

Abstract
Recent genome-wide studies have revealed that the majority of the mouse genome is transcribed as non-coding RNAs (ncRNAs) and growing evidence supports the importance of ncRNAs in regulating gene expression and epigenetic processes. However, the low efficiency of conventional gene targeting strategies has hindered the functional study of ncRNAs in vivo, particularly in generating large fragment deletions of long non-coding RNAs (lncRNAs) with multiple expression variants. The bacterial clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated 9 (Cas9) system has recently been applied as an efficient tool for engineering site-specific mutations of protein-coding genes in the genome. In this study, we explored the potential of using the CRISPR/Cas9 system to generate large genomic deletions of lncRNAs in mice. We developed an efficient one-step strategy to target the maternally expressed lncRNA, Rian, on chromosome 12 in mice. We showed that paired sgRNAs can precisely generate large deletions up to 23kb and the deletion efficiency can be further improved up to 33% by combining multiple sgRNAs. The deletion successfully abolished the expression of Rian from the maternally inherited allele, validating the biological relevance of the mutations in studying an imprinted locus. Mutation of Rian has differential effects on expression of nearby genes in different somatic tissues. Taken together, we have established a robust one-step method to engineer large deletions to knockout lncRNA genes with the CRISPR/Cas9 system. Our work will facilitate future functional studies of other lncRNAs in vivo.
Keywords: CRISPR/Cas9, large fragment deletion, Rian, imprinting, lncRNA
Introduction
Non-coding RNA has been recently discovered as a new class of factors which regulate gene expression in most complex organisms.1 They do not code for proteins, but their role in multiple biological processes has been increasingly recognized. Some evidence suggests that the number of ncRNAs may even exceed that of protein coding genes.2,3 They can be broadly classified according to transcript size. LncRNAs are defined as transcripts of more than 200 nucleotides in length which have no protein coding function.4 While small ncRNA species like miRNA, siRNA, piRNA, were shown to predominantly inhibit target gene expression by complementary base pairing,5 much less is known about the role of lncRNAs. Although the functions of some lncRNAs such as Xist6,7 have been well characterized, research on the majority of lncRNAs remains scarce due to a lack of efficient genome editing tools for deletion of large genomic regions. Recently, successful deletion of malat1, a lncRNA in zebrafish, using the TALEN system8 was reported, while the CRISPR/Cas9 system is seeing increasing use for gene editing. In this study, we aimed to explore the potential of the CRIPSR/Cas9 system for engineering large genomic deletions of lncRNAs.
The CRISPR system is a naturally occurring adaptive immune defense mechanism against foreign viruses or plasmids in bacteria.9 A short fragment of the invading virus is integrated into the cas operon and is transcribed in conjunction with another non-coding RNAs to guide the nuclease protein Cas9 to cleave foreign viral DNAs upon reinfection. Currently, three types of CRISPR systems in different bacteria have been described and the type II system from Streptococcus pyogenes has recently been modified as a genome engineering system.9,10 The Cas9 nuclease can be directed to generate site-specific DNA cleavage in the genome by an optimized complementary sgRNA. With its simplicity of manipulation and high efficiency, the CRISPR/Cas9 technology has achieved numerous successes as a robust genome engineering tool in various species.11-14 It has been used for generating site mutations,11 conditional knockout and knock-in alleles,12,13 multiple-gene knockouts,14,15 and large-scale genome modification.16 Here, we describe use of the CRISPR/Cas9 system and paired sgRNAs to generate large fragment deletions of up to 23kb of the maternally expressed lncRNA gene, Rian, in mice. We show that the deletions are precise and heritable. We also demonstrate that the use of multiple sgRNAs further increases deletion efficiency in mice.
Results
Precise deletion of a large genomic fragment using dual sgRNAs
Rian is a 57.8kb long noncoding RNA gene with alternative splice forms generated during transcript maturation.17 The spatial and temporal expression of different transcript variants have not been studied in the adult mouse.17,18 To understand the expression pattern of the various Rian transcripts, we performed quantitative RT-PCR (Q-PCR) to characterize the expression of Rian in multiple somatic tissues in mice. We designed 3 pairs of primers, named Rian-Pr-1, 2 and 3, whose amplicons include the different transcript variants of Rian (Fig S1A; Table S1). Total RNA was extracted from 11 tissues of wild-type C57BL6/J mice for Q-PCR assay. Q-PCR results showed high expression of Rian in adult brain, ovary, and heart (Fig. S1B).
