

Abstract
Trinidad and Tobago is a twin-island Republic in the Caribbean and like many developing countries, it has included generic drugs on the national drug formulary to decrease the financial burden of pharmaceutical medications. However, to ensure that medications received by patients are beneficial, generic drugs need to be interchangeable with the innovator which has demonstrated safety, efficacy, and quality. The objective of the study was to compare the dissolution profiles and weight variations for different formulations of amoxicillin, metronidazole, and zidovudine that are on the national drug formulary and marketed in Trinidad and Tobago. All the products investigated are categorized as class 1 drugs according to the Biopharmaceutics Classification System (BCS) and the dissolution profiles were assessed according to the World Health Organization (WHO) criteria for interchangeability between products. The similarity factor, f2, was used to determine sameness between the products. No generic formulation was found to be similar to Amoxil® 500-mg capsules. The two generic products for metronidazole 200-mg tablets demonstrated more than 85% drug release within 15 min in all three of the buffers; however, their 400-mg counterparts did not fulfill this requirement. The zidovudine 300-mg tablet complied with the requirements in buffer pH 4.5 and simulated gastric fluid (SGF) but not for simulated intestinal fluid (SIF). Some Class 1 pharmaceutical formulations may possess the same active ingredient and amount of drug but may show significant differences to in vitro equivalence requirements. Nevertheless, the dissolution process is suitable to detect these variations.
KEY WORDS: biopharmaceutics classification system, dissolution, generic drugs, interchangeability, in vitro equivalence
INTRODUCTION
The Drug Price Competition and Patent Term Restoration Act of 1984, also referred to as the Hatch-Waxman Act, resulted in the entry of many generics into the pharmaceutical market in the USA [1]. Trinidad and Tobago (T&T) holds membership to the World Trade Organization, hence, it abides with the international patent laws, and has done like other developing and developed countries and included generic drugs on their national drug formulary list [2,3]. Generic drugs on the national formulary are also marketed in other areas of T&T. Substitution of generic drugs for brand name drugs is allowed in T&T at the public health facilities if the prescription is written with the international non-proprietary name (INN) or the generic name of the medicine [2].
The main objective of generic drugs is to reduce the cost of medicines for patients; it therefore relieves the financial burden and the supply demand of pharmaceuticals for the population [4]. The World Health Organization (WHO) has stated that all persons have the “right to health” and should have access to essential medicines, which is defined as “… medicines that satisfy the priority health care needs of a population” [5].
Antimicrobial medications are one of the most prescribed classes of drugs and its frequent use and misuse have resulted in antimicrobial resistance, which is a problem worldwide [6, 7]. Consequently, newer antimicrobials are required to treat the resistant strains but these are few and expensive [5]. Anti-infective drugs are on the WHO Model List of Essential Medicines including amoxicillin, metronidazole, and zidovudine oral dosage forms [8,9]. Hence, it is imperative for generic drugs to be safe, effective, and of good quality for substitution or interchangeability of the innovator to adequately combat the targeted organism [6]. The Ministry of Health of Trinidad and Tobago has guaranteed that they provide medicines to its population that are safe, efficacious, and of high quality [10,11]. The Medicine Regulatory Authority of the Ministry of Health comprise of the Drug Inspectorate Division and the Chemistry Food and Drug Division, which performs duties such as inspection and registration of pharmaceutical products and a Quality Control Division of the Ministry confirms that medicines obtained, registered, and distributed throughout the two islands fulfill the requirements [2,11]. However, physicians from T&T have indicated that the generic drugs used in the public health sector are not as effective as the brand name drugs [11,12].
For products to be interchangeable, WHO states that they must be therapeutically equivalent, this includes pharmaceutically equivalent or pharmaceutically alternative formulations with similar dose, route of administration, conditions, efficacy, safety, and bioequivalence to the comparator [13]. In addition, interchangeable products must be of equivalent dosage forms, indications and directions for usage and labeling [13]. Together with product interchangeability, regulatory authorities are advised to ensure generic products possess good manufacturing practices and quality control standards [13]. It should be noted that a discrepancy exists with the definition for interchangeability as some documents prefer not to include pharmaceutically alternative products; one reason being that the description of the latter is different for various countries [13–16]. The need for harmonization then becomes imperative.
Bioequivalence studies are usually conducted to establish bioequivalence [13]. There are many methods used to determine bioequivalence. These comprise of pharmacokinetic (PK) studies; pharmacodynamic (PD) studies; comparative, clinical, bioequivalence trials; and in vitro studies [15,16]. PK studies usually require the use of healthy humans whereas PD studies verify the drug response in patients [15]. Dissolution is the main in vitro method used in quality control and of late to determine bioequivalence between certain drug products. The dissolution procedure has acquired many roles including its contribution in drug development, quality assurance, and investigation of similarity between the preparations. Furthermore, dissolution has been considered in in vitro–in vivo correlation and associations, involved in Biopharmaceutics Classification System (BCS), and has been used for biowaivers, which is the absence of clinical bioequivalence testing in humans [17]. BCS has classified the medicines under investigation to be of the class 1 category which interprets them to have very soluble and very permeable active pharmaceutical ingredients (API) and to release 85% and more of their drug in 15 min (very rapid release) in three different buffer solutions or need to be compared using f2 statistics when more than 85% are releases in 30 min (rapid release) [13].
The Pan American Network on Drug Regulatory Harmonization has explored a framework including BCS, which aims to achieve harmonization among Latin American countries in the implementation of equivalence standards for pharmaceutical products [18]. T&T is part of this region and can pursue its proposal.
The objective was to investigate whether the different immediate release oral formulations of amoxicillin, metronidazole, and zidovudine fulfill the requirements of 85% or more drug release in 15 or 30 min in all three buffers using in vitro dissolution testing; as well as, to compare weight uniformity. The products consisted of generic brands and in the case of amoxicillin, the innovator product.
