

Abstract
Aging, a natural physiological process, is characterized by a progressive loss of physiological integrity. Loss of cellular homeostasis in the aging process results from different sources, including changes in genes, cell imbalance, and dysregulation of the host-defense systems. Innate immunity dysfunctions during aging are connected with several human pathologies, including metabolic disorders and cardiovascular diseases. Recent studies have clearly indicated that the decline in autophagic capacity that accompanies aging results in the accumulation of dysfunctional mitochondria, reactive oxygen species (ROS) production, and further process dysfunction of the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome activation in the macrophages, which produce the proinflammatory cytokines. These factors impair cellular housekeeping and expose cells to higher risk in many age-related diseases, such as atherosclerosis and type 2 diabetes. In this review, we investigated the relationship between dysregulation of the inflammasome activation and perturbed autophagy with aging as well as the possible molecular mechanisms. We also summarized the natural compounds from food intake, which have potential to reduce the inflammasome activation and enhance autophagy and can further improve the age-related diseases discussed in this paper.
1. Introduction
Aging is a natural physiological process that affects a person with a progressive loss of physiological integrity over the passage of time. According to the World Health Organization (WHO), the proportion of the world's population over the age of 60 will rise to 22% by 2050 [1]. However, although everyone grows older as time passes, the degree of change of physiological function in different individuals may differ. Aging factors may have a variety of different sources, including changes in genes, cell imbalance, and organ senescence. Table 1 summarizes the cellular and molecular mechanisms related to the change process of aging. Several of the pathologies associated with aging, such as atherosclerosis and inflammation, involve uncontrolled cellular overgrowth or hyperactivity [2]. The immune system declines in reliability and efficiency with age, resulting in higher risk in the elderly for compromised pathology as a consequence of chronic inflammation such as atherosclerosis, Alzheimer's disease, and an increased susceptibility to infectious disease. Given the complexity of the issue, we attempted to elucidate and categorize the cellular and molecular mechanisms between the inflammasome activation and autophagy that occur with aging. In addition, several studies have indicated that food intake can reduce inflammation activation or increase autophagy to achieve health and longevity. We also discussed the usefulness of natural foods for promoting anti-inflammasome activity.
Table 1.
The impact of aging on lifespans.
| The molecular hallmarks of age | Age-related changes | Reference |
|---|---|---|
| Genomic instability | ||
| Nuclear DNA | Chromosomal aneuploidies and copy number variations | [71–73] |
| Mitochondrial DNA | Replication errors cause polyclonal expansion of mtDNA mutations | [74] |
| Telomere exhaustion | Shortened telomeres exhibit decreased lifespans | [75, 76] |
| Epigenetic alterations | ||
| Histone modifications | Deficiency in SIRT6 exhibits accelerated aging | [77] |
| DNA methylation | Polycomb target genes become hypermethylated with age | [78] |
| Chromatin remodeling | HP1α effects longevity in flies | [79] |
| Transcriptional alterations | Micro-RNA mir-71 is required for the lifespan extension | [80] |
| Loss of proteostasis | ||
| Chaperone-mediated protein folding and stability | HSPs decline on longevity Accumulation and aggregation of abnormal proteins occur in aged organism |
[81, 82] |
| Delay or dysfunction of autophagy | mTOR signaling in the regulation of mammalian lifespan | [83] |
| The ubiquitin-proteasome system | Enhancement of proteasome activity extends replicative lifespan in yeast | [84] |
| Deregulated nutrient sensing | ||
| The insulin- and IGF-1-signaling pathway | Levels decline and dysfunction of GH/IGF-1 signaling pathway | [39, 85] |
| mTOR and AMPK | Inhibition of mTOR/DR pathway extends lifespan | [86, 87] |
| Mitochondrial dysfunction | ||
| ROS | Amphibious effects of ROS on aging | [88–91] |
| Mitochondrial Integrity and Biogenesis | Reduced efficiency of telomerase activation with aging | [92] |
| Cellular senescence | ||
| The INK4a/ARF Locus | Ink4a/ARF expression increases aging | [93, 94] |
| Stem cell attrition | Hematopoiesis declines with age resulting in a diminished production of adaptive immune cells Reduced in cell-cycle activity of hematopoietic stem cells (HSCs) on aged mice |
[7, 95] |
| Inflammation | Activation of the NLRP3 inflammasome leading to increased production of IL-1β, TNF, and interferons | [36, 96] |
SIRT6: sirtuin-6; HP1α: heterochromatin protein 1α; HSPs: heat shock proteins; mTOR: mammalian target of rapamycin; IGF-1: insulin/insulin growth factor 1; DR: dietary restriction; ROS: reactive oxygen species; AMPK: AMP-activated protein kinase; HSCs: hematopoietic stem cells; NLRP3: nucleotide-binding domain, leucine rich family (NLR), pyrin containing 3; IL-1β: interleukin-1β; TNF: tumor necrosis factor.
2. The Innate Immune System and Aging
The immune system, which protects an organism from disease, comprises two branches: innate and acquired immunity, including phagocyte lineages, such as macrophages, monocytes, dendritic cells (DCs), neutrophils and natural killer (NK) cells in an innate part, and B and T lymphocytes in an acquired part. Table 2 summarizes the process of aging-related changes in the immune system.
Table 2.
Alterations in the immune system associated with aging.
| Immune system | Age-related changes | References |
|---|---|---|
| Innate immunity | ||
| Monocytes or macrophages | Reduced levels of MHC class II complexes, reduced phagocytic capacity, and enhanced oxidative stress | [97, 98] |
| Neutrophils | Reduction in phagocytosis ability, impaired free radical production, and decreased rescue from apoptosis | [99] |
| Dendritic cells | Reduced antigen presentation and impaired phagocytic capability to clean apoptotic cells | [100] |
| Natural killer cells | Increased number of NK cells, reduced cytotoxicity, and impaired proliferation ability in response to IL-2 stimulation | [98] |
| Acquired immunity | ||
| T cells | Reductions in T-cell thymopoiesis, accumulated highly differentiated memory T cells, loss of CD28 antigen and CD69 antigen for T cell activation and signal transduction, and reduced CD8+ cell proliferation in response to antigen stimulation | [101, 102] |
| B cells | Reductions in B-cell lymphopoiesis, increased memory B cells and fewer naive B cells, impaired antibody response to vaccination, and increased production of low-affinity antibodies due to decreased isotype switching | [98] |
MHC II: major histocompatibility complex class II.
