

Abstract
Vinorelbine (VLB) is a semi-synthetic Vinca alkaloid which is currently used in treatment of different cancer types mainly advanced breast cancer (ABC) and advanced/metastatic non-small cell lung cancer (NSCLC). However, its marketed formulation has been reported to have serious side effects, such as granulocytopenia, which is the major dose-limiting toxicity. Other unwanted effects include venous discoloration and phlebitis proximal to the site of injection, as well as localized rashes and urticaria, blistering, and skin sloughing. Our long-term aim in synthesizing a novel nanomicellar vinorelbine formulation is to reduce or even eliminate these side effects and increase drug activity by formulating the drug in a lipid-based system as a nanomedicine targeted to the site of action. To this end, the purpose of this study was to prepare, characterize, and determine the in vitro efficacy of vinorelbine-loaded sterically stabilized, biocompatible, and biodegradable phospholipid nanomicelles (SSM; size, ∼15 nm). Our results indicated that vinorelbine incorporate at high quantities and within the interface between the core and palisade sections of the micelles. Incorporation ratio of drug within sterically stabilized micelles increased as the total amount of drug in the system increased, and no drug particles were formed at the highest drug concentrations tested. The nanomicellar formulation of vinorelbine was ∼6.7-fold more potent than vinorelbine dissolved in DMSO on MCF-7 cell line. Collectively, these data indicate that vinorelbine-loaded SSM can be developed as a new, safe, stable, and effective nanomedicine for the treatment of breast and lung cancers.
KEY WORDS: DSPE-PEG 2000, nano drug delivery, sterically stabilized micelles, targeted drug delivery, vinorelbine
INTRODUCTION
Vinorelbine (Fig. 1) is a semi-synthetic derivative of the Vinca alkaloid vinblastine (Catharanthus roseus G. Don.). Vinorelbine was discovered by Pierre Potier and his team at Centre National de la Recherche Scientifique (CNRS) in France in the 1980s and was approved in the same country in 1989 under the brand name Navelbine® IV (vinorelbine bitartrate) for treatment of bronchial cancer. It gained approval to treat non-small cell lung cancer in 1991 (1).
Fig. 1.
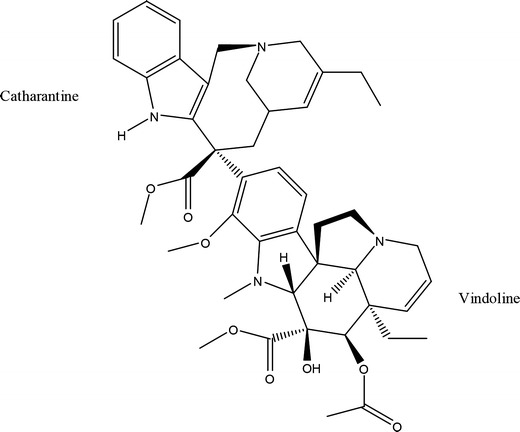
The structure of vinorelbine
A chemical modification of the catharanthine ring of vinblastine, converting it from a nine-membered ring to an eight-membered ring, generates vinorelbine (VLB) (Fig. 1), which exhibits increased lipophilicity and membrane permeability compared with other compounds in the Vinca alkaloid family (2,3). This alteration in structure resulted with an altered toxicity profile, differential effects on microtubule populations and dynamics, altered pharmacokinetic properties and a higher therapeutic index compared with other Vinca alkaloids (4,5).
Vinorelbine is now primarily used in combination with other cancer chemotherapeutic drugs for the treatment of a variety of cancers, including leukemias, lymphomas, advanced testicular cancer, as well as Kaposi’s carcinoma (6,7). It is especially active in advanced breast cancer (ABC) and advanced/metastatic non-small cell lung cancer (NSCLC) (8). Vinorelbine inhibits mitotic microtubule formation and induces a blockade of cells at metaphase (9). However, its marketed product, Navelbine® IV (vinorelbine bitartrate) does not have an optimal drug formulation, since it is prepared at low pH ∼3.5 causing venous irritation and phlebitis when directly administered intravenously as an aqueous solution (10). Most probably this is due to pH change and instant precipitation of the salt form of VLB at the site of injection. Furthermore, when given in a free form, drug distributes widely and causes dose-limiting hematologic side effects such as granulocytopenia and phlebitis. Thus, a new strategy is needed for aqueous injections of vinorelbine. Recently, different laboratories have been developing sterically stabilized liposomal formulations of vinorelbine, but stable high drug loading with high PEGylation has been difficult to achieve (11,12). Here, we describe a new lipid-based formulation of VLB using PEGylated phospholipid micelles, which should overcome the loading and stability problems of stealth liposomal formulations of VLB. Furthermore, due to smaller size (15 nm) of micelles, passive targeting of the nanomedicine to the tumors should be more efficient than liposomes. Our attempt in preparing nanomicellar formulation of vinorelbine is to achieve three important aims: 1—increase the amount of drug molecules delivered to tumor tissues by taking the advantage of the enhanced permeability and retention (EPR) effect that is the property by which certain sizes of particles (typically liposomes, nanoparticles, and macromolecular drugs) tend to accumulate in tumor tissue much more than they do in normal tissues (13). Targeting will not only increase drug efficacy but also significantly decrease toxicity; 2—provide sustained release of drug within the tumor tissue. Since VLB is a cell cycle phase specific antimitotic agent (5,14,15), prolonged exposure of cells to VLB is important for optimal activity (16–18); and 3—reduce the side effects at the site of injection and phlebitis, as observed with the current marketed product.
Compared to conventional formulations of chemotherapeutic agents, nanomedicines prepared using lipid- or polymer-based drug delivery systems have the advantage of improving the pharmacokinetic and therapeutic properties of cytotoxic drugs (19,20).