METHODOLOGY
Reagents
The reference standards for amoxicillin, metronidazole, and zidovudine were received from the US Pharmacopeia (USP, Rockville, MD). Acetonitrile, sodium acetate trihydrate, potassium phosphate monobasic, potassium hydroxide, and sodium hydroxide were purchased from Caledon Laboratories Limited (Georgetown, Ontario). Items from Fisher Scientific (Bridgewater, NJ) included hydrochloric acid NF/FCC grade, o-phosphoric acid 85% HPLC grade, glacial acetic acid USP/FCC/EP/BP, sodium hydroxide, and sodium chloride USP/FCC/EP/BP/JP granular. The water utilized was produced in the laboratory using an Elga® Maxima Ultra Pure Water system.
All of the drug formulations were purchased from distributors in Trinidad, West Indies. Product information for each of the dosage forms used in the study is listed in Table I.
Table I.
Properties of Amoxicillin, Metronidazole, and Zidovudine Formulations of This Study
| Drug name | Brand name | Manufacturer | Country | Dosage form | Expiry date | Lot number | Excipients | Is/was on formulary |
|---|---|---|---|---|---|---|---|---|
| Metronidazole USP | Gendazole® 200 mg | Genethics Pharmaceutical Limited | Republic of Trinidad and Tobago | Film-coated tablet | October 2012 | MNO-905 | Maize starch, microcrystalline cellulose, maize starch for paste, gelatine, povidone, methyl hydroxybenzoate sodium, propyl hydroxybenzoate sodium, purified water, magnesium stearate, purified talc, colloidal anhydrous silica, sodium starch glycollate, coating materials: instacoat white (color: titanium dioxide), isopropyl alcohol, dichloromethane | Yes |
| Metronidazole B.P. | Metrogyl® 200 mg | Unique Pharmaceutical Labs | India | Film-coated tablet | September 2014 | B.No.AM69006 | Lactose, disodium edentate, ethyl cellulose, sodium starch glycollate, silica-colloidal anhydrous, guar gum, magnesium stearate | Yes |
| Metronidazole USP | Gendazole F® 400 mg | Genethics Pharmaceutical Limited | Republic of Trinidad and Tobago | Film-coated tablet | January 2014 | MoZ-11029 | Maize starch, microcrystalline cellulose, maize starch for paste, gelatine, povidone, methyl hydroxybenzoate sodium, propyl hydroxybenzoate sodium, purified water, magnesium stearate, purified talc, colloidal anhydrous silica, sodium starch glycollate, coating materials: instacoat white (color: titanium dioxide), isopropyl alcohol, dichloromethane | Yes |
| Metronidazole B.P. | Metrogyl® 400 mg | Unique Pharmaceutical Labs. | India | Film-coated tablet | August 2013 | B.No. AMA8003 | Lactose, disodium edentate, ethyl cellulose, sodium starch glycollate, silica-colloidal anhydrous, guar gum, magnesium stearate, quinoline yellow ci 47005 (e104) | Yes |
| Zidovudine | Aviro-Z® 300 mg | Ranbaxy Laboratories Limited | India | Film-coated tablet | May 2013 | 2307512 | Information for excipients was not available | Yes |
| Amoxicillin trihydrate BP | Imox®-500 mg | Ipca Laboratories Limited | India | Capsule | March 2015 | B. No. LNo21004 | Information for excipients was not available | No |
| Amoxicillin trihydrate BP | Imox® 250 mg | Ipca Laboratories Limited | India | Capsule | June 2015 | B. No. PU031004 | Information for excipients was not available | Yes |
| Amoxicillin trihydrate BP | Pulmoxyl®-250 mg | Micro Labs Limited | India | Capsule | May 2014 | PXTB0082 | Information for excipients was not available | Yes |
| Amoxicillin B.P. | Ospamox® 250 mg | Sandoz | Austria | Capsule | October 2012 | 159920 | Gelatin, magnesium stearate, microcrystalline cellulose | Yes |
| Amoxicillin trihydrate | Ospamox® 500 mg | Sandoz GmbH | Austria | Film-coated tablet | September 2014 | BD3103 | Magnesium stearate, povidine, carboxymethyl starch, microcrystalline cellulose, titanium dioxide (e-171), talc, hydroxypropylmethyl cellulose | No |
| Amoxicillin trihydrate BP | Ranoxyl® 500 mg | Ranbaxy | India | Capsule | January 2014 | 2250897 | Information for excipients was not available | No |
| Amoxicillin trihydrate BP | Ranoxyl® 250 mg | Ranbaxy | India | Capsule | September 2013 | B. No. 2211609 | Information for excipients was not available | Yes |
| Amoxicillin B.P. | Amoxil® 500 mg | SmithKline Beecham plc | UK | Capsule | February 2015 | 473620 | Information for excipients was not available | No |
Weight Variation
The weight variation test was performed according to the International Pharmacopeia volume 4, 3rd edition and was executed to ensure weight uniformity among the dosage units. This test was added as an additional test to be able to link possible deviations in dissolution to potential differences in the weight of the test units. Twenty to 30 tablets and capsules of each formulation investigated were weighed, recorded, and statistically analyzed using Minitab® version 16 software.
Media Preparation
Buffer solutions pH 1.2 (simulated gastric fluid, SGF), pH 4.5 (acetate buffer), and pH 6.8 (simulated intestinal fluid, SIF) were prepared without enzymes as stated in the USP under “Test Solutions”.
The media and the mobile phase were deaerated by filtering through 0.45-μm Millipore® nylon membrane filters into a glass bottle that was immersed in a Branson® 8200 Ultrasonic water bath under vacuum.