Innate immunity is the first line of host defense against microbe infection through diverse germline-encoded pattern-recognition receptors (PRRs) in phagocytes, such as Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain- (NOD-) like receptors (NLRs), which recognize pathogen-associated molecular patterns (PAMPs) from pathogens or danger-associated molecular patterns (DAMPs) from damaged tissue in the body [3, 4].
Macrophages, one of the phagocytic cell lineages presented in most tissues, contribute to the innate immunity and are responsible for numerous inflammatory, immunological, and metabolic processes [5]. Macrophages play an important role in the recognition of danger signaling and the initiation of inflammatory responses, including clearance of pathogens via direct phagocytosis of invading pathogens or indirect release of cytokines and chemokines, which can activate and recruit other inflammatory cells to damaged sites. They also activate acquired immunity through the procession of antigens and presentation of peptides to T lymphocytes [5]. Aging-related changes to the macrophages contribute to the aging process via a functional shift toward a proinflammatory phenotype, which constitutively produces more interleukin- (IL-) 6, IL-1beta (IL-1β), and tumor necrosis factor (TNF) and reduced phagocytic function [6, 7]. Elevated plasma concentrations of IL-6, IL-1β, and TNF have been described in elderly populations and are postulated as predictive markers of functional disability, frailty, and mortality. The macrophage populations of the elderly appear to have reduced levels of major histocompatibility complex (MHC) class II, which contribute to poor CD4+ T lymphocyte responses [8]. Furthermore, it has been found that the phagocytic function of macrophages in aging individuals declines. Aging also results in reduced secretion levels of chemokines, such as macrophage inflammatory protein- (MIP-) 1alpha (MIP-1α), MIP-1β, MIP-2, and eotaxin [9]. A significant decrease in macrophage precursors and macrophages in the bone marrow of elderly individuals has been described previously [10]. These results suggest that an age-related decline in macrophage function may reduce both innate and adaptive immunities.
Neutrophils are short-lived immune cells that play an important role in the antimicrobial host defense that protects the individual from both bacterial and fungal infections [11]. Phagocytosis, chemotaxis, and ROS production of neutrophils could be changed with aging [12]. Studies measuring phagocytosis of bacteria by the neutrophils have found a significant reduction in phagocytic ability in the elderly population [13, 14]. Larbi et al. [15] demonstrated that the age-related decline in neutrophil functions can be partially explained by the reduced Fc-gamma receptor expression. It has been shown that Fc-gamma receptor-mediated free radical generation and phagocytosis are altered with aging, which is clearly the result of changes in p42/p44 mitogen-activated protein kinase (MAPK) signaling pathways.
DCs play a critical role in linking the innate and the adaptive immune system. DCs are the most potent antigen-presenting cells that can prime naive CD4+ T cells via antigen presentation. After Toll-like receptors (TLRs) stimulation such as TLR7 and TLR9, for example, the plasmacytoid dendritic cells (pDCs) produce type I interferon to defend against viral infections and activate NK cells to amplify the host response and help to clear the virus [16–18]. Myeloid dendritic cells (mDCs) are professional antigen-presenting cells to T lymphocytes. They express TLRs and C-type lectin receptors (CLRs) for the detection of viruses. They also produce cytokines, such as IL-12, to induce cytotoxic T-cell responses, which clear virus-infected cells [19]. Epidermal Langerhans cells (LCs), originally described as epidermal DCs, maintain immune homeostasis in the skin by activating skin-resident regulatory T cells. These contain langerin, large granules capable of phagocytosis [20]. LCs are impaired in their phagocytic ability to induce ovalbumin- (OVA-) specific CD4+ and CD8+ T-cell proliferation in aged mice [20]. Besides presenting the antigens, they also provide the costimulatory signals for optimal activation of NK cells and produce the cytokine IL-17, which is known to recruit neutrophils [21].
3. Inflammasome and Aging
In mammals, the inflammasome is a group of cytosolic receptors that recognize not only intracellular PAMPs, but also host-derived signal DAMPs. They control the production of proinflammatory cytokines, such as proinflammatory cytokines IL-1β and IL-18. The inflammasome is a multiprotein complex that contains at least two distinct classes of the NLR family or the pyrin domain (PYD) and HIN domain-containing (PYHIN) family. The inflammasome mediates the activation of caspase-1, leading to pro-IL-1β and pro-IL-18 processing [22]. In addition to the production of IL-1β and IL-18, the inflammasome/caspase-1 complexes may target different effector molecules to regulate diverse physiological functions, such as pyroptosis and tissue repair [23]. During inflammasome activation, NLRP3 can oligomerize through the central nucleotide-binding domain and then recruit an adaptor protein apoptosis-associated speck-like protein containing CARD (ASC) with PYD and an amino-terminal caspase-recruitment-and-activation domain (CARD domain). NLRP3 would interact with the PYD domain of ASC through its own PYD domains, whereas the CARD domain of ASC recruits procaspase-1. Assembly of the inflammasome initiates self-cleavage of caspase-1 and the formation of the active heterotetrameric caspase-1. Active caspase-1 further proteolytically processes pro-IL-1β and pro-IL-18 to their mature forms [24]. At least six inflammasome complexes of the NLR family, including NACHT, LRR and PYD domains-containing protein (NLRP)1, NLRP3, CARD domain containing (NLRC)4, NLRP6, NLRP12, and PYHIN family, absent in melanoma 2 (AIM2), have been characterized [4].