Micelles composed of PEGylated phospholipid 1,2-distearoyl-sn-glycero-3-phosphatidylethanolamine-N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG 2000) are well-established lipid-based drug delivery systems for hydrophobic anticancer drugs due to their long-circulating, sterically stabilized, biocompatible, and biodegradable properties (21–23). These so-called sterically stabilized micelles (SSM) are simple to prepare, form spontaneously above their critical micellar concentration (0.5–1 μM), relatively stable in vivo (blood serum) (24,25) and upon dilution in aqueous environment, and can be lyophilized without cryo- and lyo-protectants for long-term storage (26,27). The PEG on the surface of the micelles renders them sterically stabilized, preventing opsonization and reticular endothelial system uptake. In addition, because of their small size (∼15 nm), these carrier systems can provide targeted delivery to cancer or other injured tissues by selective extravasation through leaky vasculature (23). Recently published data indicate that antiangiogenesis-associated cancer therapy with nano drug delivery system is more effective when the nanoparticle’s size is around 12 nm (28). Based on these data, SSM have potential for in combination with antiangiogenesis agents for cancer therapy.
In our previous studies, we reported enhanced aqueous solubility and therapeutic efficacy of several anticancer drugs when incorporated in phospholipid micelles (22,23,29). In this work, we report synthesis and characterization of a novel lipid-based nanomedicine of vinorelbine (VLB-SSM), which is fully PEGylated and has a much smaller size than partially PEGylated liposomal formulations of VLB, currently under evaluation. Consequently, we show the enhancement of cytotoxic activity of this new formulation VLB-SSM compared to free vinorelbine.
MATERIALS AND METHODS
Materials
Poly(ethylene glycol-2000)-conjugated distearoylphosphatidylethanolamine (DSPE-PEG 2000) was obtained from Lipoid Lipids (MW2810, Cat # PE 18:0/18:0-PEG 2000, Lot # 882032-01/907), vinorelbine was purchased from Altan Biochemicals (Cat# AL-4176, MW: 778.932). MCF-7 cells (#HTB-22), fetal bovine serum, trypsin-EDTA and Eagle’s minimum essential medium (EMEM) with Earle’s balanced salt system (BSS) were obtained from American Type Culture Collection (Manassas, VA). Amicon® Ultra-4 Centrifugal Filter Tubes, 3,000 MWCO (Millipore) and Spectra/Por 3 Molecularporous Dialysis Membrane, (Spectrum, MWCO 6000–8000, part no. 132720) were purchased from Fisher Scientific. All used solvents were HPLC grade and were obtained from Fisher Scientific or Sigma-Aldrich. Phosphate Buffer saline (1×PBS, pH = 7.4) was prepared by dissolving 8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, 0.24 g KH2PO4 in 800 ml of distilled H2O, and pH was adjusted to 7.4 with HCl. Final volume was fixed to 1 L by adding purified H2O and subsequently sterilized by autoclave.
Instruments
A Schimadzu Prominence HPLC unit with an HPLC Column: Zorbax-300SB, C18, 4.6 × 250 mm, 5 μm, 300A, and (lot #WSB 0752003, Agilent Technologies) was used. For centrifuging filter tubes, a Beckman Coulter GS-6R Centrifuge and for centrifuging regular tubes Fisher Scientific Micro Centrifuge 235 C was used. The used instrument in differential scanning calorimetry (DSC) was a PerkinElmer, Jade DSC Instrument. Dynamic Light Scattering (DLS) assays were carried out in a NICOMP 380 Submicron Particle Sizer (Santa Barbara, CA, USA), and lyophilization was carried out using a Labconco FreeZone® 6 liter Freeze Dry System (Kansas, MO).
Preparation of SSM-VLB Samples
Vinorelbine solubilized in DSPE-PEG (2000) (VLB-SSM) was prepared by co-precipitation and reconstitution as described before (26). Briefly, vinorelbine and poly (ethylene glycol-2000)-grafted distearoylphosphatidylethanolamine (DSPE-PEG (2000)) (SSM) were dissolved in methanol. The solvent was then removed by vacuum rotary evaporation under a stream of argon to form a dry film. This dry film was further dried under vacuum overnight to remove any traces of remaining solvent. The dried film was rehydrated with PBS buffer, pH 7.4 to form micelles. The micellar solution was then flushed with argon, sealed, and equilibrated for 2 h at room temperature. Empty sterically stabilized micelles (empty SSM) were prepared following above procedure without using drug.
Particle Size Determination VLB-SSM
Particle size distribution and mean diameter of the prepared aqueous dispersions of VLB, with increasing VLB in 1 mM SSM, were determined by quasi-elastic light scattering using a NICOMP 380 Submicron Particle Sizer (Santa Barbara, CA, USA) equipped with a 100-mW helium-neon laser at 658 nm and a temperature controlled cell holder as described previously (30). Data were analyzed by volume and intensity-weighted distributions (31). Particle size determination assay was repeated after 3 days for all prepared formulations.
Determination of the Amount of the Drug Associated with SSM Using Centrifuge Process
This trial was done in two ways, first using regular centrifuge tubes and second using centrifugal filter tubes. First, VLB-SSM dispersions with different concentrations of vinorelbine (50, 150, 250, 500, 750, and 1,000 μg/ml) in 1 mM DSPE-PEG 2000 each were prepared using above mentioned method and centrifuged for 10 min at 15,000 RCF (g) in regular centrifuge tubes. Samples from supernatants were collected and the amount of VLB in formulation was measured using HPLC method (see below). As second trial, centrifuge tubes with filters were used (32). Dispersions of VLB (50, 150, 250, 500, 750, and 1,000 μg/ml) in 1 mM SSM (1 ml) were prepared and transferred to centrifuge tubes with filters. The optimum centrifugation time was calculated based on the radius of the centrifuge tube holder. A short period, 2 min at 2,700 RCF (g) was used. This only allowed free VLB dissolved in PBS pass through filter and the VLB associated with micelle stay stable in the supernatant compartment. Filtrates were analyzed for drug content by HPLC as described below.
HPLC Method Optimized for Vinorelbine in Base Form
Applied general parameters were as follows: detection wavelength 267 nm, sample concentration 100 μg/ml, and injection volume 20 μl. A gradient of MeOH in water starting from %20 achieving %100 in 20 min was applied to the system as mobile phase. TFA (%0.05) was added to both solvents (total %0.1 TFA in mobile phase (v/v)). Retention time was 13.8 min. Calibration curves were used to determine the exact amount of VLB in different assays. The curve was linear over the tested concentration range, and there was no interference of the phospholipid with the assay (33,34).