Dissolution Test
VK 7020 Vankel® Dissolution Equipment with six vessels was connected to a VK 8000 Varian® System Monitor Sampling Station. USP Apparatus 2 (paddle) was used for the tablets. USP sinker baskets as described in USP 36, chapter <711> were employed for the capsule dosage forms. All tests were performed at 75 rpm for 1 h. The media were prepared, degassed, pre heated, and then approximately 900 g of the media was weighed into each of the six vessels with care not to include air within the media. The vessels were subsequently returned to the water bath of the dissolution equipment. When the media attained a temperature of 37 ± 0.5°C, the tablets/capsules were placed into the vessels, and then the VK 7020 apparatus was initiated. The autosampler withdrew and filtered (Varian® Full Flow 70-μm filters) 1.25 ml of the media from each vessel at times 10, 15, 20, 30, 45, and 60 min without replacement but only 1 ml was placed into 1.5-ml amber colored vials discarding any remainders and 10 μL of these samples were tested using the HPLC method. The results were reported in percent of drug dissolved and were corrected by calculation for the withdrawn volume. If 85% or more of the active ingredients were not obtained at 15 min (very rapidly dissolving according to WHO terminology), the test was repeated with an additional six tablets/capsules.
Apparatus
High-pressure liquid chromatography (HPLC) equipment was used to quantify the amount of drug dissolved at each sample time point. The HPLC components comprised of two LC-10AS Shimadzu liquid chromatography pumps, a SPD-M10Avp Shimadzu Diode Array Detector, SIL-10A Shimadzu Auto Injector, Shimadzu Sample Cooler, CBM-20A Prominence Communications Bus Module, Shimadzu Syringe, and the EZStart 7.4 data acquisition software (Shimadzu, Columbia, MS).
Other equipments comprise of AB204 Mettler Toledo scale, Accumet excel XL20 pH/Conductivity Meter, and micropipettes were from Fisher Scientific and a Denver Instrument APX-1502 scale to weigh the vessels and the media.
Amoxicillin
The HPLC assay used in this study varied from that in the USP in order to improve linearity and attain reduced retention time over a concentration range of 3.75% to 120% of the labeled drug content in medium volume of 900 ml. Li Chrospher 100RP 18 column (125 mm × 4.0 mm) (Merck, Darmstadt, DE) of particle size 5 μm with guard column, detected the drug at 219-nm wavelength using a mobile phase of 5% acetonitrile and phosphate buffer, which was adjusted to pH 5.0 ± 0.1 with 45% (w/w) KOH. A suitability test was performed with each buffer solution (SIF, SGF, and buffer pH 4.5). The goodness of fit coefficients for the calibration curves were at least 0.999 in each medium and the coefficient of variation were 2.43 in SGF, 2.00 in pH 4.5 buffer, and 2.56 in SIF.
Metronidazole
Analytical quantification was modified from that of the USP 36 to attain reduced retention time over a concentration range of 3.75% to 120% of the labeled drug content in a medium volume of 900 ml. LiChrospher® 60 RP select B column (Merck) (125 mm × 4.0 mm) of particle size 5 μm with guard column detected the drug at 228 nm with a mobile phase of water and acetonitrile of ratio 66:34, respectively. Suitability tests with SIF, SGF, and buffer pH 4.5 were achieved. The goodness of fit coefficients for the calibration curves were at least 0.999 in each medium and the coefficient of variation were 2.74 for SGF, 0.55 for SIF, and 0.28 for buffer pH 4.5.
Zidovudine
The HPLC procedure applied in this study varied from that of the USP 36 in order to acquire decreased retention time over a concentration range of 3.75% to 120% of the labeled drug content in medium volume of 900 ml. LiChrospher® 60 RP select B column (Merck) (125 mm × 4.0 mm) of particle size 5 μm with guard column detected the drug at 265 nm with a mobile phase of water and acetonitrile of 72:28, respectively. Suitability tests with SIF, SGF, and buffer 4.5 were achieved. The goodness of fit coefficients for the calibration curves were at least 0.999 for each medium and the coefficient of variation were 1.22 for SGF, 2.40 for SIF, and 2.46 for buffer pH 4.5.
Statistical Analyses
The data was entered and analyzed in Microsoft Office Excel 2007 and graphs were created. DDSolver® software (an Add-In for Excel) [19] was used to determine the similarity factor (f2) among the metformin preparations that did not acquire drug release of 85% and more at 15 min (very rapidly dissolving).
RESULTS
Amoxicillin
The weights of the 250-mg capsules were between 0.34 to 0.48 g while that of the 500-mg capsules ranged from 0.64 to 0.76 g. The Ospamox® 500-mg tablets weighed an average of 0.68 g.
In the SGF, all the amoxicillin formulations released 85% and more of the drug within 15 min. The maximum drug release was at 30 min, thereafter, the dissolution profiles subsequently decreased; a behavior that was not seen in buffer pH 4.5 and SIF.
Figure 1 and Table II summarize the results of the 250-mg amoxicillin capsules. Within the 1-h period, none of the capsules released 85% or more of amoxicillin in buffer pH 4.5 and SIF but their dissolution profiles continued to increase steadily during this period. Nevertheless, Fig. 2 and Table III summarize the results of the 500-mg amoxicillin formulations and in SIF, only Amoxil® 500-mg capsules had more than 85% of drug dissolved at 45 min while Ospamox® 500-mg tablets displayed a similar amount in 20 min. In buffer pH 4.5, the results were comparable to that in SIF as Amoxil® 500-mg capsules and Ospamox® 500-mg tablets were the only two products that released 85% or more of the drug at 60 and 30 min, respectively.
Fig. 1.
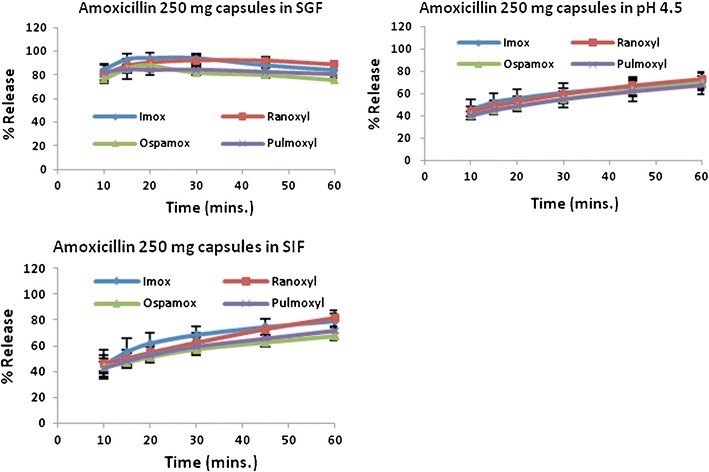
Summary of dissolution profiles for amoxicillin 250-mg capsules
Table II.