Among the numerous inflammasome complexes, the NLRP3 inflammasome has been extensively studied and shown to be activated by a large variety of activators that do not share any structural similarities [3]. The NLRP3 inflammasome is proposed to be activated through a secondary mediator, including potassium efflux, reactive oxygen species (ROS), or lysosomal proteases [4]. The NLRP3 inflammasome requires two signals for its activation in macrophages. Stimulation with lipopolysaccharides (LPS) leads to TLR4 signaling-pathway activation in a nuclear factor kappa-light-chain-enhancer of activated B cells- (NF-κB-) dependent manner. This results in the synthesis of precursor forms of proinflammatory cytokines, including pro-IL-1β and NLRP3 proteins [25]. Further stimulation of cells with ATP activates the P2X7 receptor, causing K+ efflux through membrane pores, which results in the NLRP3 inflammasome assembly. Another proposed mechanism suggests the activation of NLRP3 by cathepsin B released from ruptured lysosomes following the phagocytosis of monosodium urate and alum. It is demonstrated by using cathepsin B inhibitors and lysosome inhibitors in vitro [26, 27]. ROS was proposed to be an upstream activator of the NLRP3 inflammasome, originating from the mitochondria (mROS). In contrast, mROS generation from a series of electron transport through the mitochondrial oxidative phosphorylation complex was essential for inflammasome activation. A finding by Zhou et al. [28] reveals that mitochondrial dysfunction activates mROS production. Treatment with NLRP3 activators results in the recruitment of NLRP3 to the mitochondria-associated ER membrane (MAM) where NLRP3 recruited ASC for inflammasome activation. Nakahira et al. [29] also demonstrated that LPS together with ATP causes loss of mitochondrial membrane potential and mROS generation due to the release of mitochondrial DNA (mtDNA). Furthermore, cytosolic mtDNA levels correlate with NLRP3-dependent IL-1β production. Interestingly, the findings by Zhou et al. [28] correlated with those by Nakahira et al. [29], which also suggested a role for autophagy in attenuating IL-1β production where caspase-1 activation is limited and where the NLRP3 relocates to MAMs through the clearance of damaged mROS production. Other critical effectors of NLRP3 activation have been reported in recent years. Thioredoxin- (TRX-) interacting protein (TXNIP) upon oxidative stress has been shown to dissociate from TRX and bind to NLRP3 to promote the NLRP3 activation and to be linked to the regulation of lipid and glucose metabolism [28]. Micro-RNA-223 and ubiquitination of the NLRP3 are reported as negative regulators of the NLRP3 [30–32]. The NLRP3 inflammasome has been demonstrated to play a critical role in microbial pathogen infection [3, 33]. Nevertheless, dysregulation of the NLRP3 inflammasome activation has been associated with a variety of human diseases, including autoinflammatory diseases, Crohn's disease, type 2 diabetes, atherosclerosis, and cancers [22, 34].
A prominent age-dependent alteration is a slowly progressing proinflammatory phenotype, contributing to a long-term stimulation of the immune system. This can result in a low-grade proinflammatory status referred to as inflammaging, which accompanies the aging process in mammals [35, 36]. Several studies focused on the pattern of transcriptional factors on aging tissues found that overactivation of the I kappa B kinase- (IKK-) NF-κB pathway is one of the signatures of aging, revealing the inflammatory pathways in aging [37, 38]. More evidence shows that systemic inflammation links to inflammaging, including the accumulation of proinflammatory cytokines in metabolic organs, the overexpression of the NF-κB transcription factor in damaged tissue, or a defective autophagy-signaling pathway in phagocytes [36]. Dysregulation of the inflammatory cytokines response, such as IL-1β, TNF, and interferon, elicits pathological changes of type 2 diabetes and atherosclerosis, correlated with aging in the human population [22, 34, 39, 40].
4. Autophagy and Aging
Autophagy is considered an evolutionarily conserved cellular catabolic process, which facilitates the recycling of damaged proteins and organelles [41]. Three distinct types of autophagy coexist in most cells, including macroautophagy (usually referred to as autophagy), microautophagy, and chaperon-mediated autophagy (CMA). The three types of autophagy are well established and carry cytosolic proteins into the lysosomes for degradation. During autophagy, dysfunctional protein or organelles are sequestered into a double-membrane vesicle known as the autophagosome. The origin of the autophagosome, called the phagophore or isolation membrane, may be derived from the plasma membrane, Golgi, mitochondria, and endoplasmic reticulum (ER). Classical autophagy initiation is induced by nutrient deprivation followed by the inhibition of the mammalian target of rapamycin (mTOR), which recruits the UNC-51-like kinase (ULK) complex from the cytosol to the isolation membrane. This leads to the nucleation of the isolation membrane through the assembly of ATG14, Beclin 1, vacuolar protein sorting (VPS)15, class III phosphatidylinositol-3-OH kinase (PI(3)K), and VPS34 complexes. Additional proteins, such as Ambra 1, double FYVE-containing protein (DFCP)1, ATG9, and WD-repeat domain phosphoinositide-interacting (WIPI) protein, also regulate the nucleation step of autophagosome formation [41]. The next step is the elongation of the isolation membrane, which requires two ubiquitin-like protein conjugation systems. The first is the conjugation of ATG12-ATG5, which is covalently linked by ATG7 (E1-like) and ATG10 (E2-like) enzymes, and serves as a dimer complex that associates with ATG16L1. This multiple protein complex is crucial in autophagosome formation. The second is the conjugation of phosphatidylethanolamine- (PE-) microtubule-associated