

Abstract
Coronaviruses (CoV) have recently emerged as potentially serious pathogens that can cause significant human morbidity and death. The severe acute respiratory syndrome (SARS)-CoV was identified as the etiologic agent of the 2002-3 international SARS outbreak. Yet how SARS evades innate immune responses to cause human disease remains poorly understood. Here, we show that a protein encoded by SARS-CoV designated as open reading frame-9b (ORF-9b) localizes to mitochondria and causes mitochondrial elongation by triggering ubiquitination and proteasomal degradation of dynamin-like protein (DRP1), a host protein involved in mitochondrial fission. Also, acting on mitochondria ORF-9b targets the mitochondrial-associated adaptor molecule MAVS signalosome by usurping poly(C)-binding protein 2 (PCBP2) and the HECT domain E3 ligase AIP4 to trigger the degradation of MAVS, TRAF3, and TRAF6. This severely limits host cell interferon responses. Reducing either PCBP2 or AIP4 expression substantially reversed the ORF-9b mediated reduction of MAVS and the suppression of anti-viral transcriptional responses. Finally, transient ORF-9b expression led to a strong induction of autophagy in cells. The induction of autophagy depended upon ATG5, a critical autophagy regulator, but the inhibition of MAVS signaling did not. These results indicate that SARS-CoV ORF-9b manipulates host cell mitochondria and mitochondrial function to help evade host innate immunity. This study has uncovered an important clue to the pathogenesis of SARS-CoV infection and illustrates the havoc that a small open reading frame can cause in cells.
Keywords: mitochondria, innate immunity, SARS, coronavirus, interferon, ubiquitin
Introduction
Severe acute respiratory syndrome (SARS) is caused by a novel coronavirus (SARS-CoV) (1-3). An international outbreak of SARS began in November 2002 and ended in July 2003. It afflicted more than 8,000 people and left 774 patients dead (4, 5). Prior to the 2003 SARS outbreak coronaviruses had not been recognized as a significant pathogen. However, the subsequent documentation of a large pool of coronaviruses that circulate in bats and other animals has portended the possible emergence of other highly pathogenic coronaviruses as major threats to human health (6, 7). And recently, a SARS-like virus designated HCoV-EMC has been detected in patients, whose clinical courses have resembled that of SARS patients although with an even higher mortality rate (8, 9).
Shortly after the initial SARS outbreak the genome sequence and organization of SARS-CoV were reported (10, 11). It is a large single positive-strand RNA virus. The virus genome encodes eight accessory proteins designated ORF-3a, 3b, 6, 7a, 7b, 8a, 8b and 9b. Several have identified functions. ORF-3a triggers cellular apoptosis, ORF-7a activates nuclear factor-κB (NF-κB), ORF3b upregulates the expression of several cytokines and chemokines, ORF-6 reduces interferon production; ORF-8a triggers cellular apoptosis, and ORF-8b induces cellular DNA synthesis (12). However, the function of ORF-9b is largely unknown. Synthesized from an alternative complete reading frame within the viral N gene, the 98 amino acid protein has no known homolog. A resolved crystal structure revealed a dimeric tent-like β structure with an amphipathic surface and a central hydrophobic cavity that likely mediates membrane attachment (13). The protein has been detected in tissues from infected patients and in in vitro infected cells (14, 15). Two previous studies have addressed the localization of ORF-9b in Vero cells, a cell line derived from African green monkey kidney epithelial cells (16, 17). In the first study it was reported to be exported from the nucleus and to co-localize with an endoplasmic reticulum marker (16) and in a more recent study to shuttle between the nucleus and the cytosol via an interaction with Crm1 (17).
SARS-CoV infected cells have an impaired interferon response suggesting that the virus disrupts the normal host cell interferon response (18). Yet interferon therapy has also been suggested to be efficacious for SARS patients (18, 19). The activation of the type 1 interferon pathway is crucial for the control of many viral infections. Following viral invasion, host cells detect the presence of viral RNA by endosomally localized toll-like receptors (TLR) and cytosolic sensors of the RIG-I-like receptor (RLR) pathway, retinoic-inducible gene-I (RIG-I) and melanoma differentiation associated gene 5 (MDA5) (20). RIG-I and MDA5 consist of two N-terminal caspase activation and recruitment domains (CARDs), a central DEAD box helicase/ATPase domain, and a C-terminal regulatory domain. Although RIG-I and MDA5 differ in the types of viral RNA that they recognize they share a common signaling pathway, which utilizes the adaptor protein, mitochondrial antiviral signaling protein MAVS (also known as ISP-1/VISA/Cardiff). The N-terminal MAVS CARD domain mediates the interaction with RIG-I and MDA5 as well with downstream effectors, an adjacent proline-rich region also recruits downstream signaling molecules, and the C-terminal transmembrane domain anchors the protein to the mitochondrial membrane. MAVS recruits the E3 ligases TRAF3 and TRAF6 facilitating the activation of interferon regulatory factors (IRFs), nuclear factor κB (NF-κB), and the induction of a host antiviral state.
However, RNA viruses have evolved strategies to antagonize the type I interferon signaling pathways. MAVS and MAVS signaling are common targets. For example, the human metapneumovirus M2-2 protein inhibits cellular immunity by inhibiting MAVS signaling. The M2-2 protein interacted with MAVS; however, the specific mechanisms by which M2-2 functioned was not discerned (21). Enterovirus 71 employs a protease 2Apro to cleave and release MAVS from mitochondria, thereby deactivating the pathway and promoting viral immune evasion (22). Hepatitis C utilizes a similar mechanism as the viral protease NS3/4A also cleaves MAVS efficiently degrading mitochondria-associated membrane (MAM)-associated MAVS (23, 24). The MAM is a specialized membrane subcompartment that links mitochondria to the endoplasmic reticulum (ER)-peroxisome network. The Hepatitis B X protein also causes the degradation of MAVS by triggering its ubiquitination on Lys136 (25). Finally, Respiratory syncytial virus (RSV) uses two structural proteins, NS1 and NS2, to assemble a degradasome that also preferentially targets MAVS signaling (26). How SARS-CoV subverts the host interferon response is poorly understood.
In this study we investigate the functional role of ORF-9b in cells and find that ORF-9b alters host cell mitochondria morphology and disrupts mitochondria anti-viral signaling. ORF-9b localizes to the outer mitochondria membrane. Likely as a consequence mitochondria elongate and mitochondrial anti-viral signaling is disturbed. Furthermore, either as a direct or indirect consequence autophagosome formation in acutely ORF-9b expressing cells is enhanced.
Material and Methods
Reagents and antibodies
Hoechst 33342 (Invitrogen) was used at a concentration of 100 ng/ml. MG-132 (Cayman Chemical) was used at 10 μM and 3 Methyladenine (Sigma-Aldrich) at 5 nM. Primary antibodies used for immunofluorescence experiments were the following: anti-MAVS (Cell Signaling, 1:500), anti-Flag (Sigma, 1:500), anti-Tomm20 (Santa Cruz, 1:1,000), and anti-DRP1 (BD Biosciences, 1:200). The secondary antibodies used for immunofluorescence were the following: polyclonal anti-mouse Ig Alexa488 or Alexa568 conjugated (Invitrogen, 1:1,000), polyclonal anti-rabbit Ig Alexa488, Alexa568, or Alexa647 conjugated (Invitrogen, 1:1,000), polyclonal goat anti-rabbit Ig ATTO647N (Active Motif, 1:1,000), and polyclonal goat anti-mouse IgG MegaRed 520 (Sigma, 1:100). Primary antibodies used for immunoblotting were the following: rabbit anti-MAVS, ATG5 and DRP1, mouse anti-GFP, and IFN-α (Cell Signaling), mouse anti-PCPB2 and anti-AIP4 (Santa Cruz Biotechnology), monoclonal anti-Myc (Clontech Laboratories), mouse anti-Flag and rabbit anti-LC3 (Sigma-Aldrich), rabbit anti-K48-linked polyubiquitin chains (Millipore), mouse monoclonal anti-IFN-β (Biolegend), and mouse anti-actin conjugated to horseradish peroxidase (HRP, Sigma-Aldrich). For immunoprecipitations monoclonal mouse anti-Myc, anti-PCPB2 or anti-GFP and Protein G PLUS–Agarose (Santa Cruz Biotechnology) were used.