Summary of Similarity Factor, f 2, for amoxicillin 250-mg capsules
| pH 4.5 | Imox® | Ranoxyl® | Ospamox® | Pulmoxyl® |
|---|---|---|---|---|
| Imox® | 100 | 81.4 | 62.9 | 62.9 |
| Ranoxyl® | 81.4 | 100 | 67.8 | 66.6 |
| Ospamox® | 62.9 | 67.8 | 100 | 95.4 |
| Pulmoxyl® | 62.9 | 66.6 | 95.4 | 100 |
| SIF | Imox® | Ranoxyl® | Ospamox® | Pulmoxyl® |
| Imox® | 100 | 66.3 | 49.3 | 55.3 |
| Ranoxyl® | 66.3 | 100 | 55.3 | 63.6 |
| Ospamox® | 49.3 | 55.3 | 100 | 77.4 |
| Pulmoxyl® | 55.3 | 63.6 | 77.4 | 100 |
Similarity factor (f 2) ≥50 represents similarity while <50 represents dissimilarity
Fig. 2.
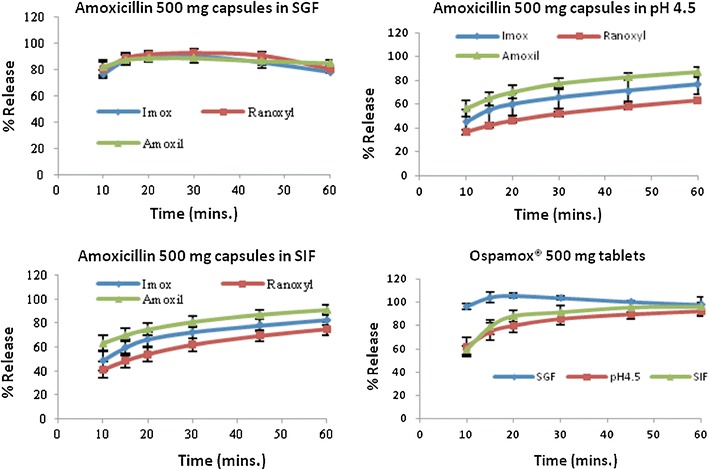
Summary of dissolution profiles for amoxicillin 500-mg formulations
Table III.
Summary of Similarity Factor, f 2, for Amoxicillin 500-mg Formulations Compared to Amoxil® reference
| pH 4.5 | Amoxil® |
|---|---|
| Imox® | 47.5 |
| Ranoxyl® | 31.3 |
| SIF | Amoxil® |
| Imox® | 49.8 |
| Ranoxyl® | 35.3 |
Similarity factor (f 2) ≥50 represents similarity while <50 represents dissimilarity
The value of f2 for Imox® and Ranoxyl® 500-mg capsules were less than 50 for both SIF and buffer pH 4.5.
Metronidazole
The weights of the 200-mg metronidazole tablets were 0.25–0.36 g while that of the 400 mg were 0.46–0.64 g. According to the International Pharmacopeia, the percent deviation of weight for the metronidazole 200- and 400-mg tablets were within ±5%.
Figures 3 and 4 and Table IV summarize the results of the 200- and 400-mg metronidazole formulations, which were generic products of Flagyl® (the innovator). The 200-mg products showed 85% and more drug release in 15 min for all three buffers and their profiles were almost superimposable. Hence, no f2 calculations were required. However, the profiles of the two metronidazole 400-mg formulations were distinct from one another in each of the three buffers. In SGF, Metrogyl F® released 85% and more dissolved drug in 15 min but showed similar amount in about 30 min for buffers pH 4.5 and SIF; nevertheless, Gendazole F® approached 85% and more in all the buffers at approximately 60 min.
Fig. 3.
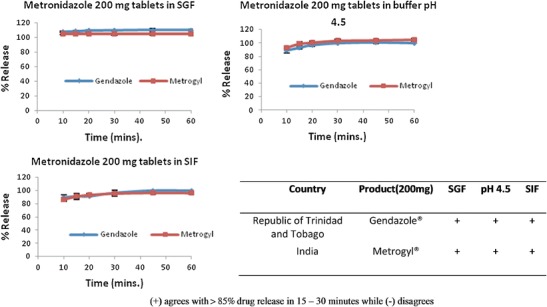
Summary of dissolution profiles for metronidazole 200-mg formulations
Fig. 4.
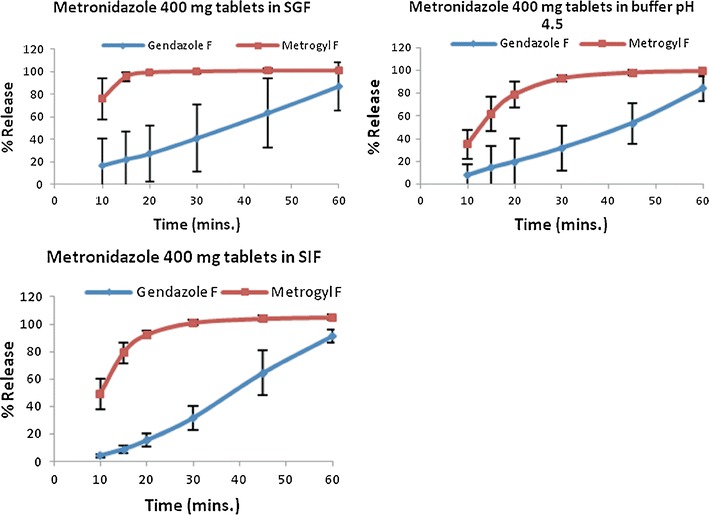
Summary of dissolution profiles for metronidazole 400-mg formulations
Table IV.