protein 1 light chain 3 (LC3), which is covalently linked by ATG7 (E1-like) and ATG3 (E2-like) enzymes. The ATG5-ATG12–ATG16 complex serves as the E3-like enzyme to generate PE-LC3 (LC3-II), which is incorporated into both the inner and outer membranes of the autophagosome. Autophagy is originally considered to be a nonselective bulk degradation process. Several lines of evidence suggest that selective autophagy occurs through the recognition of autophagy substrates, such as degradation of intracellular bacteria and viruses (xenophagy), regulation of the turnover of mitochondria (mitophagy), the clearance of polyubiquitinated protein aggregates (aggrephagy), and regulation of lipid metabolism (lipophagy). Increasing evidence has revealed that autophagy plays an important role in regulating immune responses and inflammation [41]. The engagement of TLR4 by LPS recruits the Toll-receptor-associated activator of interferon (TRIF) and the myeloid differentiation factor 88 (MyD88) adaptor. This leads to enhanced TRAF6 E3 ligase activity, which results in the K63-linked ubiquitination of Beclin 1 and the oligomerization of Beclin 1. This promotes the activation of PI(3)K and helps the formation of autophagosomes [42, 43]. Another study [44] reported that heat shock protein 90 (HSP90) plays an important role in mediating TLR4-induced autophagy. HSP90 mediates TLR4-induced autophagy through interaction with Beclin 1 and protects Beclin 1 from proteasome-mediated degradation. In addition, HSP90 and the kinase-specific cochaperone Cdc37 interact with ULK1 and promote its stability and activation. This in turn plays an important role in autophagy-mediated mitochondrial clearance [45]. Both TLRs and NLRs can induce autophagy through the activation of Beclin 1. NLRP4, on the other hand, displays an ability to inhibit autophagy [46]. Several studies have reported that autophagy and/or autophagy-related proteins play an important role in regulating mitochondria integrity, ROS generation, and the subsequent NLRP3 activation. Macrophages treated with 3-methyladenine (3MA), a chemical inhibitor of autophagy, or macrophages with the deletion of several autophagic components, including ATG16L1, ATG7, Beclin 1, and LC3, impair mitochondrial homeostasis and further induce more caspase-1 activation and IL-1β secretion in response to solely LPS or LPS+NLRP3 agonists [29, 47]. These data strongly suggest that autophagy negatively regulates the NLRP3 inflammasome activity. Autophagy is also a critical regulator of the organelles' homeostasis, particularly for aggregated protein and mitochondria in cells [41]. Damaged mitochondria that have lost their membrane potential and are more likely to release toxic apoptotic mediators and ROS serve as signaling to recruit selective autophagy (mitophagy) [29]. The aging process causes the deficient maintenance of proteostasis (see Table 1), resulting in the accumulation of damaged cellular components in old cells; for example, lipofuscin would destroy lysosome function, thus failing to clear the dysfunctional mitochondria [48]. In particular, dysfunction of mitochondrial homeostasis can increase mROS production and stimulate the NLRP3 inflammasome activation. Thus, autophagy declines with aging, enhancing the inflammaging process (Figure 1). Several regulatory mechanisms indicate that the age-related deficiency of autophagy can enhance the appearance of the inflammation phenotype in cells. It is well known that several redox-sensitive protein kinases, phosphatases, and proinflammatory cytokines can stimulate IKK-NF-κB signaling and ROS production, and the increased levels of ROS can feedback-activate NF-κB signaling. All of them can produce prime activation of the inflammasome. Moreover, declines in autophagy can result in the loss of control activity of the NF-κB complex, which is degraded via selective autophagy [49, 50]. The loss of clearance function of autophagy with aging generates a comfortable situation for stimulating NF-κB signaling directly or indirectly, resulting in inflammasome-dependence in an age-related proinflammatory phenotype manner.
Figure 1.
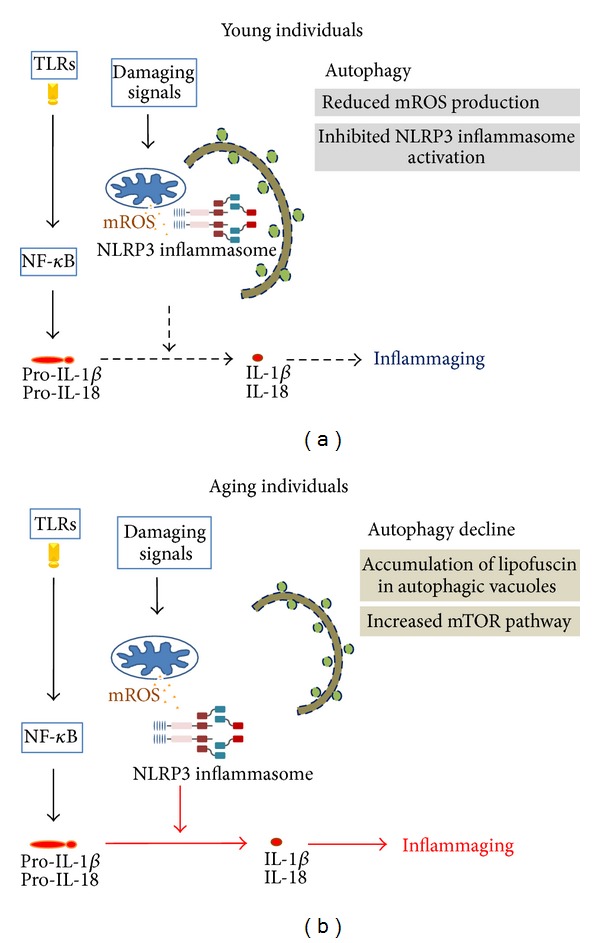
The schematic diagrams represent an overview of the signaling pathways between autophagy and inflammaging. In young individuals (left), autophagy may contribute to maintain the innate physiological lifespan through distinct mechanisms in clearing intracellular mitochondrial ROS (mROS) and NLRP3 inflammasome resulting in decreased inflammaging (black dashed lines), whereas dysfunction of autophagy homeostasis during aging results in increased inflammaging (red lines). However, autophagy protects against the NLRP3 inflammasome-dependent aging process. Aging is only one of the consequences that regulate the depicted signal transduction pathways.
5. The Impact of Natural Compounds on Inflammaging
Aging in humans is associated with a high incidence of some chronic diseases and inflammaging-associated diseases, such as type 2 diabetes, atherosclerosis, and Alzheimer's disease. There is a need to develop a preventive strategy for prolonging the period of healthy life and preventing the pathogenesis of these inflammaging-associated diseases. Interestingly, inflammaging-associated diseases are highly related to NLRP3 activation or a decline in autophagy, which increases metabolic and oxidative stress and elevates a low-grade inflammation, which increases the levels of damage.
Food intake including vegetables, fruits, tea, and wine can reduce the development of age-related diseases [51, 52]. Emerging studies suggest that some phytochemical compounds have potential as inflammation inhibitors to impair NLRP3 activation or enhance autophagy. Four main categories of phytochemical compounds may improve inflammaging-related diseases through impaired NLRP3 activation or enhanced autophagy (Table 3).
Table 3.