Cells and Plasmids
A549 and HEK 293 cells were obtained from the American Type Culture Collection (ATCC). A549 and HEK 293 cells were maintained in Dulbecco's Modified Eagle Medium supplemented with 10% FBS (Invitrogen). The A549 Tet-inducible 9b-GFP cells were made by transfecting A549 cells with pTet-on (Clontech) followed by G418 selection for 4 weeks. Single clones were picked, expanded, and transfected with pTRE2 pur-9b-GFP (Clontech vector). These cells were selected with G418 and puromycin for 4 weeks. After which the cells lines were treated with tetracycline, GFP positive cells were sorted and allowed to re-expand in the absence of tetracycline. Low frequency constitutively expressing 9b-GFP cells were also sorted and expanded. THP-1 cells expressing GFP or 9b-GFP were made transfecting the cells with the appropriate construct and selecting cells for 4 weeks with G418. The top 5% of GFP positive cells were sorted after 4 weeks and 6 weeks of selection. The cell lines were maintained in G418. Atg5 and control siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The LC3-GFP plasmid was a gift from Dr. Noboru Mizushima (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan), which GFP was replaced by RFP as the autophagosome probe. Myc-MAVS full length and its truncated constructs and full length RIG-I and RIG-I1-250 were gifts from Dr. Takumi Kushiba (Kyushu University, Fukuoka, Japan). Myc-MAVS (503-540) and Flag-Traf3 were gift from Dr. Hiscott J (Lady Davis Institute, Quebec, Canada). Mito-Cherry Red was a gift from Dr. Chen Q (Chinese Academy of Sciences, Beijing, China). The Interferon-β luciferase reporter was a gift from Dr. Saori Suzuki (University of Tokyo, Tokyo, Japan). The NF-κB-luciferase reporter was kindly provided by Dr. Ulrich Siebenlist (National Institutes of Health). SARS-CoV accessory ORF-9b DNA was generated by PCR from K14 and J17 cDNA clones, which were produced from the SARS-CoV genome Tor2 isolate (Michael Smith Genome Sciences Centre, Vancouver, Canada). The PCR primers were corresponding to SARS-CoV accessory gene sequences in accession number NC_004718 (http://www.ncbi.nlm.nih.gov/gene/). Recombinant GFP-tagged SARS-CoV ORF-9b was generated using pEGFP-N1 vector (BD Bioscience). The ORF-9b constructs were verified by DNA sequencing. The plasmids were transiently transfected into the cells using Lipofectamine 2000 (Invitrogen) following the manufacture's protocol.
Luciferase assay
HEK 293 cells (2 × 105 cells per well) were seeded in 24-well plates. The following day the cells were co-transfected with 25 ng of a luciferase reporter plasmid, 2.5 ng of the Renilla luciferase internal control vector phRL-TK (Promega), and each of the indicated plasmids with Lipofectamine 2000 (Invitrogen). Empty vector pcDNA3 was used to maintain equivalent amounts of DNA in each well. The transfection of Poly(I:C) was performed 6 h before cell harvest. The reporter gene assay was performed 24 h after transfection and analyzed by a dual-luciferase reporter assay on the Mithras LB 840 Multimode Microplate Reader (Berthold). Each experiment was replicated a minimum of three times.
RNA-mediated interference
The siRNA pools targeting human ATG5 (sc-41445) or control siRNA (sc-37007) with scrambled sequence were purchased (Santa Cruz Biotechnology). The shRNAs against human PCPB2 and AID4 were designed on published targeting sequences (27). The synthetic DNA oligomers targeting PCPB2, AID4 or scrambled sequence control were cloned into the pSIREN-RetroQ vector (Clontech). The DRP1 shRNA was a kind gift of Dr. Shigehisa Hirose (Tokyo Institute of Technology, Japan). For the knock-downs in A549 cells, the cell seeded for 4 h were transfected with siRNAs targeting ATG5 or control siRNA 30nM once, and on the following day, 10 nM siRNA was transfected again 8 h prior to harvesting the cells. The shRNAs were transfected into HEK 293 cells overnight, and then the cells were lysed for analysis. The transfections were performed using Lipofectamine 2000 (Invitrogen).
GFP-LC3 assay
These assays were done as described previously (28). A minimum of 50–100 GFP+ cells per sample were surveyed and the number of GFP-LC3+ dots/cell counted. Cells were considered positive if they had more than three large GFP-LC3+ dots. Data are presented as a percentage of the total number of GFP+ cells visualized.
Immunoblot analysis and immunoprecipitation
For standard immunoblotting the cells were lysed in a buffer of 20 mM HEPES, pH 7.4, 50 mM β-glycerophosphate, 1 mM Na3VO4, 0.5% (vol/vol) Triton X-100, 0.5% (vol/vol) CHAPS (3-[(3-cholamidopropyl)-dimethylammonio]-1-propane sulfonate hydrate), and 10% (vol/vol) glycerol with a protease inhibitor ‘cocktail’ tablet. For the LC3 immunoblot analysis, the cells were lysed with the same buffer plus 0.25% SDS. The lysates were separated by SDS-PAGE and transferred to nitrocellulose membrane by iBLOT Gel Transfer System (Invitrogen). The membrane was incubated with 5% nonfat milk w/v in TBS buffer (25 mM Tris-HCl, pH 7.5; 150 mM NaCl; 0.1% Tween-20) for 1 h, and then reacted with the primary antibody in TBS buffer with 2.5 % nonfat milk or 5% BSA w/v for overnight by shaking at 4 degree. The appropriated second antibodies conjugated to HRP were used to detect the protein of interest via enhanced chemoluminescence (ECL). To immunoprecipitate Myc-MAVS or ORF8b-GFP, the cells were lysed in the same buffer and incubated for 2 h at 4 °C with the appropriate antibodies at which point Protein G PLUS-Agarose was added, followed by an additional incubation of 1 h at 4 °C. The immunoprecipitates were collected and washed eight times with lysis buffer, separated by SDS-PAGE, and analyzed by immunoblotting. To immunoprecipitate PCPB2 1% CHAPS was used in the above buffer. To analyze ubiquitination 2% SDS was added to the lysates prior to heating them to 95 °C for 10 minutes. Subsequently, the cell lysates were diluted 10 fold with the extraction buffer, and then incubated with antibodies overnight. Membranes were stripped using OneMinute®Plus (GM Bioscience) following the manufacturer's protocol. Blots were scanned and imported into Photoshop as unmodified tagged image file format files. Quantification of band intensity was done using Photoshop.
Immunofluorescence
A549 cells were grown on 35-mm plates (MatTek) for confocal microscopy or on .15 mm coverslips (Corning) for STED imaging. After which the cells were fixed in methanol for 5 min. After blocking for 1 h in 5 % BSA in PBS-0.05% Tween 20, the cells were incubated for 1h with the appropriated primary antibody at 4 °C washed 3 times in PBS and incubated with appropriate secondary antibodies for 30 min at 4 °C. Hoescht33342 was applied for the last 5 min only for confocal experiments. After an extensive wash in PBS, cells were mounted in Prolong antifade reagent (Invitrogen), and were imaged on a Leica DMIRBE inverted microscope equipped with a TCS SP5 confocal scanner and a 63× 1.32 NA (Numerical Aperture) objective (Leica Microsystems).
High resolution light microscopy
For stimulated emission depletion (STED) microscopy and the corresponding confocal microscopy a LEICA SP5-STED microscope was used. The fluorophores ATTO647N (Activemotif) or MegaRed (Sigma) were excited with an 80 MHz pulsed diode laser at 635 nm or 531 nm respectively (Pico-Quant), (pulse width < 100 ps). STED depletion was achieved using a mode-locked titanium sapphire laser (Spectra Physics Mai Tai HP laser) operating at 770 nm with a repetition rate of 80 MHz. The delay between the excitation and STED pulses was adjusted electronically to optimize depletion. The excitation and the STED beams were focused by a 100× oil immersion objective (NA 1.4 PL APO STED, 100×; Leica Microsystems). The fluorescence signal was collected by the same objective and detected confocally between 650 and 690 nm for ATTO647N and between 540 and 595 nm for Megared with an APD detector. Using this set up, a resolution of ˜250 nm in the confocal images and 50–70 nm in the STED images was achieved. Except for contrast stretching and level adjustment no further image processing was applied.