Summary of Similarity Factor, f 2, for Metronidazole 400-mg Tablets
| Gendazole F vs. Metrogyl F | |
|---|---|
| SGF | 12.5 |
| pH 4.5 | 17.1 |
| SIF | 12.3 |
Similarity factor (f 2) ≥50 represents similarity while <50 represents dissimilarity
In addition, the dissolution process was performed with two tablets, in each of the metronidazole 200-mg products, to compare the results to that of the 400-mg dosage form. The results of the two tablets were similar to that of when one 200-mg tablet was investigated with the dissolution method.
No f2 comparison was done for the generic metronidazole formulations compared to the Flagyl® 200- and 400-mg tablets as the innovators were not available at the time of the study.
Zidovudine
Aviro-Z® 300-mg tablet weighed 0.38–0.39 g and the percent deviation was within ±5%.
The dissolution profile for the zidovudine 300-mg product was above 85% in 15 min and was almost superimposable in buffer pH 4.5 and SGF, see Fig. 5. However, in SIF, the dissolution graph did not attain the 85% or more criteria; it showed no sign of increasing and appeared plateau throughout the 60-min period.
Fig. 5.

Summary of dissolution profiles for zidovudine 300-mg formulation
No f2 comparison was done for zidovudine as the Retrovir® 300-mg innovator product was unavailable at the time of the study.
DISCUSSION
Amoxicillin
Amoxicillin is not stable in acid and its beta lactam ring opens when placed in a neutral or basic environment or when acted upon by beta lactamase enzymes, to produce an inactive substance [20,21]. In an acidic pH of 1.2, amoxicillin trihydrate released maximum amoxicillin drug at 30 min then steadily decreased. Nevertheless, at pH 4.5 and 6.8, which is considered the range of lowest solubility (pH 4 to 6), the hydrated amoxicillin dissolved slower and drug dissolution increased with time [20]. A study by Löbenberg et al. investigated amoxicillin in the Americas and concluded that all products fulfill the requirements in the SGF showing a decline after obtaining a peak at about 10 to 15 min [22]. Nevertheless, some of the products in Löbenberg’s study showed similar results to our study and did not release 85% or more of amoxicillin in 15 to 30 min or failed f2 comparison in buffers 4.5 and SIF. Hence, the generics were not in vitro equivalent to the comparator.
All the amoxicillin products investigated were capsules except for Ospamox® 500-mg tablet. Most guidance documents indicate that the generic product must be the same dosage form as the innovator when interchangeability is being considered—pharmaceutical equivalent as per US FDA definition [13,23]. However, some agencies agree to different dosage forms but same route of administration such as tablets, capsules, and liquids [24,25]. Consequently, this demonstrates the need for standardization of definitions to be globally accepted.
In addition, many other terms including generic drugs and pharmaceutically alternative products, need to be revised so that clarity and uniformity exists [12–15]. For reasons such as these, no similarity factor calculation was computed between Amoxil® 500-mg capsules, the reference drug, and Ospamox® 500-mg tablets. The f2 factor was calculated for Amoxil® 500 mg and the other amoxicillin 500-mg products, resulting in a f2 value that was less than 50 for Imox® and Ranoxyl® 500-mg capsules; indicating no similarity to the innovator product. Olanrewaju et al. stated that API, excipients, and manufacture methods are factors that affect bioequivalence of a pharmaceutical product [26]. These may be the basis why the other amoxicillin products did not show equivalence to the innovator. A study conducted in Nigeria investigated seven brands of amoxicillin/clavulanic acid tablets and found that one of them was not bioequivalent to the innovator [16]. The “bioinequivalent” brand passed the uniformity of weight and friability tests but did not comply with the hardness, disintegration, dissolution, and f2 analyses, suggesting a substandard therapeutic response [16]. Saptarini et al. compared the content and dissolution of three brands of 500-mg amoxicillin manufactured by the Indonesian government with three brands produced by private companies and found no significant difference among the six preparations [27]. The latter study identified that a rapid dissolution will produce a good absorption of the active ingredient and that the dissolution process is a better surrogate for absorption than disintegration.
Metronidazole
Metronidazole is a nitroimidazole compound that is soluble in dilute acids; hence, its preference for SGF [28]. The USP 36 indicated the quality control dissolution medium for metronidazole should be 0.1-N hydrochloride solution, which is similar to SGF [29]. All the products, except Gendazole F®, achieved the SGF the USP release requirement. Rediguieri et al. indicated that certain excipients such as sorbitol, sodium lauryl sulfate, and propylene glycol has been known to alter the bioavailability of some metronidazole formulations [30]. Magnesium stearate, which was present in some of the products investigated, can also affect the dissolution profile of the finished products [31]. Excipients for most of the products investigated in our study were not available on the packages or inserts when items were purchased. Requests were made to the companies for the information to be provided, so the manufacturer and distributor for Gendazole® and distributor for Metrogyl® responded; the former via e-mail listing the inactive substances and the latter stated to check the item on the internet. As seen in Table I, Gendazole® contains Instacoat®. This series of coating materials includes pH sensitive substances and this might be the reason for the poor dissolution at pH 1.2 and 4.5; however, it was not possible to identify which type of Instacoat® product was used for this metronidazole formulation. [32,33] Distributors of the other brand of metronidazole indicated the latter response as they, too, had no idea of the excipients of the products they carry. For regulatory purposes, labeling of all excipients on the insert is not mandated unless it is known to cause a pharmaceutical effect [13, 34]. For the Gendazole® products, the excipients listed did not specify which type of povidone was used to manufacture the formulations (i.e., 30 or 90) which can affect the finished product [35]. Hence, it is possible that the presence of specific excipients may have contributed to the poor performance of the 400-mg products in the different buffers. In T&T, the regulatory authorities indicate that excipients for pharmaceutical products should be included on the request for submission form [36]. Ibezim et al. performed physiochemical evaluation, including dissolution, on ten different metronidazole 200-mg tablets where only two brands showed evidence of possible bioequivalence [37]. In our research, the two metronidazole 200-mg tablets released 85% and more of the API in 15 to 30 min for all buffers but only one 400-mg formulation achieved this goal. Besides the use of excipients, the 200-mg tablets appeared to be manufactured distinct to that of the 400-mg formulations because they demonstrated different dissolution profiles in each of the three buffers. Manufacturing process for the API or finished products can occur via different methods and have been known to alter the performance of the product [38]. Manufacturing a product using the dose proportionality concept is usually considered for products of different strengths. Hence, knowledge of whether the dose proportionality concept or another manufacturing procedure was utilized for the 400- and 200-mg metronidazole formulations can assist in understanding the behavior of these products [39].