Classification of compounds from food sources associated with anti-NLRP3 inflammasome.
| Category | Compounds | Molecular mechanism | Resources | References | |
|---|---|---|---|---|---|
| Stilbenoids | Resveratrol | Inhibited NLRP3 activation Induced autophagy |
Impaired caspase-1 and IL-1β expression Reduced the acetylation of cytoplasmic proteins |
Veratrum album | [57, 58] |
| Flavonoids | |||||
| Flavonols | Quercetin | Suppressed renal NLRP3 activation | Impaired caspase-1 and IL-1β expression | Quercetum | [64, 65] |
| Flavones | Luteoloside | Inhibited NLRP3 activation |
Reduced ROS accumulation Impaired NLRP3, caspase-1, and IL-1β expression |
Honeysuckle | [61] |
| Flavan-3-ols | Catechins | Inhibited NLRP3 activation Enhanced autophagy |
Impaired caspase-1 and IL-1β expression Enhanced Beclin 1 expression |
Green tea | [37, 38] |
| EGCG | Inhibited NLRP3 activation Enhanced autophagy |
Reduced ROS accumulation, NF-κB activation, and NLRP3 expression Impaired caspase-1 and IL-1β expression |
Green tea | [103–105] | |
| Other phenolic compounds | Creosol | Impaired NLRP3 activation | Reduced iNOS expression and NO levels Decreased ROS production Impaired IL-1β expression |
Bamboo vinegar (BV) | [59] |
| Propolis extracts | Inhibited NLRP3 activation | Reduced the IL-1β secretion | Brazilian propolis | [60] | |
iNOS: inducible nitric oxide synthase; ROS: reactive oxygen species; IL: interleukin; MAPK: mitogen-activated protein kinase; EGCG: epigallocatechin-3-gallate; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells.
Resveratrol is a stilbenoid compound that exists in many plant-derived foods such as grapes and red wine [53]. Several discoveries provide evidence demonstrating that a significant amount of resveratrol in the diet has beneficial effects on various chronic diseases and aging. It has been found that resveratrol can protect against type 2 diabetes, heart disease, and Alzheimer's disease [54–56]. In humans, there is still no solid evidence that resveratrol intake can extend lifespan. However, it has been found that resveratrol can protect against NLRP3 inflammasome activation and enhance autophagy [57, 58], which may be able to suppress oxidative stress and inflammation and point to a promising antiaging process.
Other phenolic compounds are described as being linked to anti-inflammatory activity. These compounds include creosol in bamboo vinegar [59] and propolis extracts in Brazilian propolis [60]. They are demonstrated potentially to inhibit NLRP3 activation through the reduction of MAPK and NF-κB activation, decrease ROS production, and impair IL-1β expression. All of them are suggested to improve the aging process [52].
Luteoloside and quercetin, naturally occurring flavonoids in food, exhibit health-beneficial properties and an antiaging effect for humans [52]. Luteoloside, isolated from the medicinal plant Gentiana macrophylla, has been demonstrated to show an anticancer effect against hepatocellular carcinoma (HCC) cells through its effect of inhibiting the NLRP3 inflammasome through inhibiting proliferation, invasion, and metastasis of HCC cells in a mouse lung metastasis model [61]. Quercetin isolated from herbal foods has been reported previously to exhibit potential for anti-inflammation and antihyperlipidemia [62, 63]. Recent studies [64, 65] have demonstrated that quercetin could impair NLRP3 inflammasome activation to improve renal inflammation.
Catechins and epigallocatechin-3-gallate (EGCG) are abundant in teas derived from the tea plant Camellia sinensis. These products show the effect of ameliorating a variety of human diseases such as cancers, atherosclerotic lesions, and Alzheimer's disease [66–70]. Recent studies [37, 38] have shown that they also attenuate the Helicobacter pylori-triggered caspase-1 signaling pathway, oxidative stress, and apoptosis in the gastric mucosa of the Helicobacter pylori-infected mouse model.
6. Conclusion
A healthy lifestyle to avoid premature aging is achievable through maintaining a happy, relaxed mood, engaging in regular sports and exercise, not smoking or drinking, and following a nutrient-rich, low-calorie diet. Studies have shown that people who do not have a healthy lifestyle and do not adhere to a nutritious diet are at high risk of age-related diseases such as type 2 diabetes, cancer, and cardiovascular disease. In this review, we summarize the relationship between inflammaging and autophagy. There are indications that autophagic capacity is dysfunctional in age-related diseases. Autophagy declines with aging, triggering NLRP3 activation, and enhancing the inflammaging process. Decreased NLRP3 activation and increased autophagy can extend the lifespan. In this respect, the effective function of autophagic uptake in the clearance of dysfunctional mitochondria reduced oxidative stress and impaired NLRP3 activation is critical to maintaining cell homeostasis. Growing evidence shows that some foods containing natural compounds, such as resveratrol, catechins, EGCG, propolis extracts, creosol, and luteoloside, are categorized as antiaging molecules [52]. There is suggestion that dietary intake of these compounds may promote health and extend the lifespan via multiple mechanisms, including the reduction of oxidative stress, induction of autophagy, and suppression of NLRP3 activation. This can lead to a healthier and longer lifespan.
Acknowledgments
The authors are grateful to the financial support by Chang Gung University and Chang Gung Memorial Hospital (Grant nos. EMRPD1D901 and CMRPD1B0332).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Shih-Yi Chuang and Chih-Hung Lin are contributed equally to this paper.