Stability of Vinorelbine Associated with DSPE-PEG After Lyophilization
The amounts of vinorelbine associated with SSM and the size of micelles before and after lyophilization were measured to determine the stability of formulation. In this assay, 1 ml of VLB-SSM formulations with 5 mg VLB and 10 mM DSPE-PEG (optimum drug to lipid ratio, as will be explained later) were prepared. The samples were frozen overnight at −20°C, followed by freezing in liquid nitrogen for 3 min before overnight lyophilization. Amount of vinorelbine incorporated with SSM was measured using above mentioned HPLC method. The stability of micellar size was determined using DLS. The latter assay was repeated after 2, 3, 5, 7, and 15 days storage at room temperature in dark as dry form following rehydration.
Drug Release Studies of VLB-SSM by Dialysis
To perform dialysis, 3 ml of VLB-SSM with 15 mg VLB and 30 mM DSPE-PEG with optimum drug to lipid ratio, as will be explained later, was transferred into dialysis membrane tubing, which was then placed into 300 ml of dialysis medium (PBS buffer, pH 7.4) at 37°C under constant slow stirring and kept in dark throughout the experiment. Two milliliters of dialysis medium were withdrawn at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 20, 22, 24, 28, and 48 h of the experiment. Samples were also taken from the dialysis membrane tubing before and after the experiment. Release studies were conducted for 48 h without any change or replacement of dialysis medium (35,36).
Studying the Association of the Drug with Drug Delivery System by Differential Scanning Calorimetry (DSC)
Ideally, the study of association of VLB with SSM should be done in aqueous system where the drug delivery system is in micellar form. However, the heavy ingredients of the system, including buffer salts, drug delivery system, drug, and most importantly, water itself, cover a very high ratio of the mass of total system, leaving the amount of SSM and VLB relatively low. So it was hard to observe significant and good peaks in aqueous system. To overcome this challenge using the method mentioned above, we prepared ten times higher concentrations of the most stable optimum formulation, (5,000 μg VLB with 10 mM DSPE-PEG in 1 ml PBS) and consequently, we lyophilized the samples to eliminate water effect. Previously, we had shown that freeze-drying did not make any changes in the structure and composition of drug-SSM formulations (27). The DSC thermograms of SSM with and without VLB were measured. Also, the thermogram of pure drug was measured and compared with VLB associated with SSM.
The recorded peaks are obtained by controlled heating and cooling process (10 and 5°/min).
In Vitro Cytotoxicity of VLB-SSM
The cell line used to evaluate the in vitro activity of the formulations was MCF-7. The cell line was maintained in RPMI 1640 medium containing 10% fetal bovine serum and 1.0% antibiotics (penicillin and streptomycin) in a 5% carbon dioxide humidified atmosphere at 37°C.
Solutions of VLB-SSM with optimum drug/lipid molar ratio (0.036) chosen from the solubilization studies were used as the test solutions. Sterile PBS was used to rehydrate co-precipitated VLB-SSM prior to cytotoxicity assays. A 10% dimethyl sulfoxide (DMSO) solution of vinorelbine sterilized by filtration through 200 nm syringe filters also was tested as a positive control. Drug-free SSMs in 1×PBS buffer, pH 7.4, were used as negative controls. Empty SSM was tested at the highest concentration used in the formulations. All the samples were prepared and tested in triplicate. The procedure used to test the in vitro cytotoxic activity of the formulation was as previously described (21). Briefly, samples were prepared as described earlier, and serial dilutions were made to obtain final VLB concentrations ranging from 16 to 8 × 10−4 μg/ml using the respective solvent that is either PBS buffer, 10% DMSO or 1 mM SSM, a thousand times above CMC. One hundred ninety microliters of cell suspension at a density of 7 × 103 cells/well was plated in a 96-well plate. After that, 8 μl/well of the test solutions and 10 μl/well controls were added to the microtiter plates. Before adding VLB-delivery system, drug-free SSM have been applied to well plates in amount of 2 μl to keep the drug delivery system above CMC. The plates were then incubated for 3 days in a 5% CO2 humidified atmosphere at 37°C (22,23,29). MTT assay was applied at the concentration of 1 mg/ml in the volume of 100 μl to each well to determine the cell survival.
Before applying MTT, the medium was aspirated from wells and all wells were washed by PBS for three times (37,38). The readings obtained for the solvent controls were used to define 100% growth after correcting for the value obtained for the zero day control. These values were then expressed as % survival and IC50 values calculated using nonlinear regression analysis (percent survival vs. concentration) (21).
RESULTS
Particle Size Analysis of VLB-SSM
Aqueous dispersions of DSPE-PEG (2000) containing VLB, for all drug concentrations studied, indicated the presence of only one population by quasi-elastic light scattering analysis (χ2 values >1,000 for Gaussian analysis). Previously, it was demonstrated in our and other laboratories that DSPE-PEG (2000) above 0.8–1 μM concentration forms micelles with an average diameter of about 15 nm (26,39,40). Our previous studies have shown that different drugs incorporate in SSM at different quantities (41,42), and co-precipitation of excess amounts of drug caused the formation of sterically stabilized crystals (SSCs), made by coating of the surface of the hydrophobic drug particles by DSPE-PEG 2000 as monolayers (21). The latter particles could be identified as a second population in DLS particle size graphs, with diameters mostly above 100 nm. However, by adding excess amount of VLB (even up to 3,000 μg) no other population of particles was observed in addition to that reported for SSM. However, a slight reduction in the size of the micelles was observed (Table I). This size decrease was reversed after three days, and the size of VLB-SSM became ∼14 nm for all drug concentrations. The size distribution of 500 μg VLB in 1 mM SSM formulation is shown in Fig. 2. The size distribution of all other formulations was very similar to Fig. 2 with no extra peaks. Also, there was no significant difference in the distribution of particles at the first and third days.
Table I.