Löbenberg et al. also looked at some metronidazole formulations and reported that the generics examined were not comparable to the innovator product, Flagyl® [22]. Furthermore, there were two products labeled with the trade name Flagyl®; one from Pharmacia, a subsidiary of Pfizer, which was used as the comparative pharmaceutical product according to the Orange Book [16], and the other from Sanofi Aventis. The Flagyl® product from Sanofi Aventis was found not to be in vitro equivalent to the Pfizer product. Products with the same trade names may have manufacturing and/or marketing authorizations from the innovator company; therefore, these products should definitely comply with the requirements and standards for equivalence and interchangeability. This is because patients who prefer to purchase a product of a particular trade name cannot tell whether there is any difference to the innovator because, on their part, they believe that they are receiving the “original” or innovator medicine [13]. Furthermore, the strengths of the products used are important; as in the case of metronidazole, the Orange Book US FDA database has Flagyl® 250- and 500-mg products as their reference drug [16] while the electronic Medicines Consortium of the UK has Flagyl® 200- and 400-mg products, the latter strengths are used in T&T. Patients also need to be aware that such variations exist. Appeals for an international reference product (innovator) to which all countries can refer may alleviate such problems. [23]. Comparison with the innovator was not done for our study because the innovator product was not available at that time. However, about 1 year later, it was observed that a product of trade name, Flagyl®, of 400-mg strength was available in Trinidad and was manufactured by Sanofi Aventis Pakistan Limited. No test was done on this product.
Zidovudine
Zidovudine comprises of a thymidine structure and shows solubility in water, diluted acid, acetate buffer, and phosphate buffer, in the order of more soluble to least soluble [40,41]. Zidovudine in SIF showed a steady release of drug as the profile appeared flat over the 60 min period. One study by Ochekpe et al. investigated the dissolution profile of Zidovudine combination with Lamivudine in the Nigerian market and demonstrated zidovudine to release less than 85% of drug in SIF (pH 6.8 buffer) at 30 min, which is the cut-off time for rapidly dissolving products [42]. Hence, it was concluded that Combivir® was not similar to the other products and was therefore not considered to be interchangeable with the other formulations. Our study also showed less than 85% release in buffer pH 6.8 (SIF) at a similar time, hence, agreeing with the Nigerian study.
The innovator form for Zidovudine is known as Retrovir® 300-mg film coated tablets from GlaxoSmithKline Beecham, which was unavailable at the time of study [43,44]. The innovator and the generic product investigated are of same strength and dosage form, hence, it would be appropriate for similarity testing.
Different zidovudine formulations compared to that in our study, were examined in the research done by Löbenberg et al. and all products were demonstrated to be similar to the innovator product identified, which was Retrovir® 300-mg capsules, from USA; including another capsule product labeled Retrovir®, from Mexico [22].
CONCLUSION
The study showed that some BCS class 1 drugs did not fulfill the requisite for the dissolution similarity and the reasons suggested included variation of excipients and manufacturing processes of the different formulations. The examination of these products highlighted differences in defining terms such as generic drug and pharmaceutically alternative products; as for example, the US FDA has described generic drugs as being identical to the innovator, which can be difficult to be determined in a global setting due to different innovators in different parts of the world and variations in definitions. Hence, harmonization of pharmaceutical terms and reference products is important to remove many variations that can occur among medicinal formulations. If products fail in vitro dissolution comparison criteria does not automatically mean that such products are substandard; however, bioequivalence to an appropriate reference product must be established using an in vivo study.
In addition, the research suggested for the in vitro equivalence process to be implemented in T&T for specific generic products; consequently confirming therapeutic equivalence to their innovator counterparts. The use of biowaivers can increase the quality and availability of generics
Acknowledgments
We wish to thank the University of Alberta International, the Drug Development and Innovation Centre, Edmonton, Alberta, Canada, and the University of the West Indies, St. Augustine, Trinidad, for providing assistance to perform the study.
REFERENCES
- 1.Schutt CE. Regulating the pharmaceutical industry. The Wharton School, University of Pennsylvania. [Internet] 2007 Oct 4 [cited 2013 Feb 9]. Available from: http://www.princeton.edu/~actin/documents/RegulatingthePharmaceuticalIndustry.pdf
- 2.The Ministry of Health of Trinidad and Tobago, Pan American Health Organization, World Health Organization. Trinidad and Tobago Pharmaceutical Country Profile. [Internet] 2012 [cited 2013 Feb 12]. Available from: http://www.who.int/medicines/areas/coordination/PSCP_TRT_en.pdf
- 3.Pinto Pereira LM, Fraser H, Burnett F. Assessment of generic drugs in the Caribbean. Drug Inf J. [serial on the Internet]. 1998 [cited 2013 Feb 12]; 32:145-50. Available from: http://www.diahome.org/productfiles/8357/diaj_12481.pdf
- 4.Venkatesh M, Bairavi VG, Sasikumar KC. Generic antibiotic industries: Challenges and implied strategies with regulatory perspectives. J Pharm Bioallied Sci [serial on the Internet]. 2011 [cited 2013 April 18]; Jan-Mar; 3(1). Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3053506/?report=printable. [DOI] [PMC free article] [PubMed]
- 5.World Health Organization. Medicines: essential medicines. [Internet] 2010 [cited 2013 Apr 4]. Available from: http://www.who.int/mediacentre/factsheets/fs325/en/.