References
- 1.Larbi A, Rymkiewicz P, Vasudev A, et al. The immune system in the elderly: a fair fight against diseases? Aging Health. 2013;9(1):35–47. [Google Scholar]
- 2.Blagosklonny MV. Aging: ROS or TOR. Cell Cycle. 2008;7(21):3344–3354. doi: 10.4161/cc.7.21.6965. [DOI] [PubMed] [Google Scholar]
- 3.Davis BK, Wen H, Ting JP-Y. The Inflammasome NLRs in immunity, inflammation, and associated diseases. Annual Review of Immunology. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rathinam VAK, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nature Immunology. 2012;13(4):333–342. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chow A, Brown BD, Merad M. Studying the mononuclear phagocyte system in the molecular age. Nature Reviews Immunology. 2011;11(11):788–798. doi: 10.1038/nri3087. [DOI] [PubMed] [Google Scholar]
- 6.Sadeghi HM, Schnelle JF, Thomas JK, Nishanian P, Fahey JL. Phenotypic and functional characteristics of circulating monocytes of elderly persons. Experimental Gerontology. 1999;34(8):959–970. doi: 10.1016/s0531-5565(99)00065-0. [DOI] [PubMed] [Google Scholar]
- 7.Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM. Aging of the innate immune system. Current Opinion in Immunology. 2010;22(4):507–513. doi: 10.1016/j.coi.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang CQ, Udupa KB, Xiao H, Lipschitz DA. Effect of age on marrow macrophage number and function. Aging. 1995;7(5):379–384. doi: 10.1007/BF03324349. [DOI] [PubMed] [Google Scholar]
- 9.Swift ME, Burns AL, Gray KL, DiPietro LA. Age-related alterations in the inflammatory response to dermal injury. Journal of Investigative Dermatology. 2001;117(5):1027–1035. doi: 10.1046/j.0022-202x.2001.01539.x. [DOI] [PubMed] [Google Scholar]
- 10.Ogawa T, Kitagawa M, Hirokawa K. Age-related changes of human bone marrow: a histometric estimation of proliferative cells, apoptotic cells, T cells, B cells and macrophages. Mechanisms of Ageing and Development. 2000;117(1–3):57–68. doi: 10.1016/s0047-6374(00)00137-8. [DOI] [PubMed] [Google Scholar]
- 11.Mócsai A. Diverse novel functions of neutrophils in immunity, infammation, and beyond. Journal of Experimental Medicine. 2013;210(7):1283–1299. doi: 10.1084/jem.20122220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nature Reviews Immunology. 2013;13(12):875–887. doi: 10.1038/nri3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khanfer R, Carroll D, Lord JM, Phillips AC. Reduced neutrophil superoxide production among healthy older adults in response to acute psychological stress. International Journal of Psychophysiology. 2012;86(3):238–244. doi: 10.1016/j.ijpsycho.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 14.Wenisch C, Patruta S, Daxböck F, Krause R, Hörl W. Effect of age on human neutrophil function. Journal of Leukocyte Biology. 2000;67(1):40–45. doi: 10.1002/jlb.67.1.40. [DOI] [PubMed] [Google Scholar]
- 15.Larbi A, Douziech N, Fortin C, Linteau A, Dupuis G, Fulop T., Jr. The role of the MAPK pathway alterations in GM-CSF modulated human neutrophil apoptosis with aging. Immunity and Ageing. 2005;2, article 6 doi: 10.1186/1742-4933-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agrawal A, Gupta S. Impact of aging on dendritic cell functions in humans. Ageing Research Reviews. 2011;10(3):336–345. doi: 10.1016/j.arr.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romagnani C, Della Chiesa M, Kohler S, et al. Activation of human NK cells by plasmacytoid dendritic cells and its modulation by CD4+ T helper cells and CD4+ CD25hi T regulatory cells. European Journal of Immunology. 2005;35(8):2452–2458. doi: 10.1002/eji.200526069. [DOI] [PubMed] [Google Scholar]
- 18.Lande R, Gilliet M. Plasmacytoid dendritic cells: key players in the initiation and regulation of immune responses. Annals of the New York Academy of Sciences. 2010;1183:89–103. doi: 10.1111/j.1749-6632.2009.05152.x. [DOI] [PubMed] [Google Scholar]
- 19.Kawai T, Akira S. Signaling to NF-κB by Toll-like receptors. Trends in Molecular Medicine. 2007;13(11):460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Seneschal J, Clark RA, Gehad A, Baecher-Allan CM, Kupper TS. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity. 2012;36(5):873–884. doi: 10.1016/j.immuni.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stout-Delgado HW, Du W, Shirali AC, Booth CJ, Goldstein DR. Aging promotes neutrophil-induced mortality by augmenting IL-17 production during viral infection. Cell Host and Microbe. 2009;6(5):446–456. doi: 10.1016/j.chom.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nature Immunology. 2012;13(4):343–351. doi: 10.1038/ni.2224. [DOI] [PubMed] [Google Scholar]
- 23.Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunological Reviews. 2011;243(1):206–214. doi: 10.1111/j.1600-065X.2011.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nature Reviews Immunology. 2013;13(6):397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guarda G, Zenger M, Yazdi AS, et al. Differential expression of NLRP3 among hematopoietic cells. Journal of Immunology. 2011;186(4):2529–2534. doi: 10.4049/jimmunol.1002720. [DOI] [PubMed] [Google Scholar]
- 26.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 27.Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nature Immunology. 2008;9(8):847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nature Immunology. 2010;11(2):136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 29.Nakahira K, Haspel JA, Rathinam VAK, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature Immunology. 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, Hornung V. NLRP3 inflammasome activity is negatively controlled by miR-223. Journal of Immunology. 2012;189(8):4175–4181. doi: 10.4049/jimmunol.1201516. [DOI] [PubMed] [Google Scholar]
- 31.Haneklaus M, Gerlic M, Kurowska-Stolarska M, et al. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1β production. The Journal of Immunology. 2012;189(8):3795–3799. doi: 10.4049/jimmunol.1200312. [DOI] [PubMed] [Google Scholar]
- 32.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. Journal of Biological Chemistry. 2012;287(43):36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nature Immunology. 2012;13(4):325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang C-S, Shin D-M, Jo E-K. The role of NLR-related protein 3 inflammasome in host defense and inflammatory diseases. International Neurourology Journal. 2012;16(1):2–12. doi: 10.5213/inj.2012.16.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Franceschi C, Bonafè M, Valensin S, et al. Inflamm-aging: an evolutionary perspective on immunosenescence. Annals of the New York Academy of Sciences. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 36.Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging. 2012;4(3):166–175. doi: 10.18632/aging.100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang J-C, Yang H-C, Shun C-T, Wang T-H, Chien C-T, Kao JY. Catechins and sialic acid attenuate helicobacter pylori -triggered epithelial caspase-1 activity and eradicate helicobacter pylori infection. Evidence-Based Complementary and Alternative Medicine. 2013;2013:13 pages. doi: 10.1155/2013/248585.248585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang JC, Shun CT, Chien CT, Wang TH. Effective prevention and treatment of Helicobacter pylori infection using a combination of catechins and sialic acid in AGS cells and BALB/c mice. Journal of Nutrition. 2008;138(11):2084–2090. doi: 10.3945/jn.108.090985. [DOI] [PubMed] [Google Scholar]