The Reduction in Micelle (1 mM SSM/1 ml PBS) Size with Adding VLB (μg/ml) to System
| VLB conc. | Particle size (nm) (n = 3) day 1 | Particle size (nm) (n = 3) day 3 |
|---|---|---|
| 0 | 15 ± 0.027 | 15 ± 0.036 |
| 50 | 14 ± 0.044 | 14 ± 0.130 |
| 100 | 14 ± 0.054 | 14 ± 0.054 |
| 200 | 14 ± 0.044 | 14 ± 0.230 |
| 350 | 14 ± 0.054 | 14 ± 0.240 |
| 500 | 14 ± 0.07 | 14 ± 0.207 |
| 1,000 | 13 ± 0.324 | 14 ± 0.164 |
| 2,000 | 12 ± 0.044 | 14 ± 0.304 |
| 3,000 | 11 ± 0.054 | 14 ± 0.248 |
Fig. 2.
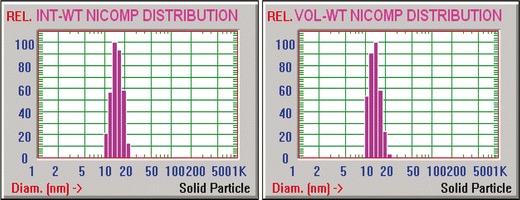
NICOMP size distribution analysis of 500µg VLB in 1 mM SSM, (INT-WT (left) VOL-WT (right))
Determination of the Amount of the Drug Associated with SSM Using Centrifuge Process
Using regular centrifuge tubes, there was no significant difference in the concentration of VLB at the upper and bottom parts of centrifuge tubes. This result confirmed the size analysis that VLB either did not make any aggregates detectable by light scattering or big enough to precipitate by centrifugation.
To separate the free VLB from SSM-associated VLB, we used centrifugation tubes with filters. The results obtained by centrifugal filter tubes are shown in Fig. 3.
Fig. 3.
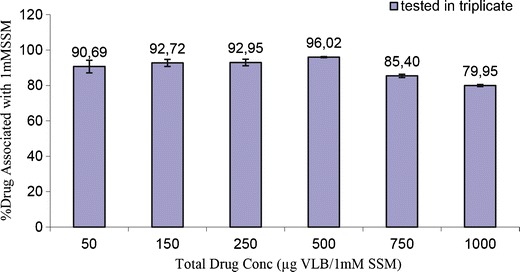
% VLB associated with 1mM DSPE-PEG followed by centrifuge assay
The percentage of drug associated with SSMs increased slightly, as the drug concentration increased up to 500 μg. Above this concentration the drug association with the micelle decreased, making the 500 μg VLB and 1 mM DSPE-PEG in 1 ml PBS buffer, the optimum composition.
Stability of Vinorelbine Associated with DSPE-PEG After Lyophilization
When proposing a new lipid-based formulation of vinorelbine to the pharmaceutical industry, it is important to consider its stability during storage. We suggest to lyophilize the formulation before storing and to rehydrate it before using at bedside. Lyophilization of SSM incorporated with VLB resulted in obtaining a very fluffy material at the bottom of container. This fluffy material was kept under room temperature in dark for 2, 3, 5, 7, and 15 days and rehydrated. The percentage difference of particle size and/or decrease in the amount of drug incorporated with SSM was measured before and after lyophilization, and there was no significant difference (Fig. 4). Therefore, our polymeric micellar nano-VLB formulation is stable enough to be stored in freeze-dried form and rehydrated prior to use.
Fig. 4.

VLB (5 mg) association with DSPE-PEG (10mM) before and after lyophilization
Drug Release Studies on SSM-VLB by Dialysis
As it could be seen in Fig. 5, the rate of drug release from VLB-SSM formulation was slow enough to keep drug stable for long hours, indicating that VLB-SSM may reach the tumors with high drug load. The amount of drug associated with SSM before and after dialysis assay was measured by sampling the formulation from inside dialysis tubing. The measured concentrations were in complete agreement with those shown on release spectrum, Fig. 5. The SSMs were stable in dialysis membrane, and there was no change in size of the micelles at the sample obtained from inside of the membrane at the end point of the release study.
Fig. 5.

Release of VLB from SSM by dialysis
Characterization of VLB-SSM Formulation Using Differential Scanning Calorimetry (DSC)
Differential scanning calorimetry (DSC) is a thermodynamic technique suitable for studying phase transitions of phospholipid bilayers with and without inserted drug molecules. It has been extensively used to investigate the thermal changes caused by the incorporation of the drugs into the phospholipid bilayers (6,43–47). We used this method to show the incorporation of VLB within DSPE-PEG 2000 micelles.
PEG 2000 showed a melting temperature at 56°C (data not shown). This is due to the stacks of folded PEG chains which also are called lamella (46). However, this melting temperature decreased to 52°C when PEG 2000 was conjugated to lipid DSPE and analyzed as unprocessed powder form (Fig. 6a). The peak at 48°C was related to DSPE. Probably, the DSPE part was also making some stacks possibly not as organized as PEG but ordered enough to give a melting thermogram.
Fig. 6.
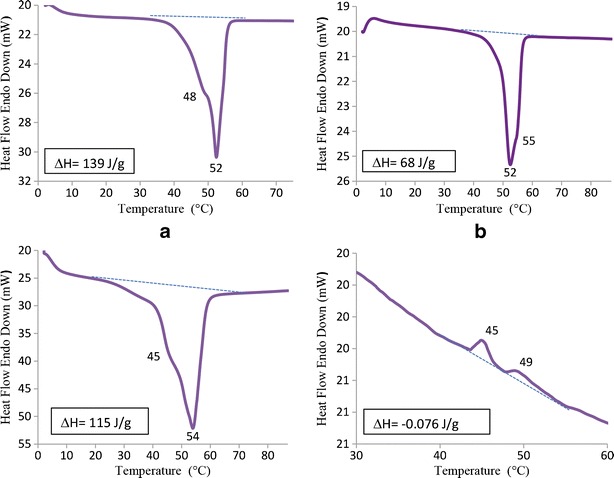
Characterization of VLB-SSM Formulation Using (DSC), (a): DSC thermogram of solid DSPE-PEG 2000 (commercially obtained powder), (b): DSC Thermogram of solid DSPE-PEG 2000 after making SSM, (c): Thermogram of 500µg VLB associated with SSM after lyophilization, (d): The thermogram of solid Vinorelbine
After preparing micelles of DSPE-PEG 2000 in aqueous media by following the previously described procedure and freeze drying, a very interesting result showed up in the thermogram of DSPE-PEG (Fig. 6b). The melting thermogram of DSPE, after micelle formation and lyophilization appears to be lost. This is probably related to the amorphous shape of the hydrophobic DSPE gained after freeze drying, since amorphous materials do not show melting temperature. Another possibility is the shielding of DSPE melting thermogram by PEG layer. Other interesting result which is obtained from the latter thermogram is the second peak at 55°C. The melting peak at 52°C is the same with DSPE-PEG (shown in Fig. 6a), and we hypothesize that the shoulder at 55°C is related to PEG stacks that found the opportunity of making organized lamellas again after lyophilization around the micelle.
