylation. Mol Endocrinol. 2010;24(2):436–446. doi: 10.1210/me.2009-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bakker WH, Krenning EP, Breeman WA, Koper JW, Kooij PP, Reubi JC, et al. Receptor scintigraphy with a radioiodinated somatostatin analog—radiolabeling, purification, biologic activity, and invivo application in animals. J Nucl Med. 1990;31(9):1501–1509. [PubMed] [Google Scholar]
- 129.Bushnell D, Menda Y, O’Dorisio T, Madsen M, Miller S, Carlisle T, et al. Effects of intravenous amino acid administration with Y-90 DOTA-Phe1-Ty3-octreotide (SMT487 [OctreoTher (TM)]) treatment. Cancer Biother Radiopharm. 2004;19(1):35–41. doi: 10.1089/108497804773391658. [DOI] [PubMed] [Google Scholar]
- 130.Antunes P, Ginj M, Walter MA, Chen J, Reubi J-C, Maecke HR. Influence of different spacers on the biological profile of a DOTA-somatostatin analogue. Bioconjug Chem. 2007;18(1):84–92. doi: 10.1021/bc0601673. [DOI] [PubMed] [Google Scholar]
- 131.Eberle AN, Mild G. Receptor-mediated tumor targeting with radiopeptides Part 1. General principles and methods. J Recept Signal Transduct. 2009;29(1):1–37. doi: 10.1080/10799890902732823. [DOI] [PubMed] [Google Scholar]
- 132.Schottelius M, Wester H-J. Molecular imaging targeting peptide receptors. Methods. 2009;48(2):161–177. doi: 10.1016/j.ymeth.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 133.Reubi JC, Maecke HR. Peptide-based probes for cancer imaging. J Nucl Med. 2008;49(11):1735–1738. doi: 10.2967/jnumed.108.053041. [DOI] [PubMed] [Google Scholar]
- 134.Vaidyanathan G, Affleck DJ, Norman J, O’Dorisio S, Zalutsky MR. A radloiodinated MEBG-octreotate conjugate exhibiting enhanced uptake and reten tion in SSTR2-expressing tumor cells. Bioconjug Chem. 2007;18(6):2122–2130. doi: 10.1021/bc700240r. [DOI] [PubMed] [Google Scholar]
- 135.Zhang Y, Zhang H, Wang X, Wang J, Zhang X, Zhang Q. The eradication of breast cancer and cancer stem cells using octreotide modified paclitaxel active targeting micelles and salinomycin passive targeting micelles. Biomaterials. 2012;33(2):679–691. doi: 10.1016/j.biomaterials.2011.09.072. [DOI] [PubMed] [Google Scholar]
- 136.Niu J, Su Z, Xiao Y, Huang A, Li H, Bao X, et al. Octreotide-modified and pH-triggering polymeric micelles loaded with doxorubicin for tumor targeting delivery. Eur J Pharm Sci. 2012;45(1–2):216–226. doi: 10.1016/j.ejps.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 137.Guo Y, Ferdani R, Anderson CJ. Preparation and biological evaluation of Cu-64 labeled Tyr(3)-octreotate using a phosphonic acid-based cross-bridged macrocyclic chelator. Bioconjug Chem. 2012;23(7):1470–1477. doi: 10.1021/bc300092n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dai W, Jin W, Zhang J, Wang X, Wang J, Zhang X, et al. Spatiotemporally controlled co-delivery of anti-vasculature agent and cytotoxic drug by octreotide-modified stealth liposomes. Pharm Res. 2012;29(10):2902–2911. doi: 10.1007/s11095-012-0797-2. [DOI] [PubMed] [Google Scholar]
- 139.Xiao Y, Jaskula-Sztul R, Javadi A, Xu W, Eide J, Dammalapati A, et al. Co-delivery of doxorubicin and siRNA using octreotide-conjugated gold nanorods for targeted neuroendocrine cancer therapy. Nanoscale. 2012;4(22):7185–7193. doi: 10.1039/c2nr31853a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lecolle K, Begard S, Caillierez R, Demeyer D, Grellier E, Loyens A, et al. Sstr2A: a relevant target for the delivery of genes into human glioblastoma cells using fibermodified adenoviral vectors. Gene Ther. 2013;20(3):283–297. doi: 10.1038/gt.2012.39. [DOI] [PubMed] [Google Scholar]