- 39.Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes. 2012;61(6):1315–1322. doi: 10.2337/db11-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nature Reviews Immunology. 2010;10(1):36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27(1):135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi C-S, Kehrl JH. Traf6 and A20 differentially regulate TLR4-induced autophagy by affecting the ubiquitination of Beclin 1. Autophagy. 2010;6(7):986–987. doi: 10.4161/auto.6.7.13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu C, Liu J, Hsu L-C, Luo Y, Xiang R, Chuang T-H. Functional interaction of heat shock protein 90 and Beclin 1 modulates Toll-like receptor-mediated autophagy. The FASEB Journal. 2011;25(8):2700–2710. doi: 10.1096/fj.10-167676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joo JH, Dorsey FC, Joshi A, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Molecular Cell. 2011;43(4):572–585. doi: 10.1016/j.molcel.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jounai N, Kobiyama K, Shiina M, Ogata K, Ishii KJ, Takeshita F. NLRP4 negatively regulates autophagic processes through an association with Beclin1. The Journal of Immunology. 2011;186(3):1646–1655. doi: 10.4049/jimmunol.1001654. [DOI] [PubMed] [Google Scholar]
- 47.Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. 2008;456(7219):264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 48.Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial Turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxidants and Redox Signaling. 2010;12(4):503–535. doi: 10.1089/ars.2009.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang CP, Su YC, Hu CW, Lei HY. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death and Differentiation. 2013;20(3):515–523. doi: 10.1038/cdd.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paul S, Kashyap AK, Jia W, He Y-W, Schaefer BC. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-kappaB. Immunity. 2012;36(6):947–958. doi: 10.1016/j.immuni.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Everitt AV, Hilmer SN, Brand-Miller JC, et al. Dietary approaches that delay age-related diseases. Clinical Interventions in Aging. 2006;1(1):11–31. doi: 10.2147/ciia.2006.1.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Si H, Liu D. Dietary antiaging phytochemicals and mechanisms associated with prolonged survival. The Journal of Nutritional Biochemistry. 2014;25(6):581–591. doi: 10.1016/j.jnutbio.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gu X, Creasy L, Kester A, Zeece M. Capillary electrophoretic determination of resveratrol in wines. Journal of Agricultural and Food Chemistry. 1999;47(8):3223–3227. doi: 10.1021/jf981211e. [DOI] [PubMed] [Google Scholar]
- 54.Frankel EN, Waterhouse AL, Kinsella JE. Inhibition of human LDL oxidation by resveratrol. The Lancet. 1993;341(8852):1103–1104. doi: 10.1016/0140-6736(93)92472-6. [DOI] [PubMed] [Google Scholar]
- 55.Smoliga JM, Baur JA, Hausenblas HA. Resveratrol and health—a comprehensive review of human clinical trials. Molecular Nutrition and Food Research. 2011;55(8):1129–1141. doi: 10.1002/mnfr.201100143. [DOI] [PubMed] [Google Scholar]
- 56.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nature Reviews Drug Discovery. 2006;5(6):493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 57.Yang SJ, Lim Y. Resveratrol ameliorates hepatic metaflammation and inhibits NLRP3 inflammasome activation. Metabolism-Clinical and Experimenta. 2014;63(5):693–701. doi: 10.1016/j.metabol.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 58.Pietrocola F, Mariño G, Lissa D, et al. Pro-autophagic polyphenols reduce the acetylation of cytoplasmic proteins. Cell Cycle. 2012;11(20):3851–3860. doi: 10.4161/cc.22027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ho CL, Lin CY, Ka SM, et al. amboo vinegar decreases inflammatory mediator expression and NLRP3 inflammasome activation by inhibiting reactive oxygen species generation and protein kinase C-alpha/delta activation. PLoS ONE. 2013;8(10) doi: 10.1371/journal.pone.0075738.e75738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hori JI, Zamboni DS, Carrão DB, Goldman GH, Berretta AA. The inhibition of inflammasome by Brazilian propolis (EPP-AF) Evidence-Based Complementary and Alternative Medicine. 2013;2013:11 pages. doi: 10.1155/2013/418508.418508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fan SH, Wang YY, Lu J, et al. Luteoloside suppresses proliferation and metastasis of hepatocellular carcinoma cells by inhibition of NLRP3 inflammasome. PLoS ONE. 2014;9(2) doi: 10.1371/journal.pone.0089961.e89961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Egert S, Bosy-Westphal A, Seiberl J, et al. Quercetin reduces systolic blood pressure and plasma oxidised low-density lipoprotein concentrations in overweight subjects with a high-cardiovascular disease risk phenotype: a double-blinded, placebo-controlled cross-over study. The British Journal of Nutrition. 2009;102(7):1065–1074. doi: 10.1017/S0007114509359127. [DOI] [PubMed] [Google Scholar]
- 63.Zhu JX, Wang Y, Kong LD, Yang C, Zhang X. Effects of Biota orientalis extract and its flavonoid constituents, quercetin and rutin on serum uric acid levels in oxonate-induced mice and xanthine dehydrogenase and xanthine oxidase activities in mouse liver. Journal of Ethnopharmacology. 2004;93(1):133–140. doi: 10.1016/j.jep.2004.03.037. [DOI] [PubMed] [Google Scholar]
- 64.Wang C, Pan Y, Zhang Q-Y, Wang F-M, Kong L-D. Quercetin and allopurinol ameliorate kidney injury in STZ-treated rats with regulation of renal NLRP3 inflammasome activation and lipid accumulation. PLoS ONE. 2012;7(6) doi: 10.1371/journal.pone.0038285.e38285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu Q-H, Zhang X, Pan Y, Li Y-C, Kong L-D. Allopurinol, quercetin and rutin ameliorate renal NLRP3 inflammasome activation and lipid accumulation in fructose-fed rats. Biochemical Pharmacology. 2012;84(1):113–125. doi: 10.1016/j.bcp.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 66.Arts ICW, Jacobs DR, Jr., Harnack LJ, Gross M, Folsom AR. Dietary catechins in relation to coronary heart disease death among postmenopausal women. Epidemiology. 2001;12(6):668–675. doi: 10.1097/00001648-200111000-00015. [DOI] [PubMed] [Google Scholar]
- 67.Babu PVA, Liu D. Green tea catechins and cardiovascular health: an update. Current Medicinal Chemistry. 2008;15(18):1840–1850. doi: 10.2174/092986708785132979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Higdon JV, Frei B. Tea catechins and polyphenols: health effects, metabolism, and antioxidant functions. Critical Reviews in Food Science and Nutrition. 2003;43(1):89–143. doi: 10.1080/10408690390826464. [DOI] [PubMed] [Google Scholar]