- 6.Zuluaga A, Agudelo M, Rodriguez C, Vesga O. Application of microbiological assay to determine pharmaceutical equivalence of generic intravenous antibiotics. BMC Clin Pharmacol. 2009;9(1):1. doi: 10.1186/1472-6904-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baptiste-Cornelis T. Statement by Senator, the Honourable Therese Baptiste-Cornelis World Health Day 2011 Press Conference, 3rd floor Conference Room: Government of the Republic of Trinidad and Tobago. [Internet] 2011 April [cited 2013 April 18]. Available from: http://www.news.gov.tt/index.php?news=7599.
- 8.World Health Organization. WHO model list of essential medicines—18th list. [Internet] 2013 April [cited 2013 July 12]. Available from: http://www.who.int/medicines/publications/essentialmedicines/18th_EML_Final_web_8Jul13.pdf.
- 9.Kar SS, Pradhan HS, Mohanta GP. Concept of essential medicines and rational use in public health. Indian J of Community Med [serial on the Internet]. 2010 [cited 2013 Jul 12]; 35(1). Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2888334/. [DOI] [PMC free article] [PubMed]
- 10.Ministry of Health of the Government of the Republic of Trinidad and Tobago. Drug safety. [Internet] 2012 [cited 2013 Feb 12]. Available from: http://www.health.gov.tt/news/newsitem.aspx?id=323.
- 11.Health Ministry: Hospital drugs safe and effective. Trinidad Express. [Internet] 2012 [cited 2013 Feb 12]. Available from: http://www.trinidadexpress.com/news/Health_Ministry__Hospital_drugs_safe_and_effective-139168444.html.
- 12.Lambie I. Truth about CDAP drugs. Newsday. [Internet] 2007 Jul 5 [cited 2013 Feb 12]. Available from: http://www.newsday.co.tt/news/0,60026.html.
- 13.World Health Organization. World Health Organization Expert Committee on specifications for pharmaceutical preparations, 40th report Geneva: World Health Organization. [Internet] 2006 [cited 2012 Nov 12]. Available from: http://apps.who.int/prequal/info_general/documents/TRS937/WHO_TRS_937_eng.pdf#page=359. [PubMed]
- 14.European Medicines Agency. Guideline on the investigation of Bioequivalence. London. [Internet] 2010 Jan 10 [cited 2013 Feb 24]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf.
- 15.Government of India New Delhi. Guidelines for bioavailability and bioequivalence studies [Internet] 2005 [cited 2013 Feb 9]. Available from: http://cdsco.nic.in/html/be%20guidelines%20draft%20ver10%20march%2016,%2005.pdf.
- 16.United States Food and Drug Administration. Orange book Preface development and approval process (Drugs). 32nd ed. [Internet] 2012 [cited 2013 Feb 9]. Available from: http://www.fda.gov/drugs/developmentapprovalprocess/ucm079068.htm.
- 17.Krämer J, Grady LT, Gajendran J. Historical development of dissolution testing [Internet]. In: Dressman J, Krämer J, editors. Pharmaceutical Dissolution Testing. Taylor and Francis Group, LLC; 2005. pp 1 - 37. doi:10.1201/9780849359170.ch1 Taylor and Francis Group, LLC. [Internet] 2005 [cited 2013 Feb 9]. Available from: www.crcnetbase.com/doi/pdf/10.1201/9780849359170.ch1
- 18.Pan American Health Organization. Framework for implementation of equivalence requirements for pharmaceutical products. [Internet] 2008 [cited 2013 Jul 8]. Available from: http://www2.paho.org/hq/dmdocuments/2008/WG-BE_document_approved_V_conference.pdf.
- 19.Zhang Y, Huo M, Zhou J, Zou A, Li W, Yao C, et al. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J [serial on the Internet]. 2010 Sept [cited 2012 Sept 25]; 12(3):263-71. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2895453 [DOI] [PMC free article] [PubMed]
- 20.Kaur SP, Rao R, Nanda S. Amoxicillin: a broad spectrum antibiotic. International J Pharm Pharm Sci [serial on the Internet]. 2011[cited 2013 Jul 3]; 3(3). Available from: http://www.ijppsjournal.com/Vol3Issue3/2249.pdf.
- 21.Katzung BG, Masters SB, Trevor AJ. Basic and clinical pharmacology. 11. New York: McGraw-Hill; 2009. [Google Scholar]
- 22.Löbenberg R, Chacra N, Stippler E, Shah V, DeStefano A, Hauck W, et al. Toward Global standards for comparator pharmaceutical products: case studies of amoxicillin, metronidazole, and zidovudine in the Americas. AAPS J [serial on the Internet]. 2012 [cited 2012 Sept 1]; 14(3). Available from: http://dx.doi.org/10.1208/s12248-012-9350-9. [DOI] [PMC free article] [PubMed]
- 23.Mei-Ling C, Shah VP, Crommelin DJ, Shargel L, Bashaw D, Bhatti M, et al. Harmonization of regulatory approaches for evaluating therapeutic equivalence and interchangeability of multisource drug products: workshop summary report. AAPS J [serial on the Internet]. 2011 [cited 2013 Jul 3]; 13(4). Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3231855/pdf/12248_2011_Article_9294.pdf. [DOI] [PMC free article] [PubMed]
- 24.College of Pharmacists of British Columbia. Drug interchangeability update—amended from the August 2004 Vol. 3 No.1 FYI Newsletter. [Internet] 2011 [updated 2011 Oct 3; cited 2013 Jul 5]; 2011.1. Available from: http://library.bcpharmacists.org/D-Legislation_Standards/D-4_Drug_Distribution/5016-FYI-Drug_Interchangeability_Update.pdf.