Electron microscopy (EM)
For standard EM HEK 293 cells were transfected with a control vector or 9b-Flag. The following day the cells were double-fixed in PBS-buffered glutaraldehyde (2.5%) and osmium tetroxide (0.5%), dehydrated, and embedded with Spurr's epoxy resin. Ultrathin sections (90 nm) were made and double-stained with uranyl acetate and lead citrate, and viewed in a JEOL JEM-1010 transmission electron microscope. For immunoelectron microscopy HEK 293 cells were transfected with a control vector or 9b-Flag. The following day the cells were fixed in formaldehyde, pelleted in 1.5% low melting point agarose, washed with PBS (3X, 5 min.), dehydrated in an ethanol series, and embedded in LR White (SPI). Ultrathin sections were mounted on 150-mesh uncoated nickel grids. Grids were floated on blocking solution (PBS, 0.1% Tween 20, 0.5% cold-water fish gelatin (Ted Pella, Inc.) for 20 min, incubated for 1 h with the primary antibodies, rinsed in blocking buffer for 5 min, blocked with 2% goat serum, rinsed with blocking buffer, then incubated with 10 nm and 20 nm gold-conjugated secondary goat antibody accordingly (Ted Pella, Inc.), rinsed in PBS, and air dried. Sections were stained with aqueous uranyl acetate (5%) and examined with a JEOL JEM-1010 electron microscope.
Flow cytometry
A549 cells stably expressing 9b-GFP or GFP were harvested and fixed with PFA 4% for 20 min. After 3 washes in PBS 1% BSA the cells were permeablized with ice cold methanol for 5 min. After blocking for 1 h in PBS 1 % BSA, the primary antibody (DRP1) was incubated for 3 h and secondary Alexa647 conjugated antibody was incubated for 40 min. Data were acquired with a FACS CANTO (Becton-Dickinson) and analyzed using FlowJo software.
Statistical analysis
Prism (GraphPad software) was used for all statistical analyses. All experiments were repeated a minimum of three times.
Results
SARS-CoV ORF-9b localizes to mitochondria and results in reduced DRP1 levels and mitochondrial elongation
While SARS-CoV infects multiple organs, the pulmonary epithelium and immune cells are major targets (29, 30). To investigate the functions of ORF-9b, we first expressed an ORF-9b GFP fusion protein (9b-GFP) in A549 cells, a cell line used as a model of type II pulmonary epithelial cells. We found that ORF-9b localized to mitochondria and that those cells that expressed the fusion protein had more elongated mitochondria compared to control cells (Fig. 1A & B, Video 1). Similar results were found following expression of 9b-GFP in HEK 293 cells (Fig. 1A). We also verified the mitochondrial morphology changes in a permanent transfection model using THP-1 cells, a human monocyte like cell line that can be differentiated to macrophages following treatment with phorbol 12-myristate 13-actetate (PMA). Immunostaining 9b-GFP expressing THP-1 cells for the mitochondrial protein Tomm20 revealed numerous elongated mitochondria that co-localized with 9b-GFP (Fig. 1C). Mitochondrial fusion and fission maintain mitochondrial number, morphology, and function. In the initial step of fission, the dynamin related protein DRP1 is recruited to mitochondria where it assembles into a fission complex (31). As reduction in DRP1 favors mitochondrial fusion, we checked whether ORF-9b affected DRP1 levels. Expression of 9b-GFP in HEK 293 cells reduced DRP1 levels by approximately 70% via a mechanism sensitive to proteasome inhibition, but not autophagic degradation inhibition (Fig. 1D). We found similar results when we used a 9b-Flag construct (Fig. 1E). The sensitivity to proteasome inhibition suggested that DRP1 underwent ubiquitination. Consistent with that possibility the immunoblotting of DRP1 immunoprecipitates revealed the presence of K48-linked ubiquitin (Fig. 1F). We also examined DRP1 levels in the permanently transfected THP-1 cells and again found a marked decrease in DRP1 levels (Fig. 1G). Further imaging and flow cytometry studies confirmed the reduction of DRP1 in 9b-GFP expressing cells (Fig. 2A & 2B). The imaging data showed that some DRP1 is recruited to the 9b-GFP delineated mitochondria. As transfection of 9b-GFP into A549 cells led to varying levels of 9b-GFP expression in individual cells, we checked the endogenous DRP1 expression in cells as a function of 9b-GFP levels (Fig. 2A). We found that those cells with higher levels of 9b-GFP had lower amounts of DRP1 and less DRP1 associated with mitochondria. Consistent with the presence of DRP1 and ORF-9b in the same molecular complex, endogenous DRP1 immunoprecipitates contained small amounts of 9b-GFP, but not GFP (Fig. 2C). These data show that ORF-9b localizes to mitochondria and likely causes mitochondrial elongation by triggering DRP1 ubiquitination and its proteasomal destruction.
Figure 1.
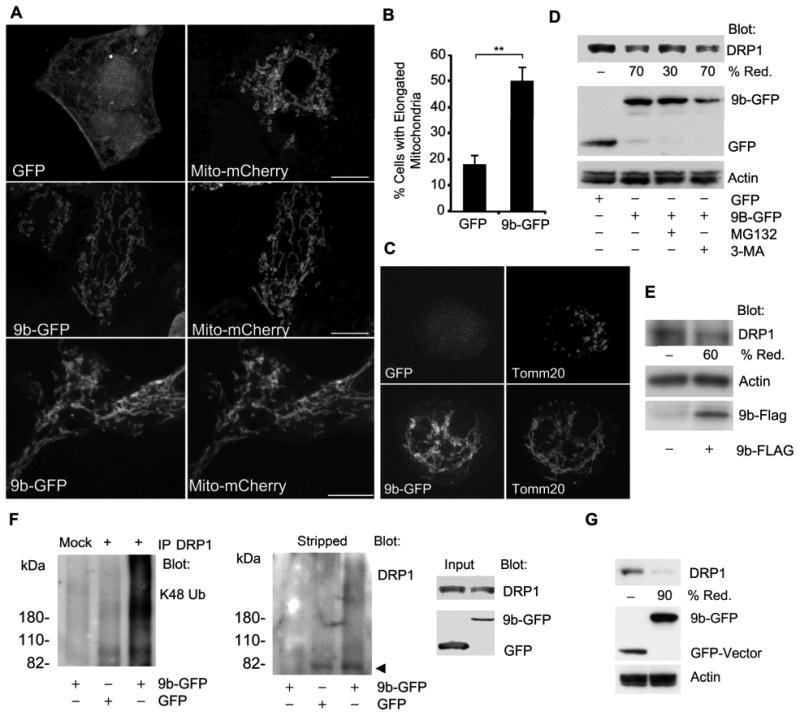
ORF-9b expression elongates mitochondria and enhances the degradation of DRP1. (A) Confocal microscopy of A549 cells expressing GFP (top) or 9b-GFP (middle) and mito-mCherry or HEK 293 cells expressing 9b-GFP and mito-mCherry (lower). Individual images are shown. Scale bar: 10 μm. (B) Quantitation of percentage of cells with elongated mitochondria in A549 cells expressing GFP or 9b-GFP and Mito-mCherry. Co-expressing cells (100 each) were imaged and % of cells with obviously elongated mitochondria enumerated. (C) Confocal microscopy of THP-1 cells permanently expressing 9b-GFP or GFP and immunostained for Tomm20. (D) Immunoblot of cell lysates from HEK 293 cells expressing GFP or 9b-GFP to detect DRP1. In some instances the cells were treated with MG-132 (10 μM)) or 3-MA (5 mM) for 4 h. (E) Immunoblots of cell lysates from HEK 293 cells transfected with Flag or 9b-Flag for indicated proteins. (F) Immunoblots of DRP1 immunoprecipitates (+) and cell lysates from HEK 293 cells transfected with GFP or 9b-GFP. The mock lane is a control antibody immunoprecipitation. Following the K48 ubiquitin (Ub) immunoblot the membrane was stripped and re-blotted for DRP1. The cells were treated with MG-132 for the last 4 h prior to cell lysis. (G) Immunoblot of cell lysates from THP-1 cells permanently expressing either 9b-GFP or GFP to detect DRPl.
Figure 2.