It is noteworthy that the methods used in the preparation of self-assembly systems from amphiphilic materials have a significant effect on the structure of amphiphilic chains at the end of preparation process. For this reason, it is possible to see different melting transitions in reported data (46).
Figure 6c is the thermogram of lyophilized VLB loaded SSM which shows a very significant change both in ΔH (115 J/g for 500 μg VLB-SSM and 68 J/g for the free SSM) and in melting temperature. This clearly indicated the association of VLB with SSM. This interaction defiantly occurred at or close to the core of micelle since there was an obvious change in DSPE thermogram at 48°C. The disappeared thermogram of DSPE part in Fig. 6b did show up very significantly in Fig. 6c at 45°C due to the tight interdigitation of VLB with DSPE part of the micelle. As we mentioned above, PEG layer shows a weak shoulder at 55°C in Fig. 6b. This shoulder changes to a very intense peak in Fig. 6c at 54°C. This indicates that the PEG chains have folded in a more organized manner causing the increase in the intensity of their melting point. This result is further confirmation of reduced size of the SSMs due to folding of PEG chains after incorporation with drug molecules.
As we will discuss further in Fig. 10, incorporation of drug at the interphase of core and corona parts of SSM between DSPE units gives the PEG layer enough space to fold down and make stacked layers.
Fig. 10.

Localization of VLB in between 2 DSPE-PEG unimers and observation of a decrease in size of particle
To ensure that vinorelbine’s DSC spectrum does not interfere with SSM’s thermogram, we also recorded thermal behavior of the free drug. Interestingly, no substantial crystallization or melting peaks were obtained in VLB’s DSC thermogram as shown in Fig. 6d. However, the very small and irregular exothermic peaks (ΔH = −0.076 J/g) shown in this figure can be attributed to the partially overlapping of phenyl groups of the drug resulting from the increasing mobility of the molecules with increasing temperature. We did not see these exothermic peaks in the thermogram of VLB-SSM samples. This is probably due to the protection of the drug by the protective shell of SSM. If this assumption is correct, this evidence again confirms the incorporation of VLB within or close to the core part of the micelle and the stabilization of the drug in SSM. The settlement of VLB in shell should not protect the drug molecules from overlapping and we would see the exothermic peak of the drug in the sample thermograms.
On the other hand, this thermogram showed us that vinorelbine had an amorphous shape. It was not in crystalline form in molecular level or in SSM.
In Vitro Cytotoxicity
As a next step, we investigated whether the interaction between the drug and SSM in the formulation affected the drug’s bioactivity. To determine the cytotoxic activity of VLB in SSM, the formulations were tested against MCF-7, a human breast cancer cell line which has been previously used in studying the anticancer activity of vinorelbine using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (48). Micellar formulation had cytotoxic activities toward cultured MCF-7 cells 6.78 times better than the similar activity of free drug in 10% DMSO. Dose response curve of in vitro activity evaluation is shown in Fig. 7, and the IC50 values are summarized in Fig. 8. We also report that control vehicles SSM, %10 DMSO, and PBS did not cause any significant effects on cell survival (Fig. 9).
Fig. 7.
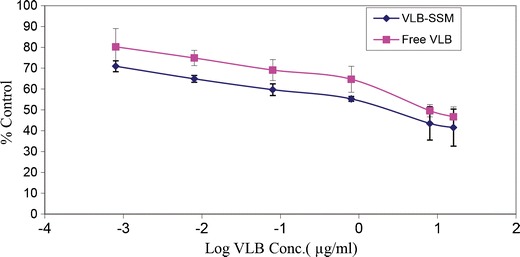
VLB Activity on MCF-7 Cells in 72 h as free drug or in SSM
Fig. 8.
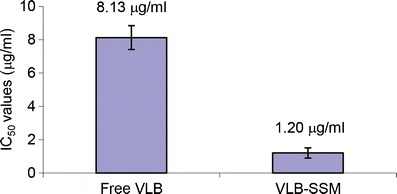
IC50 values of VLB loaded SSM and Free VLB
Fig. 9.

Cytotoxic activity of controls
DISCUSSION
In this study, we report synthesis and characterization of a novel nanomicellar vinorelbine formulation prepared using sterically stabilized phospholipid micelles, which have the perfect size (15 nm) for passive targeting to tumors, and low critical micelle concentration (1 μM) to provide in vivo stability and safety.
In this study, we have observed a different solubilization behavior of VLB in SSM. Uploading VLB in different and high concentrations to the drug delivery system did not cause the formation of any drug aggregates or sterically stabilized crystals composed of excess amount of drug, as we observed previously for other poorly soluble drugs (21,29).
This could be explained by several mechanisms. During the formation of VLB-SSM, the drug associated with SSM up to a certain concentration and the added excess amount of VLB had the ability of making its own soluble self-assemblies which were too small to be seen on size analysis. This may explain the unexpectedly high solubilization of the drug. Similar behavior was observed by others for another poorly soluble molecule, amphotericin B (49). The second possibility is that VLB molecules settle in between the DSPE core and PEG corona interface. Previous studies have shown that vinorelbine causes a change in the phase transition of phospholipid bilayers suggesting partial interdigitation of vinorelbine with mentioned bilayers (6). Vinorelbine is characterized by an extensive hydrophobic area covering both catharanthine and vindoline moieties and hydrophilic groups predominated in the vindoline moiety (hydroxyl, methyl ester, and methoxy groups) (Fig. 1). It has been found that in vinorelbine inserted phospholipid bilayers, the most favored topographical position of vinorelbine is at the interface of polar head groups and alkyl chains of lipid bilayers (6). We measured the octanol/water partition coefficient of vinorelbine as 1.18 ± 0.038 which confirms the amphiphilic behavior of this molecule.