- 69.Shankar S, Ganapathy S, Hingorani SR, Srivastava RK. EGCG inhibits growth, invasion, angiogenesis and metastasis of pancreatic cancer. Frontiers in Bioscience. 2008;13(2):440–452. doi: 10.2741/2691. [DOI] [PubMed] [Google Scholar]
- 70.Haque AM, Hashimoto M, Katakura M, Tanabe Y, Hara Y, Shido O. Long-term administration of green tea catechins improves spatial cognition learning ability in rats. Journal of Nutrition. 2006;136(4):1043–1047. doi: 10.1093/jn/136.4.1043. [DOI] [PubMed] [Google Scholar]
- 71.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Forsberg LA, Rasi C, Razzaghian HR, et al. Age-related somatic structural changes in the nuclear genome of human blood cells. The American Journal of Human Genetics. 2012;90(2):217–228. doi: 10.1016/j.ajhg.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Faggioli F, Wang T, Vijg J, Montagna C. Chromosome-specific accumulation of aneuploidy in the aging mouse brain. Human Molecular Genetics. 2012;21(24):5246–5253. doi: 10.1093/hmg/dds375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Payne BAI, Wilson IJ, Hateley CA, et al. Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtDNA mutations. Nature Genetics. 2011;43(8):806–810. doi: 10.1038/ng.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jaskelioff M, Muller FL, Paik JH, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011;469(7328):102–107. doi: 10.1038/nature09603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boonekamp JJ, Simons MJP, Hemerik L, Verhulst S. Telomere length behaves as biomarker of somatic redundancy rather than biological age. Aging Cell. 2013;12(2):330–332. doi: 10.1111/acel.12050. [DOI] [PubMed] [Google Scholar]
- 77.Mostoslavsky R, Chua KF, Lombard DB, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124(2):315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 78.Maegawa S, Hinkal G, Kim HS, et al. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Research. 2010;20(3):332–340. doi: 10.1101/gr.096826.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Larson K, Yan S-J, Tsurumi A, et al. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genetics. 2012;8(1) doi: 10.1371/journal.pgen.1002473.e1002473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boulias K, Horvitz HR. The C. elegans MicroRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell Metabolism. 2012;15(4):439–450. doi: 10.1016/j.cmet.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Walker GA, Lithgow GJ. Lifespan extension in C. elegans by a molecular chaperone dependent upon insulin-like signals. Aging Cell. 2003;2(2):131–139. doi: 10.1046/j.1474-9728.2003.00045.x. [DOI] [PubMed] [Google Scholar]
- 82.Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: the importance of exquisite quality control. Ageing Research Reviews. 2011;10(2):205–215. doi: 10.1016/j.arr.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kruegel U, Robison B, Dange T, et al. Elevated proteasome capacity extends replicative lifespan in saccharomyces cerevisiae. PLoS Genetics. 2011;7(9) doi: 10.1371/journal.pgen.1002253.e1002253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schumacher B, van der Pluijm I, Moorhouse MJ, et al. Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genetics. 2008;4(8) doi: 10.1371/journal.pgen.1000161.e1000161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Johnson SC, Rabinovitch PS, Kaeberlein M. MTOR is a key modulator of ageing and age-related disease. Nature. 2013;493(7432):338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mair W, Morantte I, Rodrigues APC, et al. Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature. 2011;470(7334):404–408. doi: 10.1038/nature09706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harman D. The free radical theory of aging: effect of age on serum copper levels. Journal of Gerontology. 1965;20:151–153. doi: 10.1093/geronj/20.2.151. [DOI] [PubMed] [Google Scholar]
- 89.Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends in Cell Biology. 2011;21(10):569–576. doi: 10.1016/j.tcb.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van Remmen H, Ikeno Y, Hamilton M, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiological Genomics. 2003;16(1):29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- 91.Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radical Biology and Medicine. 2011;51(2):327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 92.Sahin E, DePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nature Reviews—Molecular Cell Biology. 2012;13(6):397–404. doi: 10.1038/nrm3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Krishnamurthy J, Torrice C, Ramsey MR, et al. Ink4a/Arf expression is a biomarker of aging. Journal of Clinical Investigation. 2004;114(9):1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ressler S, Bartkova J, Niederegger H, et al. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006;5(5):379–389. doi: 10.1111/j.1474-9726.2006.00231.x. [DOI] [PubMed] [Google Scholar]
- 95.Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725–729. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 96.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333(6046):1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Plowden J, Renshaw-Hoelscher M, Engleman C, Katz J, Sambhara S. Innate immunity in aging: impact on macrophage function. Aging Cell. 2004;3(4):161–167. doi: 10.1111/j.1474-9728.2004.00102.x. [DOI] [PubMed] [Google Scholar]
- 98.Weiskopf D, Weinberger B, Grubeck-Loebenstein B. The aging of the immune system. Transplant International. 2009;22(11):1041–1050. doi: 10.1111/j.1432-2277.2009.00927.x. [DOI] [PubMed] [Google Scholar]
- 99.Fulop T, le Page A, Fortin C, et al. Cellular signaling in the aging immune system. Current Opinion in Immunology. 2014;29:105–111. doi: 10.1016/j.coi.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 100.Agrawal A, Agrawal S, Gupta S. Dendritic cells in human aging. Experimental Gerontology. 2007;42(5):421–426. doi: 10.1016/j.exger.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Serra JA, Fernandez-Gutierrez B, Hernandez-Garcia C, et al. Early T cell activation in elderly humans. Age and Ageing. 1996;25(6):470–478. doi: 10.1093/ageing/25.6.470. [DOI] [PubMed] [Google Scholar]
- 102.Baylis D, Bartlett DB, Patel HP, Roberts HC. Understanding how we age: insights into inflammaging. Longevity and Healthspan. 2013;2(1, article 8) doi: 10.1186/2046-2395-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ellis LZ, Liu W, Luo Y, et al. Green tea polyphenol epigallocatechin-3-gallate suppresses melanoma growth by inhibiting inflammasome and IL-1β secretion. Biochemical and Biophysical Research Communications. 2011;414(3):551–556. doi: 10.1016/j.bbrc.2011.09.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tsai P-Y, Ka S-M, Chang J-M, et al. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radical Biology and Medicine. 2011;51(3):744–754. doi: 10.1016/j.freeradbiomed.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 105.Li W, Zhu S, Li J, et al. EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages. Biochemical Pharmacology. 2011;81(9):1152–1163. doi: 10.1016/j.bcp.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]