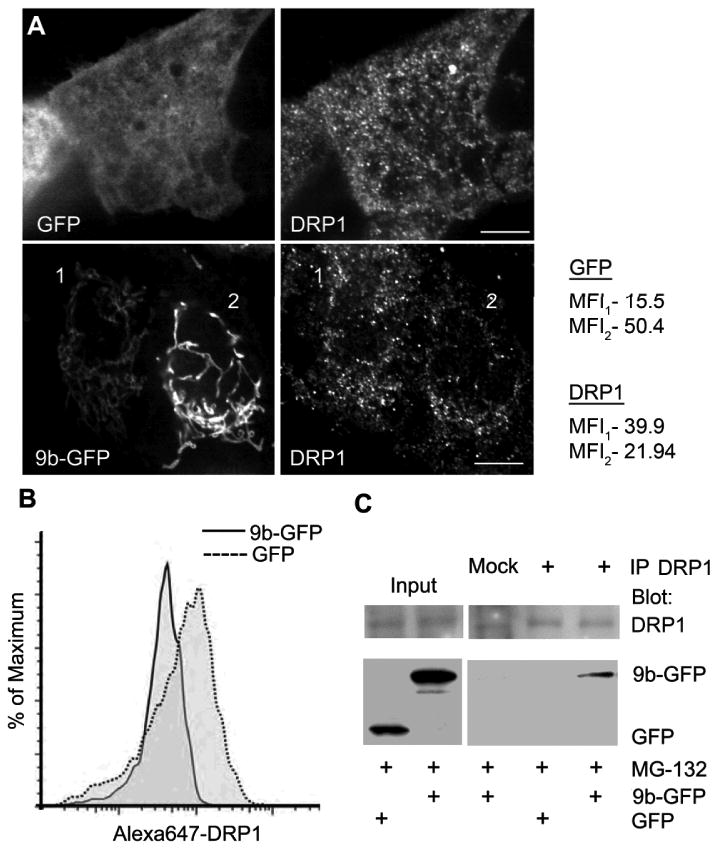
ORF-9b expression leads to less DRP1 associated with mitochondria and ORF-9b co-immunoprecipitates with DRP1. (A) Confocal microscopy of endogenous DRP1 in A549 cells transiently transfected with GFP-ORF9b or GFP vector alone. Scale bar: 10 μm. Mean fluorescence intensity (MFI) of DRP1 or GFP was quantified from the whole volume using ImageJ for cell 1 and cell 2. (B) Flow cytometry analysis of endogenous DRP1 level in A549 cells expressing either GFP or 9b-GFP. The mean fluorescent intensity of DRP1 in GFP+ cells was 186 and 46 in the 9b-GFP+ cells. (C) Immunoblots of DRP1 immunoprecipitates (+) and cell lysates from HEK 293 cells transfected with GFP or 9b-GFP for indicated proteins. Mock is a control antibody immunoprecipitation. Cells were treated with MG-132 (10 μM).
SARS-CoV ORF-9b limits host cell interferon signaling and co-immunoprecipitates with the mitochondrial adaptor protein MAVS
Besides their recognized role in energy production, mitochondria serve as a platform for host defense against RNA viruses such as SARS-CoV (32). Mitochondrial localized MAVS links mitochondria to antiviral type I interferon signaling. Since SARS-CoV infected cells have impaired interferon responses (18) and ORF9b localized to mitochondria, we checked whether ORF-9b impacted MAVS signaling. We expressed 9b-GFP in the presence of either an IFN-β or a NF-κB reporter construct and simulated an intracellular viral infection by co-transfecting Poly(I:C). As expected Poly(I:C) induced both reporter constructs, but the presence of ORF-9b significantly inhibited this response (Fig. 3A). Next, we tested where in the signaling pathway ORF-9b acted by assessing reporter gene activation following expression of a constitutively active form of RIG-I (RIG-I1-250) or by MAVS overexpression (33). In both instances ORF-9b reduced the activation of the reporters arguing that ORF-9b functions at the level of MAVS or more distally (Fig. 3A). We verified that the 9b-Flag construct also inhibited MAVS signaling (Fig. 3B). Since MAVS is required for the phosphorylation of IRF3 in the RIG-I mediated anti-viral response, we checked whether 9b-GFP expression reduced IRF3 phosphorylation, which it did (Fig. 3C). Next, we measured interferon-β levels by immunoblotting cell supernatants conditioned by THP1 cells transfected with Myc-MAVs in the presence or absence of 9b-Flag. As expected the amount of interferon-β decreased in the cell supernatant in the presence of 9b-Flag (Fig. 3D). In contrast, ORF-9b had little impact on interferon-β production in THP-1 cells triggered by either exposing the cells to a TLR3 ligand (Fig. 3E) or by transfecting the cells with an IRF-3 expression vector (Fig. 3F). Next, we determined whether ORF-9b and MAVS could be found in the same molecular complex. We found that expressed 9b-GFP consistently immunoprecipitated with endogenous MAVS (Fig. 3G). Mapping the region in MAVS needed to co-immunoprecipitate with ORF-9b suggested that the mitochondrial membrane insertion sequence in MAVS (amino acids 514-535) was necessary. However, the 503-540 MAVS construct co-immunoprecipitated less well than full length MAVS implying that another region in MAVS may be needed (Fig. 3H). We also checked whether the reduction of DRP1 noted above might contribute to the impaired induction of IFN-β. We used a previously describe shRNA to reduce DRP1 levels in HEK 293 cell (34). Lowered DRP1 levels impaired the MAVS-induced increase IFN-β reporter activity although not nearly as effectively as did the 9b-FLAG construct (Fig. 3I). The DRP1 shRNA appropriately decreased DRP1 expression, but did not impact endogenous MAVS expression (Fig. 3J). This indicates that the ORF-9b mediated reduction in DRP1 may contributes to the decrease in type I interferon signaling, however other mechanisms must be operant.
Figure 3.
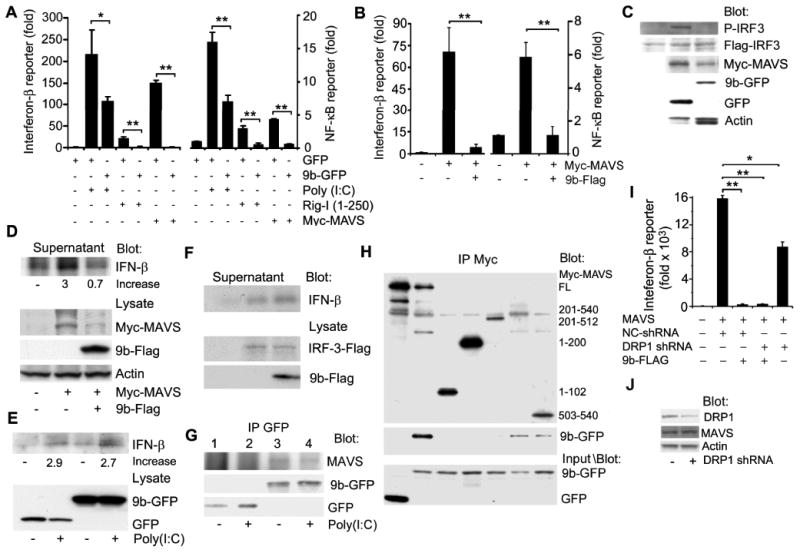
ORF-9b inhibits anti-viral type I interferon responses. (A) IFN-β and NF-κB reporter gene assays using HEK 293 cells expressing GFP or 9b-GFP and induced by transfection of Poly(I:C), RIG-I1-250, or MAVS. Luciferase activity is shown as fold induction. *p<0.05 or **p<0.01 by t test. (B) IFN-β and NF-κB reporter gene assays using HEK 293 cells expressing Flag or 9b-Flag and induced by transfection of myc-MAVS. Luciferase activity is shown as fold induction. **p<0.01 by t test. (C) Immunoblot of HEK 293 cells expressing Flag-IRF3 and Myc-MAVS and either GFP or 9b-GFP to detect phosphorylated (P)-IRF-3. (D) Immunoblot of IFN-β in cell supernatants and indicated proteins in cell lysates from THP-1 transiently transfected with Myc-MAVS and expressing 9b-Flag or not. (E) Immunoblot of IFN-β and indicated proteins in cell lysates from THP-1 permanently expressing GFP or 9b-GFP and exposed to Poly (I:C) overnight (10 μg/ml). (F) Immunoblots of IFN-β in cell supernatants and of the indicated proteins in cell lysates from THP-1 cells transiently transfected with an IRF-3 expression vector. (G) Immunoblot of GFP immunoprecipitates from HEK 293 cells expressing GFP-vector or ORF-9b-GFP for endogenous MAVS. Poly(I:C) was transfected 1 h prior to cell lysis. (H) Immunoblot of Myc-immunoprecipitates from HEK 293 cells expressing full-length or truncated Myc-MAVS and GFP or 9b-GFP. GFP and 9b-GFP expression levels were verified by immunoblotting cell lysates from the same cells. (I) IFN-β reporter assays using HEK293 cells expressing a control (NC) or a DRP1 shRNA in the presence of absence of 9b-FLAG. MAVS expression was used to induce reporter gene activity. (J) Immunoblot verifying that the DRP1 shRNA reduced DRP1 expression, but did not affect endogenous MAVS expression.