The micelle interface would provide more area to accommodate higher number of molecules than the core of the micelle. In fact, this explanation supports the decrease in size of micelle after large number of drug molecules are loaded in SSM interface, causing PEG to fold more, making the PEG layer thinner (Fig. 10) (50). We have observed similar phenomena when we included water insoluble lipid, phosphatidylcholine, into PEGylated lipid micelles (21). Also, the results obtained from DSC studies are in complete correlation with those obtained from DLS assays. The folding of PEG layer caused by incorporation of VLB with SSM between two DSPE units causes the decrease in the size of the micelle from 15 to 11 nm together with the increase in the thermogram of organized PEG layers at 54°C.
In order to clarify the abovementioned possibilities, we went ahead and determined the amount of VLB associated with SSM using a centrifuge process that can separate free drug from micelle-associated drug.
First, we calculated water solubility of drug as 250μg/ml PBS (data not shown). Then, using filtered centrifugal tubes, we showed that the drug incorporated into the micelle even below its water solubility (Fig. 3). For example when we mixed 50μg VLB with 1 mM DSPE-PEG, only 4.6μg of the drug was dissolved in PBS, and the rest was incorporated with the SSM. So in contrast with our previous reports, PBS never became saturated with the drug. This indicated that drug micelle association was more favorable than drug self-association. The percentages of SSM-associated drug increased until 500μg VLB. After this concentration which probably is the highest point of association of SSM with the drug, excess amount of drug continued to dissolve or form its own small assemblies in buffer. It is interesting to note that the percentage of incorporated drug within SSM decreased after 500μg loading. This may be is due to reversal of some type of super saturation in micelle above 500μg.
It is noteworthy that using co-precipitation and reconstitution method, we provide more drug monomers available to incorporate into micelles. This is because, by direct addition of the drug to the preformed micelles, we are limited to the solubility of the drug in PBS (250μg/ml), which can only provide the incorporation of the drug in concentration ∼200μg/ml with 1 mM SSM.
Collectively, in this study, the results obtained to characterize VLB-SSM by DLS, centrifuge and DSC studies, show that the reason for the high solubility VLB in the micelles, with no additional drug aggregate formation, is most probably because of the incorporation of drug at the interphase of core and corona parts of SSM between DSPE units. In this way, PEG units find enough space to fold down and this causes a slight decrease in particle size (Fig. 10). This property also provides the formulation a slow drug release characteristics.
Based on these results, the optimum molar ratio of VLB to DSPE-PEG 2000 for VLB-SSM preparation was found to be 0.036 which corresponded to the formulation with 500μg VLB in 1 mM SSM.
The VLB-SSM formulation shows≅7 times enhanced cytotoxic activity compared to free VLB at 72 h. Although the formulation shows a slow cumulative release (25% in 48 h) it is more effective than free VLB on MCF-7 breast cancer cell lines because the formulation shows the cytotoxic effect by internalizing the cells and releasing the drug inside of the cell not only by releasing the drug to the extracellular environment. Previously, we have shown the internalization of SSM to MCF-7 cell lines by incorporating quantum dots with micelles made of DSPE-PEG 2000 (51). More recently, we showed similar situation with another anticancer drug, paclitaxel (23).
CONCLUSION
A novel lipid-based formulation of anticancer agent, vinorelbine, was developed using phospholipid micelles, which had perfect size for passive targeting and were known to be relatively safe (21,29). Physical chemical characterization of the VLB-SSM formulations indicated that the optimum drug lipid ratio was 0.036. Optimized VLB-SSM formulation showed 6.7 times higher cytotoxic activity against MCF-7 breast cancer cells, compared to free drug. Results from this study strongly suggest that the optimized VLB-SSM formulation is a promising targeted nanomedicine to treat breast and lung cancers in the future.
ACKNOWLEDGMENTS
This study was supported, in part, by NIH grant CA121797. Most part of this investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number CO6RR15482 from the National Center for Research Resources, NIH. Also, the authors would like to thank Dr. Antonina Kuzmis for her very precious advices and assistance during the experimentations.
REFERENCES
- 1.Krikorian A, Breillout F. Vinorelbine (Navelbine ® 1). A new semisynthetic Vinca alkaloid. Onkologie. 1991;4:7–12. doi: 10.1159/000216938. [DOI] [PubMed] [Google Scholar]
- 2.Zhou XJ, Placidi M, Rahmani R. Uptake and metabolism of Vinca alkaloids by freshly isolated human hepatocytes in suspension. Anticancer Res. 1994;14(3A):1017–22. [PubMed] [Google Scholar]
- 3.Etievant C, Barret JM, Kruczynski A, Perrin D, Hill BT. Vinflunine (20′,20′-difluoro-3′,4′-dihydrovinorelbine), a novel Vinca alkaloid, which participates in P-glycoprotein (Pgp)-mediated multidrug resistance in vivo and in vitro. Investig New Drugs. 1998;16(1):3–17. doi: 10.1023/A:1006022811895. [DOI] [PubMed] [Google Scholar]
- 4.Duflos A, Jacquesy JC, Kruczynski A, Etievant C, Barret JM, Hill BT, et al. Extending the scope of Vinca alkaloids with superacid chemistry. Clin Cancer Res. 1999;5:3794S-S. [Google Scholar]
- 5.Johnson SA, Harper P, Hortobagyi GN, Pouillart P. Vinorelbine: an overview. Cancer Treat Rev. 1996;22(2):127–42. doi: 10.1016/S0305-7372(96)90032-8. [DOI] [PubMed] [Google Scholar]