- 25.Nova Scotia Department of Health. Nova Scotia criteria for interchangeability. [Internet] 2011 Feb [cited 2013 Jul 1]; 15 pages. Available from: http://www.gov.ns.ca/health/pharmacare/pubs/Nova_Scotia_Criteria_for_Interchangeability.pdf.
- 26.Olanrewaju OJ, Paul AC, Olusola AM. Quality assessment of amoxicillin-clavulanate potassium tablets in Lagos, Nigeria. J Chem Pharm Res [serial on the Internet]. 2012 [cited 2013 Apr 21]; 4(12). Available from: http://jocpr.com/vol4-iss12-2012/JCPR-2012-4-12-5032-5038.pdf.
- 27.Saptarini NM, Rusniyanti. Evaluation of content and dissolution profile of generic amoxicillin tablets marketed in Indonesia. IRJP [serial on the Internet]. 2012 [cited 2013 Apr 21]; 3(12). Available from: http://www.irjponline.com/admin/php/uploads/1531_pdf.pdf.
- 28.The Merck Index. 11 ed. New Jersey: Merck and Co., Inc; 1989.
- 29.Anand O, editor. Dissolution testing: an FDA perspective. AAPS Workshop, Physical Pharmacy and Biopharmaceutics. [Internet]. 2009 [cited 2013 Apr 21]. Available from: http://mediaserver.aaps.org/meetings/09_PPB/Wed/Track_I/Om_Anand.pdf
- 30.Rediguieri CF, Porta V, Nunes DSG, Nunes TM, Junginger HE, Kopp S, et al. Biowaiver monographs for immediate release solid oral dosage forms: metronidazole. J Pharm Sci [serial on the Internet]. 2011 May [cited 2013 Apr 21]; 100(5). Available from: http://www.fip.org/files/fip/BPS/BCS/Monographs/Metronidazole.pdf. [DOI] [PubMed]
- 31.Sinko PJ e. Martin’s physical pharmacy and pharmaceutical sciences 6th ed. Baltimore: Lippincott Williams and Wilkins; 2011.
- 32.Ideal Cures Pvt. Ltd. New E-series film coating range from Ideal Cures Pvt. Ltd. [Internet]. [cited 2013 August 15]. Available from: http://www.idealcures.co.in/downloads/Brochure%20e-series.pdf.
- 33.Ideal Cures Pvt. Ltd. Instacoat E-series. [Internet] 2011 [cited 2013 Aug 15]. Available from: http://www.idealcures.co.in/instacoat-e-series.html.
- 34.European Commission. Guidelines: medicinal products for human use—safety, environment and information. Excipients in the label and package leaflet of medicinal products for human use. [Internet] 2003 [cited 2013 Jul 8]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003412.pdf.
- 35.Block LC, Schemling LO, Couto AG, Mourao SC, Bresolin TMB. Pharmaceutical equivalence of metformin tablets with various binders. Rev de Cienc Farm Basica e Apl [serial on the Internet]. 2008 [cited 2013 Feb 9]; 29(1). Available from: http://serv-bib.fcfar.unesp.br/seer/index.php/Cien_Farm/article/view/431/414
- 36.Government of Republic of Trinidad and Tobago, Ministry of Health, Chemistry Food and Drugs Division. Government of Republic of Trinidad and Tobago, Ministry of Health, Chemistry Food and Drugs Division - New Drug Submission Form. [Internet]. [cited 2013 Jul 8]; p. 1-12. Available from: http://www.gov.tt/gortt/wcm/connect/ea0a65804d60bdd1962ab73c18a7ac93/MOH.pdf?MOD=AJPERES&CACHEID=ea0a65804d60bdd1962ab73c18a7ac93
- 37.Ibezim EC, Attama AA, Obitte NC, Onyishi V, Brown SA. In vitro prediction of in vivo bioavailability and bioequivalence of brands of metronidazole tablets in Eastern Nigerian drug market. Sci Res Essays. 2008;3(11):552–558. [Google Scholar]
- 38.Moreton RC, editor. Pharmaceutical development and manufacturing: trends in processing of oral solid dose forms. PharmSciFair; 2009 Jun 8-12; Nice, France.
- 39.Health Canada. Guidance document—conduct and analysis of comparative bioavailability studies. [Internet]. 2012 [cited 2013 Sept 8]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/applic-demande/guide-ld/bio/gd_cbs_ebc_ld-eng.pdf
- 40.Zidovudine matrix tablets and microcapsules.[Internet]. [cited 2013 Jul 3]; p. 109-79. Available from: http://shodhganga.inflibnet.ac.in/bitstream/10603/3453/16/16_chapter%205.pdf
- 41.Prakash K, Narayana RP, Shanta KK, Lakshmi NM. Solubility and dissolution rate determination of different antiretroviral drugs in different pH media using UV visible spectrophotometer. E-Journal of Chemistry [serial on the Internet]. 2008 [cited 2013 Jul 3]; 5 (S2):1159 - 1164. doi:10.1155/2008/125917. Available from: http://downloads.hindawi.com/journals/jchem/2008/125917.pdf
- 42.Ochekpe N, Owolayo H. Dissolution profiles of three brands of Lamivudine and Zidovudine combinations in the Nigerian market. Dissolut Technol [Internet]. 2006 [cited 2013 Apr 21]; 12-7]. Available from: http://www.dissolutiontech.com/DTresour/200611Articles/DT200611_A02.pdf.
- 43.World Health Organization. Guidance document—recommended comparator products: medicines for HIV/AIDS and related diseases. [Internet]. 2012 [cited 2013 Jul 8]; 1-4. Available from: http://apps.who.int/prequal/info_applicants/BE/Comparators-HIV_02February2012.pdf.
- 44.Daily Med. Retrovir. [Internet]. 2011 [cited 2013 July 8]. Available from: http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=f7214f89-5215-49b4-5491-6462a71b1f5c.