To check whether ORF-9b altered the intracellular distribution of MAVS, we expressed 9b-GFP in A549 cells and immunostained for endogenous MAVS. Standard confocal microscopy revealed that in 9b-GFP expressing cells MAVS had a more punctate appearance forming small aggregates along 9b-GFP delineated mitochondria (Fig. 4A). This is most evident in the zoomed images shown in the insets (Fig. 4A). To better visualize the distribution of MAVS in the control and ORF-9b expressing cells, we used stimulated emission depletion (STED) microscopy. In the control cell most of the MAVS distributed uniformly along mitochondria-like structures, while small, discrete MAVS puncta distributed along elongated ORF-9b delineated mitochondria (green in the inset), (Fig. 4B). While this is a subjective conclusion it is based on the examination of numerous images. We also examined the localization of MAVS and ORF-9b by immunoelectron microscopy and observed the apposition of Flag tagged ORF-9b (9b-Flag) with endogenous MAVS at the outer mitochondrial membrane (Fig. 4C & D). These data indicate that ORF-9b is in close physical association with MAVS.
Figure 4.
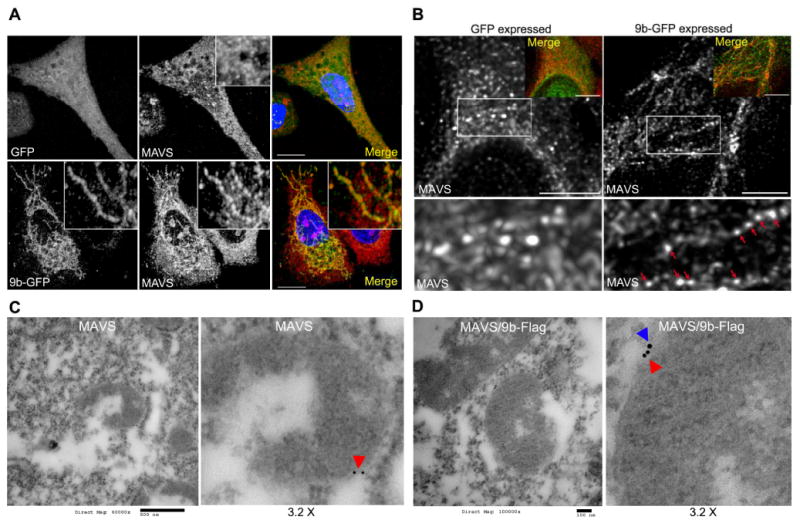
Imaging ORF-9b expression reveals ORF-9b and MAVS co-localize and increased MAVS aggregation. (A) Images from confocal microscopy of A549 cells expressing GFP or 9b-GFP immunostained for MAVS (red) and labeled with DNA dye (blue). Insert 3X electronic zoom. Scale bar: 10 μm. (B) Images from STED microscopy of A549 cells expressing 9b-GFP and immunostained for MAVS. 9b-GFP was imaged by confocal microscopy. Overlap is shown as an insert. Scale bar: 2 μm. Below each image is a 2.5X electronic zoom of the white boxed area. Red arrows indicate MAVS accumulation along 9b-GFP delineated mitochondria. (C) Immunoelectron microscopy of HEK 293 cells transfected with a control vector and immunostained for MAVS (red arrowhead). Scale bar: 500 nM. Right image is a 3.2x electronic zoom of the mitochondria in the left image. (D) Immunoelectron microscopy of HEK 293 cells transfected with 9b-Flag and immunostained for MAVS (red arrowhead) and Flag (blue arrowhead). Scale bar: 100 nM. Right image is a 3.2x electronic zoom of the mitochondria in left image.
SARS-CoV ORF-9b limits interferon signaling by degrading the MAVS signalosome
We had noted the level of the epitope tagged MAVS was reduced in the ORF-9b expressing cells compared to the level in control cells so we checked endogenous MAVS levels following 9b-GFP expression. We found that following 9b-GFP expression the level of endogenous MAVS, as determined by immunoblotting, had declined by approximately 50%. The reduction was partially reversed by a proteasomal inhibitor, but not by an autophagy inhibitor (Fig. 5A). Since MAVS recruits TRAF3 and TRAF6 to activate downstream signaling pathways, we also checked whether ORF-9b expression affected their expression. To do so we immunoblotted cell lysates prepared from HEK 293 cells expressing epitope tagged versions of TRAF3 or TRAF6 along with either GFP or 9b-GFP (Fig. 5B). Surprisingly the reductions in the epitope tagged TRAF3 and TRAF6 equaled those of Myc-MAVS. Indicating that ORF-9b likely triggers K48-linked ubiquitination of MAVS immunoblotting Myc-MAVS immunoprecipitates revealed the presence of K48-linked ubiquitin (Fig. 5C). This last experiment was done using a stringent protocol that reduces the likelihood that the immunoprecipitates will contain spurious proteins (28, 35). Stripping the membrane and re-blotting with antibodies directed at the Myc tag revealed a smeared band also suggestive of MAVS ubiquitination (Fig. 5C). We verified that 9b-Flag also reduced Myc-MAVS levels (Fig. 5D). Finally, in the THP-1 cells permanently transfected with 9b-GFP we also found a marked reduction in the endogenous level of MAVS (Fig. 5E). These results led us to search for the E3 ligase responsible for ubiquitinating MAVS following ORF-9b expression.
Figure 5.
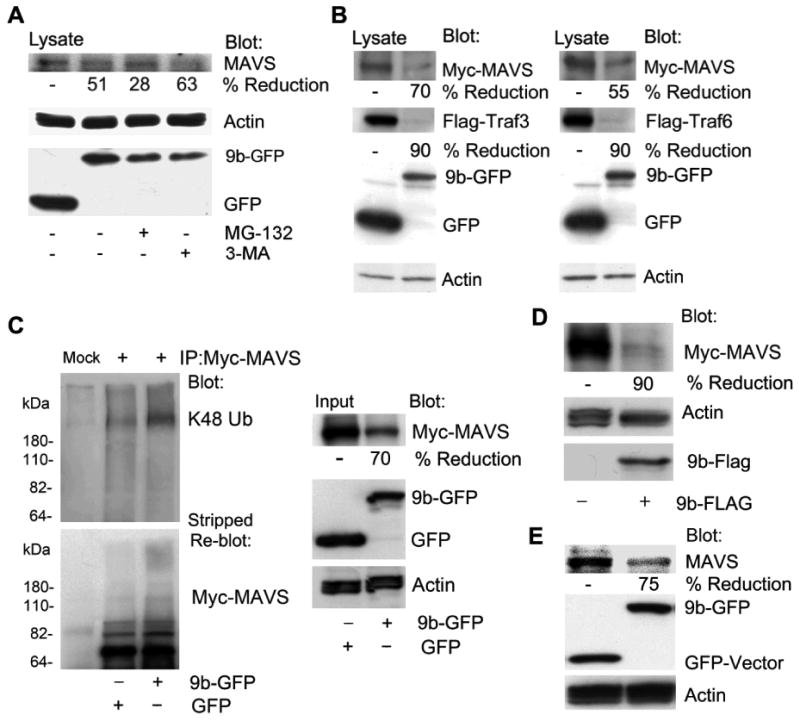
ORF-9b enhances MAVS proteasomal degradation via its K48-linked ubiquitination. (A) Immunoblots of lysates from HEK 293 cells expressing GFP-vector or 9b-GFP untreated or treated with MG-132 (10 μM) or 3-MA (5 mM) 4 h. The blots show the indicated proteins. (B) Immunoblots of lysates from HEK 293 cells expressing Flag-TRAF3 or Flag-TRAF6 and either GFP or 9b-GFP. The blots show the indicated proteins. (C) Immunoblots of Myc immunoprecipitates (+) and cell lysates from HEK 293 cells expressing Myc-MAVS and GFP or 9b-GFP. Following immunoblotting for K48 ubiquitin (Ub) the membrane was stripped and probed for Myc-MAVS. Mock is a control antibody immunoprecipation. (D) Immunoblots of lysates from HEK 293 cells expressing Myc-MAVS in the presence of either the Flag-vector or 9b-Flag. The blots show the indicated proteins. (E) Immunoblots of lysates from THP-1 cells permanently expressing GFP or 9b-GFP for endogenous MAVS. Also shown are GFP, 9b-GFP, and actin levels.