- 6.Koukoulitsa C, Kyrikou I, Demetzos C, Mavromoustakos T. The role of the anticancer drug vinorelbine in lipid bilayers using differential scanning calorimetry and molecular modeling. Chem Phys Lipids. 2006;144(1):85–95. doi: 10.1016/j.chemphyslip.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Cragg GM, Newman DJ. Plants as a source of anti-cancer agents. J Ethnopharmacol. 2005;100(1–2):72–9. doi: 10.1016/j.jep.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 8.Semple SC, Leone R, Wang J, Leng EC, Klimuk SK, Eisenhardt ML, et al. Optimization and characterization of a sphingomyelin/cholesterol liposome formulation of vinorelbine with promising antitumor activity. J Pharm Sci. 2005;94(5):1024–38. doi: 10.1002/jps.20332. [DOI] [PubMed] [Google Scholar]
- 9.Toso C, Lindley C. Vinorelbine: a novel Vinca alkaloid. Am J Health Syst Pharm AJHP Off J Am Soc Health Syst Pharm. 1995;52(12):1287–304. doi: 10.1093/ajhp/52.12.1287. [DOI] [PubMed] [Google Scholar]
- 10.Yoh K, Niho S, Goto K, Ohmatsu H, Kubota K, Kakinuma R, et al. Randomized trial of drip infusion versus bolus injection of vinorelbine for the control of local venous toxicity. Lung Cancer. 2007;55(3):337–41. doi: 10.1016/j.lungcan.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Li C, Cui J, Wang C, Zhang L, Xiu X, Li Y, et al. Encapsulation of vinorelbine into cholesterol-polyethylene glycol coated vesicles: drug loading and pharmacokinetic studies. J Pharm Pharmacol. 2011;63(3):376–84. doi: 10.1111/j.2042-7158.2010.01227.x. [DOI] [PubMed] [Google Scholar]
- 12.Drummond DC, Noble CO, Guo Z, Hayes ME, Park JW, Ou CJ, et al. Improved pharmacokinetics and efficacy of a highly stable nanoliposomal vinorelbine. J Pharmacol Exp Ther. 2009;328(1):321–30. doi: 10.1124/jpet.108.141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–92. [PubMed] [Google Scholar]
- 14.Okouneva T, Hill BT, Wilson L, Jordan MA. The effects of vinflunine, vinorelbine, and vinblastine on centromere dynamics. Mol Cancer Ther. 2003;2(5):427–36. [PubMed] [Google Scholar]
- 15.Jordan A, Hadfield JA, Lawrence NJ, McGown AT. Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Med Res Rev. 1998;18(4):259–96. doi: 10.1002/(SICI)1098-1128(199807)18:4<259::AID-MED3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 16.Georgiadis MS, Russell EK, Gazdar AF, Johnson BE. Paclitaxel cytotoxicity against human lung cancer cell lines increases with prolonged exposure durations. Clin Cancer Res. 1997;3(3):449–54. [PubMed] [Google Scholar]
- 17.Horton JK, Houghton PJ, Houghton JA. Relationships between tumor responsiveness, vincristine pharmacokinetics and arrest of mitosis in human-tumor xenografts. Biochem Pharmacol. 1988;37(20):3995–4000. doi: 10.1016/0006-2952(88)90085-8. [DOI] [PubMed] [Google Scholar]
- 18.Burris HA, Hanauske AR, Johnson RK, Marshall MH, Kuhn JG, Hilsenbeck SG, et al. Activity of topotecan, a new topoisomerase-I inhibitor, against human tumor colony-forming-units in vitro. J Natl Cancer Inst. 1992;84(23):1816–20. doi: 10.1093/jnci/84.23.1816. [DOI] [PubMed] [Google Scholar]
- 19.Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R, et al. Phase I clinical and pharmacokinetic study of PK1 N-(2-hydroxypropyl) methacrylamide copolymer doxorubicin: first member of a new class of chemotherapeutic agents—drug-polymer conjugates. Clin Cancer Res. 1999;5(1):83–94. [PubMed] [Google Scholar]
- 20.Drummond DC, Meyer O, Hong KL, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51(4):691–743. [PubMed] [Google Scholar]
- 21.Krishnadas A, Rubinstein I, Onyuksel H. Sterically stabilized phospholipid mixed micelles: in vitro evaluation as a novel carrier for water-insoluble drugs. Pharm Res. 2003;20(2):297–302. doi: 10.1023/A:1022243709003. [DOI] [PubMed] [Google Scholar]
- 22.Onyuksel H, Jeon E, Rubinstein I. Nanomicellar paclitaxel increases cytotoxicity of multidrug resistant breast cancer cells. Cancer Lett. 2009;274(2):327–30. doi: 10.1016/j.canlet.2008.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dagar A, Kuzmis A, Rubinstein I, Sekosan M, Onyuksel H. VIP-targeted cytotoxic nanomedicine for breast cancer. Drug Deliv Transl Res. 2012;2(6):454–62. doi: 10.1007/s13346-012-0107-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dagar S, Krishnadas A, Rubinstein I, Blend MJ, Onyuksel H. VIP grafted sterically stabilized liposomes for targeted imaging of breast cancer: in vivo studies. J Control Release. 2003;91(1–2):123–33. doi: 10.1016/S0168-3659(03)00242-6. [DOI] [PubMed] [Google Scholar]
- 25.Sethi V, Rubinstein I, Kuzmis A, Kastrissios H, Artwohl J, Onyuksel H. Novel, biocompatible, and disease modifying VIP nanomedicine for rheumatoid arthritis. Mol Pharm. 2013;10(2):728–38. doi: 10.1021/mp300539f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashok B, Arleth L, Hjelm RP, Rubinstein I, Onyuksel H. In vitro characterization of PEGylated phospholipid micelles for improved drug solubilization: effects of PEG chain length and PC incorporation. J Pharm Sci. 2004;93(10):2476–87. doi: 10.1002/jps.20150. [DOI] [PubMed] [Google Scholar]