- 141.Xu LP, Josan JS, Vagner J, Caplan MR, Hruby VJ, Mash EA, et al. Heterobivalent ligands target cell-surface receptor combinations in vivo. Proc Natl Acad Sci U S A. 2012;109(52):21295–21300. doi: 10.1073/pnas.1211762109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ortner A, Wernig K, Kaisler R, Edetsberger M, Hajos F, Koehler G, et al. VPAC receptor mediated tumor cell targeting by protamine based nanoparticles. J Drug Target. 2010;18(6):457–467. doi: 10.3109/10611860903508796. [DOI] [PubMed] [Google Scholar]
- 143.Liu Z, Stanojevic V, Brindamour LJ, Habener JF. GLP1-derived nonapeptide GLP1(28–36)amide protects pancreatic beta-cells from glucolipotoxicity. J Endocrinol. 2012;213(2):143–154. doi: 10.1530/JOE-11-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tafreshi NK, Huang X, Moberg VE, Barkey NM, Sondak VK, Tian H, et al. Synthesis and characterization of a melanoma-targeted fluorescence imaging probe by conjugation of a melanocortin 1 receptor (MC1R) specific ligand. Bioconjug Chem. 2012;23(12):2451–2459. doi: 10.1021/bc300549s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Falciani C, Brunetti J, Lelli B, Accardo A, Tesauro D, Morelli G, et al. Nanoparticles exposing neurotensin tumor-specific drivers. J Pept Sci. 2013;19(4):198–204. doi: 10.1002/psc.2493. [DOI] [PubMed] [Google Scholar]
- 146.Rangger C, Helbok A, Ocak M, Radolf T, Andreae F, Virgolini II, et al. Design and evaluation of novel radiolabelled VIP derivatives for tumour targeting. Anticancer Res. 2013;33(4):1537–1546. [PubMed] [Google Scholar]
- 147.Xu Y, Duggineni S, Espitia S, Richman DD, An J, Huang Z. A synthetic bivalent ligand of CXCR4 inhibits HIV infection. Biochem Biophys Res Commun. 2013;435(4):646–650. doi: 10.1016/j.bbrc.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Odorisio MS, Fleshman DJ, Qualman SJ, Odorisio TM. Vasoactive-intestinal-peptide - autocrine growth-factor in neuroblastoma. Regul Pept. 1992;37(3):213–226. doi: 10.1016/0167-0115(92)90616-3. [DOI] [PubMed] [Google Scholar]
- 149.Khondee S, Baoum A, Siahaan TJ, Berkland C. Calcium condensed LABL-TAT complexes effectively target gene delivery to ICAM-1 expressing cells. Mol Pharm. 2011;8(3):788–798. doi: 10.1021/mp100393j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Blackburn WH, Dickerson EB, Smith MH, McDonald JF, Lyon LA. Peptide-functionalized nanogels for targeted siRNA delivery. Bioconjug Chem. 2009;20(5):960–968. doi: 10.1021/bc800547c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Angata T, Fujinawa R, Kurimoto A, Nakajima K, Kato M, Takamatsu S, et al. Integrated approach toward the discovery of glyco-biomarkers of inflammation-related diseases. Glycobiology of the immune response. Ann N Y Acad Sci. 2012;1253:159–169. doi: 10.1111/j.1749-6632.2012.06469.x. [DOI] [PubMed] [Google Scholar]
- 152.Pirogova E, Istivan T, Gan E, Cosic I. Advances in methods for therapeutic peptide discovery. Design and development. Curr Pharm Biotechnol. 2011;12(8):1117–1127. doi: 10.2174/138920111796117436. [DOI] [PubMed] [Google Scholar]
- 153.Liu BA, Engelmann BW, Nash PD. High-throughput analysis of peptide-binding modules. Proteomics. 2012;12(10):1527–1546. doi: 10.1002/pmic.201100599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Levine RM, Scott CM, Kokkoli E. Peptide functionalized nanoparticles for nonviral gene delivery. Soft Matter. 2013;9(4):985–1004. [Google Scholar]