It is known that PCBP2 acts as a gatekeeper to control MAVS levels and signaling by linking the AID4 E3 ubiquitin ligase with MAVS (27). To determine whether ORF-9b usurped PCBP2 and AID4 to degrade MAVS, we reduced their levels in cells that expressed 9b-GFP. We found that the two shRNAs partially reversed the ORF-9b mediated reduction in MAVS (Fig. 6A). In addition, endogenous PCBP2 could pull-down 9b-GFP and endogenous MAVS suggesting that ORF-9b may form a tri-molecular complex (Fig. 6B). Next, we reduced PCBP2 or AID4 expression and checked Poly(I:C) induced reporter gene activation in the presence of 9b-GFP (Fig. 6C). We found that reducing either PCBP2 or AIP4 largely reversed the ORF-9b mediated inhibition of Poly(I:C) induced reporter gene activity. Together these data indicates that ORF-9b targets several members of the MAVS signalosome for destruction. It largely does so by recruiting PCBP2 and the AID4 E3 ligase. As a consequence MAVS, TRAF3, and TRAF6 levels decline in the host cell limiting the cells ability to generate a potent interferon response to viral RNA.
Figure 6.
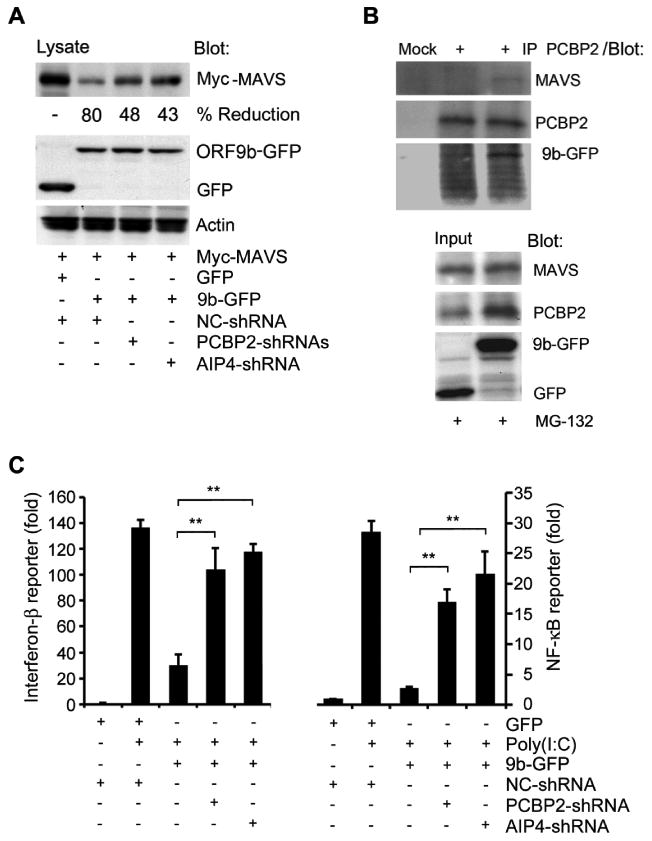
ORF-9b uses PCBP2/AIP4 to degrade MAVS and to impair type I interferon responses. (A) Immunoblot of lysates from HEK 293 cells expressing Myc-MAVS and either GFP or 9b-GFP in the presence of a control shRNA or a shRNA targeting either PCBP2 or AIP4. (B) Immunoblot of cell lysates and PCBP2 immunoprecipitates (+) from HEK 293 cells expressing GFP or 9b-GFP. Cells were treated with MG-132 (10 μM) for 2 h prior to lysis. Mock is a control antibody immunoprecipitation. (C) IFN-β and NF-κB reporter assays using HEK 293 cells expressing GFP or 9b-GFP along with a control, PCBP2, or AIP4 shRNA and induced by transfection of Poly(I:C). The luciferase activity shows in fold induction. **p<0.01, statistics analyzed by t test.
SARS-CoV ORF-9b promotes the formation of autophagosomes
Finally, since mitochondrial damage and changes in mitochondrial morphology have been linked to autophagy, and many CoV trigger autophagy (36), we checked autophagy levels in 9b-GFP expressing cells by co-expressing the autophagy marker LC3 tagged red fluorescent protein (LC3-RFP) in A549 cells. When autophagosomes form, LC3-RFP is processed and recruited to the autophagosomal membrane resulting in the accumulation of fluorescent cytoplasmic dots. Interestingly, 9b-GFP expression potently induced autophagosome formation in A549 cells (Fig. 7A, Video 2). The video shows numerous interactions between elongated mitochondria and autophagosomes. Similarly, 9b-Flag triggered autophagy in the same cells (Fig. 7B). Quantification of the imaging, electron microscopy, and LC3 immunoblotting confirmed the increase in autophagosome formation (Fig. 7C-E). The electron microscopy also revealed the mitochondrial morphology changes noted previously and a close association between mitochondria and the autophagosomes (Fig. 7D). The induction of autophagy by ORF-9b depended upon ATG5, an important regulator of autophagosome formation, as reducing its expression decreased 9b-GFP triggered LC3 processing (Fig. 7F). While ATG5-ATG12 conjugates can interact with MAVS to impair its antiviral effects (37), reducing ATG5 expression did not appreciably impact MAVS signaling in the presence or absence of 9b-GFP (Fig. 7G). This indicates that the increased autophagy in ORF-9b expressing minimally contributes to the previously noted loss of MAVS signaling. We also checked the basal autophagy levels in the THP-1 cells permanently transfected with either 9b-GFP or GFP. While we noted an increase in LC3 processing in the 9b-GFP expressing cells compared to control cells, the difference was small (Fig. 7H). This suggests that the cells had compensated for the prolonged ORF-9b expression or that some intrinsic difference exists between macrophages and the other cell types.
Figure 7.
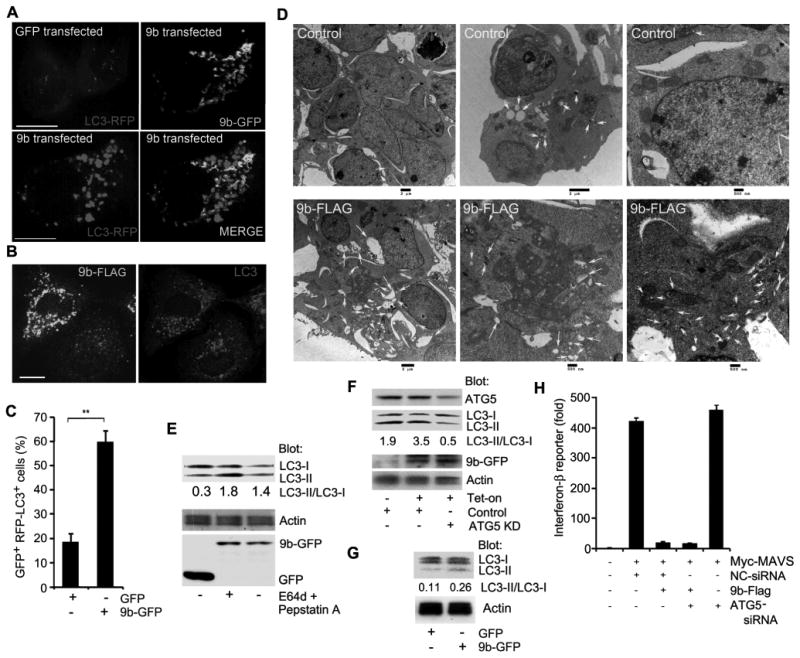
ORF-9b induces autophagy. (A) Images from confocal microscopy of A549 cells co-expressing GFP-vector or 9b-GFP with LC3-RFP. Individual and merged images are shown as indicated. Scale bar: 10 μm. (B) Images from confocal microscopy of A549 cells expressing 9b-Flag and immunostained for endogenous LC3 and Flag. Individual images are shown. Scale bar: 10 μm. (C) The number of GFP positive cells with > 10 LC3-RFP puncta in A549 cells based on the imaging from part A. **p<0.01, t test. (D) Images from electron microscopy of HEK 293 cells expressing a control (top 3 images) or 9b-Flag (bottom 3 images). Scale bars are shown. Arrows mark the autophagosomes. (E) Immunoblot of lysates from A549 cells expressing GFP or 9b-GFP for the indicated proteins. The lysosome inhibitors E64d and pepstatin A were added as indicated for the last 4h. The ratio of LC3-II/LC3-I is shown. (F) Immunoblot of cell lysates from A549 cells expressing 9b-GFP under tetracycline control expressing an ATG5 siRNA or not. The cells were tetracycline-induced for 16 h prior to cell lysis. The ratio of LC3-II/LC3-I is shown. (G) Immunoblot of cell lysates from THP-1 permanently expressing GFP or 9b-GFP. The ratio of LC3-II/LC3-I is shown. (H) IFN-β reporter assay using HEK 293 cells expressing Flag or 9b-Flag along with Myc-MAVS and control or ATG5 siRNAs. Luciferase activity is shown as fold induction.