- 27.Lim SB, Rubinstein I, Onyuksel H. Freeze drying of peptide drugs self-associated with long-circulating, biocompatible and biodegradable sterically stabilized phospholipid nanomicelles. Int J Pharm. 2008;356(1–2):345–50. doi: 10.1016/j.ijpharm.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, Kamoun WS, et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7(6):383–8. doi: 10.1038/nnano.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onyuksel H, Mohanty PS, Rubinstein I. VIP-grafted sterically stabilized phospholipid nanomicellar 17-allylamino-17-demethoxy geldanamycin: a novel targeted nanomedicine for breast cancer. Int J Pharm. 2009;365(1–2):157–61. doi: 10.1016/j.ijpharm.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vukovic L, Khatib FA, Drake SP, Madriaga A, Brandenburg KS, Kral P, et al. Structure and dynamics of highly PEG-ylated sterically stabilized micelles in aqueous media. J Am Chem Soc. 2011;133(34):13481–8. doi: 10.1021/ja204043b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banerjee A, Onyuksel H. Human pancreatic polypeptide in a phospholipid-based micellar formulation. Pharm Res. 2012;29(6):1698–711. doi: 10.1007/s11095-012-0718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markovsky E, Koroukhov N, Golomb G. Additive-free albumin nanoparticles of alendronate for attenuating inflammation through monocyte inhibition. Nanomedicine. 2007;2(4):545–53. doi: 10.2217/17435889.2.4.545. [DOI] [PubMed] [Google Scholar]
- 33.Debal V, Morjani H, Millot JM, Angiboust JF, Gourdier B, Manfait M. Determination of vinorelbine (Navelbine) in tumor-cells by high-performance liquid-chromatography. J Chromatogr Biomed Appl. 1992;581(1):93–9. doi: 10.1016/0378-4347(92)80451-U. [DOI] [PubMed] [Google Scholar]
- 34.Jehl F, Debs J, Herlin C, Quoix E, Gallion C, Monteil H. Determination of navelbine and desacetylnavelbine in biological fluids by high-performance liquid chromatography. J Chromatogr. 1990;525(1):225–33. doi: 10.1016/S0378-4347(00)83397-6. [DOI] [PubMed] [Google Scholar]
- 35.Cho YW, Lee J, Lee SC, Huh KM, Park K. Hydrotropic agents for study of in vitro paclitaxel release from polymeric micelles. J Control Release. 2004;97(2):249–57. doi: 10.1016/j.jconrel.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 36.Michalowski CB, Guterres SS, Dalla Costa T. Microdialysis for evaluating the entrapment and release of a lipophilic drug from nanoparticles. J Pharm Biomed Anal. 2004;35(5):1093–100. doi: 10.1016/j.jpba.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 37.Allahverdiyev A, Duran N, Ozguven M, Koltas S. Antiviral activity of the volatile oils of Melissa officinalis L. against Herpes simplex virus type-2. Phytomedicine. 2004;11(7–8):657–61. doi: 10.1016/j.phymed.2003.07.014. [DOI] [PubMed] [Google Scholar]
- 38.Davydov M, Volkov S, Polotsky B, Gerasimov S, Machaladze Z, Allahverdiyev A, et al. Mediastinal lymphadenectomy improves survival in surgically treated patients with non-small cell lung cancer. Int J Cancer. 2002;414–5.
- 39.Sugin Z, Yuksel N, Baykara T. Preparation and characterization of polymeric micelles for solubilization of poorly soluble anticancer drugs. Eur J Pharm Biopharm. 2006;64(3):261–8. doi: 10.1016/j.ejpb.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 40.Torchilin VP. Structure and design of polymeric surfactant-based drug delivery systems. J Control Release. 2001;73(2–3):137–72. doi: 10.1016/S0168-3659(01)00299-1. [DOI] [PubMed] [Google Scholar]
- 41.Cesur H, Rubinstein I, Pai A, Onyuksel H. Self-associated indisulam in phospholipid-based nanomicelles: a potential nanomedicine for cancer. Nanomedicine. 2009;5(2):178–83. doi: 10.1016/j.nano.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koo OM, Rubinstein I, Onyuksel H. Actively targeted low-dose camptothecin as a safe, long-acting, disease-modifying nanomedicine for rheumatoid arthritis. Pharm Res. 2011;28(4):776–87. doi: 10.1007/s11095-010-0330-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lv Q, Yu A, Xi Y, Li H, Song Z, Cui J, et al. Development and evaluation of penciclovir-loaded solid lipid nanoparticles for topical delivery. Int J Pharm. 2009;372(1–2):191–8. doi: 10.1016/j.ijpharm.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 44.Sullivan CO, Birkinshaw C. In vitro degradation of insulin-loaded poly (n-butylcyanoacrylate) nanoparticles. Biomaterials. 2004;25(18):4375–82. doi: 10.1016/j.biomaterials.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 45.Holdgate GA, Ward WHJ. Measurements of binding thermodynamics in drug discovery. Drug Discov Today. 2005;10(22):1543–50. doi: 10.1016/S1359-6446(05)03610-X. [DOI] [PubMed] [Google Scholar]
- 46.Kastantin M, Ananthanarayanan B, Karmali P, Ruoslahti E, Tirrell M. Effect of the lipid chain melting transition on the stability of DSPE-PEG (2000) micelles. Langmuir. 2009;25(13):7279–86. doi: 10.1021/la900310k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maswadeh H, Demetzos C, Daliani I, Kyrikou I, Mavromoustakos T, Tsortos A, et al. A molecular basis explanation of the dynamic and thermal effects of vinblastine sulfate upon dipalmitoylphosphatidylcholine bilayer membranes. Biochim Biophys Acta-Biomembr. 2002;1567(1–2):49–55. doi: 10.1016/S0005-2736(02)00564-3. [DOI] [PubMed] [Google Scholar]
- 48.Liu XM, Wang LG, Kreis W, Budman DR, Adams LM. Differential effect of vinorelbine versus paclitaxel on ERK2 kinase activity during apoptosis in MCF-7 cells. Br J Cancer. 2001;85:1403–11. doi: 10.1054/bjoc.2001.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor RL, Williams DM, Craven PC, Graybill JR, Drutz DJ, Magee WE. Amphotericin-B in liposomes—a novel therapy for histoplasmosis. Am Rev Respir Dis. 1982;125(5):610–1. doi: 10.1164/arrd.1982.125.5.610. [DOI] [PubMed] [Google Scholar]
- 50.Rex S, Zuckermann MJ, Lafleur M, Silvius JR. Experimental and Monte Carlo simulation studies of the thermodynamics of polyethyleneglycol chains grafted to lipid bilayers. Biophys J. 1998;75(6):2900–14. doi: 10.1016/S0006-3495(98)77732-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rubinstein I, Soos I, Onyuksel H. Intracellular delivery of VIP-grafted sterically stabilized phospholipid mixed nanomicelles in human breast cancer cells. Chem Biol Interact. 2008;171(2):190–4. doi: 10.1016/j.cbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]