Discussion
Our observations indicate the SARS-CoV ORF-9b provides a receptive intracellular environment for viral replication by targeting the mitochondrial MAVS signalosome. ORF-9b localizes to mitochondria where it promotes mitochondrial elongation by enhancing DRP1 degradation. In the presence of ORF-9b, MAVS becomes concentrated into small puncta and subject to PCBP2 and AIP4 ubiquitination. The degradation of MAVS is accompanied by a loss of TRAF3 and TRAF6, two key signaling intermediaries in anti-viral defenses. In addition, acute ORF-9b expression results in ATG5-dependent autophagosome formation.
The expression of ORF-9b in A549, HEK 293, and THP-1 cells resulted in a rather striking set of cellular changes. ORF-9b localized to mitochondria and led to mitochondrial elongation. This is in contrast to the usual mitochondrial fragmentation that occurs following viral infection and to our knowledge has not been reported with another viral open reading frame. Mitochondrial elongation has been reported following infection of cells with a defective strain of Sendai virus. The mitochondrial elongation was linked to RLR activation and enhanced MAVS signaling (38). This differed from ORF-9b expression, which also caused mitochondrial elongation, but severely limited MAVS signaling. In contradistinction to the defective virus, the wild type Sendai virus triggered mitochondrial fragmentation and aggregation (38). Similarly, Hepatitis C virus infection caused mitochondrial perinuclear clustering, the translocation of Parkin to mitochondria, and mitophagy (39). Perhaps ORF-9b counteracts the impact of other cellular stresses during SARS-CoV infection, which fragment and aggregate mitochondria, thereby helping to promote cell survival during viral replication. Mitochondrial fusion is known to protect mitochondria from starvation induced autophagosomal degradation (40, 41).
Our localization results differ from those previously reported localization of ORF-9b in Vero cells (16, 17). We observed a similar localization of ORF-9b in three different human cell types using both Flag and GFP tagged versions. We show mitochondrial localization in all three cell types and failed to see a visible accumulation of ORF-9b in the nucleus of any of three cell types. The mitochondrial localization reported here was confirmed by life cell imaging of cells co-expressing Mito-tracker and by immunostaining fixed cells for Tomm20 and MAVS. The two other studies did not look for mitochondria localization. As mitochondria are often closely associated with the endoplasmic reticulum the co-localization of ORF-9b with an endoplasmic marker may have obscured its true localization. Less likely is that the localization of ORF-9b has a cell type dependent distribution pattern.
In the ORF-9b expressing cells DRP1 levels declined as a consequence of ubiquitination and proteasomal degradation likely explaining the change in mitochondria morphology. Parkin is an E3 ligase that is known to ubiquinate DRP1 for proteasome-dependent degradation. Reducing Parkin expression in HeLa cells enhanced DRP1 levels and caused mitochondrial fragmentation (42). To test whether ORF-9b used Parkin to ubiquitinate DRP1 we reduced its expression and examined the mitochondrial morphology in the knock-down cells. The reduced Parkin expression did not alter the mitochondrial morphology changes triggered by ORF-9b expression arguing that another E3 ligase ubiquitinates DRP1 following ORF-9b expression (C. Shi, unpublished result). We also considered whether autophagosomal degradation contributed to the reduced DRP1 levels noted in ORF-9b expressing cells. Inhibitors of autophagy increased DRP1 levels in HEK 293T cells and increasing autophagy reduced DRP1 levels in neurons (43), however an autophagy inhibitor did not reverse the lowered DRP1 levels observed in the ORF-9b expressing cells.
SARS-CoV ORF-9b likely has little role in directly supporting viral replication, but rather its primary function is to inactivates the RLR pathway by triggering degradation of the MAVS/TRAF3/TFAF6 signalosome. As mentioned in the introductory section, RNA viruses have evolved several different mechanisms to disable MAVS signaling including cleaving MAVS from the mitochondria (24), assembling a degradasome (26), and usurping a host cell negative regulatory system, the AIP4-PCBP2-MAVS axis, as we describe here. PCBP2 serves as an adapter molecule recruited to mitochondrial MAVS following viral infection or MAVS overexpression (27). The c-terminal portion of MAVS is needed to recruit PCBP2 and ORF-9b. Following ORF-9b expression we found that PCBP2 immunoprecipitates contained both MAVS and ORF-9b. This suggests the localization of ORF-9b to the mitochondria had led to the translocation of PCBP2 from the nucleus to the mitochondria. Rescue of the ORF-9b phenotype by knock-down of PCBP2 also indicates that ORF-9b expression had led to PCBP2 recruitment to mitochondria. Although the physiologic signal that triggers PCBP2 nuclear export and its localization to mitochondria following viral infection is unknown, mitochondria are a logical source. Pathogenic viral infections cause mitochondrial stress. Stressed mitochondria release reactive oxygen species and a variety of other mediators, and affect intracellular calcium levels. However, mitochondrial stress typically causes mitochondrial fission, not mitochondria fusion (44) as we noted in the ORF-9b expressing cells.
Not only did we observe a strong reduction in MAVS levels following ORF-9b expression, but also in two key signaling intermediates, TRAF3 and TRAF6. Since reducing either AIP4 or PCBP2 expression largely rescued the ORF-9b induced signaling defect, the TRAF3 and TRAF6 levels are likely to have been restored. It is unlikely that TRAF3 and TRAF6 are direct targets of the PCBP2/AIP4, but rather are indirectly targeted for destruction by their association with MAVS. While TRAF3 is directly targeted by the E3 ligase Triad3, which also negatively regulates MAVS signaling, Triad3 does not cause MAVS degradation (45). The viral degradasome induced by the expression of respiratory syncytial virus (RSV) NS1 proteins results in the degradation of a number of proteins in the interferon induction and response pathway including RIG-I, TRAF3, and IRF3, but MAVS was not one of them (26). Our current studies are aimed at understanding how ORF-9b expression recruits PCBP2/AIP4 and determining how TRAF3 and TRAF6 are also degraded. Our working hypothesis is that the ORF-9b/MAVS interaction triggered a partial assembly of the MAVS/TRAF3/TRAF6 signalosome, which leads to a signal that recruits PCBP2/AIP4. As a consequence MAVS and perhaps other proteins are ubiquitinated and delivered to the proteasome for degradation.
While the observed decreases in DRP1 and MAVS/TRAF3/TRAF6 can explain the mitochondrial elongation and reduced anti-viral signaling in the ORF-9b expressing we have not found an immediately proximal cause of the increase autophagy in HEK293 and A549 cells. Given its localization to mitochondria, mitochondria involvement in the enhanced autophagy noted in the ORF-9b expressing cells seems likely. However, while mitochondrial damage and increased mitochondrial ROS production are known triggers for autophagosome formation they had no clearly defined role here. Since the mitochondrial potential in control and ORF-9b expressing cells did not differ, mitochondrial ROS production is an unlikely culprit. We also examined autophagy as assessed by LC3 immunoblotting in the THP-1 cells permanently transfected with ORF-9b. While we observed a small increase in LC3 processing in the ORF-9b expressing cells the difference was less marked than what we had observed following the acute expression of ORF-9b in A549 and HEK 203 cells. Further studies will be needed to delineate the mechanisms by which ORF-9b triggers marked autophagosome formation in some cell types and not others.
We also considered whether the enhanced autophagy in A549 and HEK 293 cells might contribute to the reduced MAVS activity we observed. Previous studies have shown that some viruses activate the autophagy pathway to reduce MAVS signaling and impair anti-viral responses (46, 47). Furthermore, the ATG5-ATG12 conjugate has been shown to negatively regulate the type I IFN production pathway by direct association with RIG-I and IFN-beta promoter stimulator 1 (37). However, in our hands ORF-9b mediated suppression of MAVS signaling did not depend upon the autophagy factor ATG5, as reducing its expression did not impact the suppression in anti-viral signaling. In addition, the pharmacological inhibition of autophagy did not reverse the loss of MAVS following ORF-9b expression. The modest increase in autophagy in the 9b-GFP expressing THP-1 despite a potent reduction in MAVS expression also argues that the induction of autophagy did not contribute to the loss of MAVS and MAVS signaling that we noted.
Finally, the ORF-9b mediated loss of MAVS may further limit host innate responses by reducing NLRP3 inflammasome activity as MAVS promotes NLRP3 mitochondrial localization and inflammasome activation (48). We are currently testing whether the expression of ORF-9b also affects inflammasome activity by targeting NLRP3 levels. In conclusion SARS-CoV uses ORF-9b to target host cell mitochondria to disable MAVS signaling. Our data supports the use interferon-α therapy as a prophylactic treatment against SARS-CoV, but suggests that it will be less effective when administered to patients with an established infection.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Anthony S. Fauci for his continued support.
This research was supported by the Intramural Research Program of the National Institutes of Health [National Institute of Allergy and Infectious Diseases].
References
- 1.Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RW, Ng TK, Yuen KY. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Peiris JS, Guan Y, Yuen KY. Severe acute respiratory syndrome. Nat Med. 2004;10:S88–97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stadler K, Masignani V, Eickmann M, Becker S, Abrignani S, Klenk HD, Rappuoli R. SARS--beginning to understand a new virus. Nat Rev Microbiol. 2003;1:209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hilgenfeld R, Peiris JS. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antiviral Res. 2013 doi: 10.1016/j.antiviral.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balboni A, Battilani M, Prosperi S. The SARS-like coronaviruses: the role of bats and evolutionary relationships with SARS coronavirus. New Microbiol. 2012;35:1–16. [PubMed] [Google Scholar]
- 8.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 9.Butler D. SARS veterans tackle coronavirus. Nature. 2012;490:20. doi: 10.1038/490020a. [DOI] [PubMed] [Google Scholar]
- 10.Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 11.Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL. The Genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 12.McBride R, Fielding BC. The Role of Severe Acute Respiratory Syndrome (SARS)-Coronavirus Accessory Proteins in Virus Pathogenesis. Viruses. 2013;4:2902–2923. doi: 10.3390/v4112902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meier C, Aricescu AR, Assenberg R, Aplin RT, Gilbert RJ, Grimes JM, Stuart DI. The crystal structure of ORF-9b, a lipid binding protein from the SARS coronavirus. Structure. 2006;14:1157–1165. doi: 10.1016/j.str.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu M, Shi Y, Guo Z, Chen Z, He R, Chen R, Zhou D, Dai E, Wang X, Si B, Song Y, Li J, Yang L, Wang J, Wang H, Pang X, Zhai J, Du Z, Liu Y, Zhang Y, Li L, Sun B, Yang R. Antibody responses to individual proteins of SARS coronavirus and their neutralization activities. Microbes Infect. 2005;7:882–889. doi: 10.1016/j.micinf.2005.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan WS, Wu C, Chow SC, Cheung T, To KF, Leung WK, Chan PK, Lee KC, Ng HK, Au DM, Lo AW. Coronaviral hypothetical and structural proteins were found in the intestinal surface enterocytes and pneumocytes of severe acute respiratory syndrome (SARS) Mod Pathol. 2005;18:1432–1439. doi: 10.1038/modpathol.3800439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moshynskyy I, Viswanathan S, Vasilenko N, Lobanov V, Petric M, Babiuk LA, Zakhartchouk AN. Intracellular localization of the SARS coronavirus protein 9b: evidence of active export from the nucleus. Virus Res. 2007;127:116–121. doi: 10.1016/j.virusres.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma K, Akerstrom S, Sharma AK, Chow VT, Teow S, Abrenica B, Booth SA, Booth TF, Mirazimi A, Lal SK. SARS-CoV 9b protein diffuses into nucleus, undergoes active Crm1 mediated nucleocytoplasmic export and triggers apoptosis when retained in the nucleus. PLoS One. 2011;6:e19436. doi: 10.1371/journal.pone.0019436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spiegel M, Pichlmair A, Martinez-Sobrido L, Cros J, Garcia-Sastre A, Haller O, Weber F. Inhibition of Beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J Virol. 2005;79:2079–2086. doi: 10.1128/JVI.79.4.2079-2086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cinatl J, Jr, Michaelis M, Scholz M, Doerr HW. Role of interferons in the treatment of severe acute respiratory syndrome. Expert Opin Biol Ther. 2004;4:827–836. doi: 10.1517/14712598.4.6.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J Biochem. 2007;141:137–145. doi: 10.1093/jb/mvm032. [DOI] [PubMed] [Google Scholar]
- 21.Ren J, Wang Q, Kolli D, Prusak DJ, Tseng CT, Chen ZJ, Li K, Wood TG, Bao X. Human metapneumovirus M2-2 protein inhibits innate cellular signaling by targeting MAVS. J Virol. 2012;86:13049–13061. doi: 10.1128/JVI.01248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, Jin Q, Zhao Z. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 2013;9:e1003231. doi: 10.1371/journal.ppat.1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horner SM, Liu HM, Park HS, Briley J, Gale M., Jr Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A. 2011;108:14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei C, Ni C, Song T, Liu Y, Yang X, Zheng Z, Jia Y, Yuan Y, Guan K, Xu Y, Cheng X, Zhang Y, Wang Y, Wen C, Wu Q, Shi W, Zhong H. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J Immunol. 2010;185:1158–1168. doi: 10.4049/jimmunol.0903874. [DOI] [PubMed] [Google Scholar]
- 26.Goswami R, Majumdar T, Dhar J, Chattopadhyay S, Bandyopadhyay SK, Verbovetskaya V, Sen GC, Barik S. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res. 2013;23:1025–1042. doi: 10.1038/cr.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z, Jiang Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol. 2009;10:1300–1308. doi: 10.1038/ni.1815. [DOI] [PubMed] [Google Scholar]
- 28.Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3:ra42. doi: 10.1126/scisignal.2000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, Zou W, Zhan J, Wang S, Xie Z, Zhuang H, Wu B, Zhong H, Shao H, Fang W, Gao D, Pei F, Li X, He Z, Xu D, Shi X, Anderson VM, Leong AS. Multiple organ infection and the pathogenesis of SARS. J Exp Med. 2005;202:415–424. doi: 10.1084/jem.20050828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farcas GA, Poutanen SM, Mazzulli T, Willey BM, Butany J, Asa SL, Faure P, Akhavan P, Low DE, Kain KC. Fatal severe acute respiratory syndrome is associated with multiorgan involvement by coronavirus. J Infect Dis. 2005;191:193–197. doi: 10.1086/426870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013;1833:1256–1268. doi: 10.1016/j.bbamcr.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Jacobs JL, Coyne CB. Mechanisms of MAVS Regulation at the Mitochondrial Membrane. J Mol Biol. 2013 doi: 10.1016/j.jmb.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura N, Hirose S. Regulation of mitochondrial morphology by USP30, a deubiquitinating enzyme present in the mitochondrial outer membrane. Mol Biol Cell. 2008;19:1903–1911. doi: 10.1091/mbc.E07-11-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maier HJ, Britton P. Involvement of autophagy in coronavirus replication. Viruses. 2012;4:3440–3451. doi: 10.3390/v4123440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, Ishii KJ, Kawai T, Akira S, Suzuki K, Okuda K. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A. 2007;104:14050–14055. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castanier C, Garcin D, Vazquez A, Arnoult D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010;11:133–138. doi: 10.1038/embor.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim SJ, Syed GH, Siddiqui A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 9:e1003285. doi: 10.1371/journal.ppat.1003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, Zhou J, Chen Q. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem. 2011;286:11649–11658. doi: 10.1074/jbc.M110.144238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Purnell PR, Fox HS. Autophagy-mediated turnover of dynamin-related protein 1. BMC Neurosci. 14:86. doi: 10.1186/1471-2202-14-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakhaei P, Mesplede T, Solis M, Sun Q, Zhao T, Yang L, Chuang TH, Ware CF, Lin R, Hiscott J. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 2009;5:e1000650. doi: 10.1371/journal.ppat.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin R, Zhu W, Cao S, Chen R, Jin H, Liu Y, Wang S, Wang W, Xiao G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS One. 2013;8:e52909. doi: 10.1371/journal.pone.0052909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009;106:2770–2775. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153:348–361. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.