Conventional gene targeting by homologous recombination is neither convenient nor efficient for the generation of large fragment deletions. These limitations represent the major obstacle to generating a Rian knockout mouse model in which all the transcript variants would be inactivated. CRISPR/Cas9 technology has been recently proven to be a robust genome engineering system. Therefore, we tested the use of the CRISPR/Cas9 system with two sgRNAs, N-sgRNA1 and C-sgRNA1, to induce a 23kb deletion in the Rian gene (Fig. 1A). The region for deletion was chosen to avoid inadvertent knockout of three miRNA genes located at the N-terminal region of the Rian gene, miR-1188, miR-370 and miR-341, which could complicate phenotype analysis. Cas9 and sgRNA mRNAs were transcribed in vitro and co-injected into one-cell stage mouse embryos. The dosage and number of injected embryos are shown in Table 1. In total, 25 pups were obtained from 190 transferred embryos. Primers Rian-F1 and Rian-R2 (Fig. 1A; Table S1) were designed to validate the predicted large fragment deletions. The PCR results indicated that 4 of the 25 founders (4/25, 16%) harbored the desired large fragment deletion (Fig. 1B; Table 2). PCR products were cloned into a TA cloning vector and sequenced for further confirmation (Fig. 1C). All 4 founders showed deletion of 23 kb between the two targeting sites, with founder #20 having an extra 81bp insertion complementary to the sequence downstream to N-sgRNA1. PCR and T7EN1 assays were performed to detect genome modifications at the target sites in other founders (Fig. S2A). We designed one pair of PCR primers (F1/R1 and F3/R3 respectively) for each of the two N-sgRNA targeting sites (Fig. 1A). Only one pair of primers (F2/R2) was used for two C-sgRNAs as the two sites are very close (Fig. 1A). In most founders, we detected gene modifications in at least one of the two targeting sites (Fig. S2A). Some founders had small deletions or insertions, such as #6, #7, #15 and #16 (Fig. S2A). From these, we demonstrated successful generation of precise large genomic decisions in the Rian locus in mice with the CRISPR/Cas9 system and dual sgRNAs.

Figure 1. Dual sgRNA:Cas9-mediated Rian gene mutations. (A) Schematic diagram of the 4 sgRNA target sites at the Rian gene locus. Rian exons are indicated by black rectangles. SgRNA target sites are indicated by red rectangles and the sequences are highlighted in red. PAM sites are underlined and highlighted in green. N-sgRNA1 is located between exon 7 and 8, N-sgRNA2 is on the exon 8, and the C-sgRNAs are beyond the last exon (Exon 21). F1/2/3 and R1/2/3 represent primer pairs used for PCR and the sequences can be found in Table S1. The scale bar represents 4 kb. (B) Demonstration of 23kb deletions in the Rian gene in founder animals from co-injections of Cas9 and dual sgRNAs. Four of the 25 founders showed a 23kb deletion in the Rian gene. The primers used were Rian-F1 and R2. M, DNA marker. WT, wild-type control. (C) Sequences of mutant alleles present in the 4 mutant founder animals. The PAM sites are underlined and highlighted in green; the target sequences are red; the mutations are in blue, lower case; deletions (-), insertions (+). WT, wild-type control.
Table 1. Summary of embryo injections.
| Injection mixture | Embryos injected | Embryos transferred | No. Recipients | Pups Born | |
|---|---|---|---|---|---|
| 1st | 20 ng/μl Cas9 mRNA. | 260 | 190 | 7 | #1-#25 |
| N-sg1,C-sg1, 5 ng/μl each. | |||||
| 2nd | 20 ng/μl Cas9 mRNA. | 230 | 121 | 5 | #26-#34 |
| 4 sgRNAs, 5 ng/μl each. |
Table 2. Summary of the pups with 23kb deletion and their mutation transmission.
| Total No.Pups | Pups with 23kb deletion | mutant transmission | |||
|---|---|---|---|---|---|
| No. (%) | Serial No. | No. (%) | Serial No. | ||
| 2 sgRNAs | 25 | 4 (16%) | #10, #17, #20, #22 | 1 (25%) | #22 |
| 4 sgRNAs | 9 | 3 (33.3%) | #29, #31, #34 | 3 (100%) | #29, #31, #34 |
Increased targeting efficiency using multiple sgRNAs
To further improve the system, we tested whether the use of multiple sgRNAs could increase the efficiency of generating large genomic deletions and we designed two more sgRNAs (N-sgRNA2 and C-sgRNA2) (Fig. 1A) for this purpose. The 4 sgRNAs and Cas9 mRNAs were transcribed and co-injected as described. We obtained 9 pups from 121 transferred embryos (Table 1) and 3 of them had large fragment deletions (3/9, 33%) (Fig. 2A). Sequencing results confirmed the presence of the desired deletions similar to the first 4 founders generated by dual sgRNA (Fig. 2B). PCR and T7EN1 assay were also performed to detect gene modifications from N-sgRNA1/2 and C-sgRNAs. We found that every founder had a gene modification in at least one targeting site (Fig. S2B). We designed another primer (Rian-R1; Table S1) residing in the deleted sequence and used it with Rian-F1 to test whether the 7 mutant founders were heterozygous or homozygous for the 23kb deletion. The result showed that all 7 founders were heterozygous (Fig. S2C). In conclusion, we generated mice heterozygous for a 23kb deletion in the Rian locus using CRISPR/Cas9 with paired sgRNAs. Furthermore, we doubled the deletion efficiency from 16% to 33.3% through use of multiple sgRNAs (Table 2).
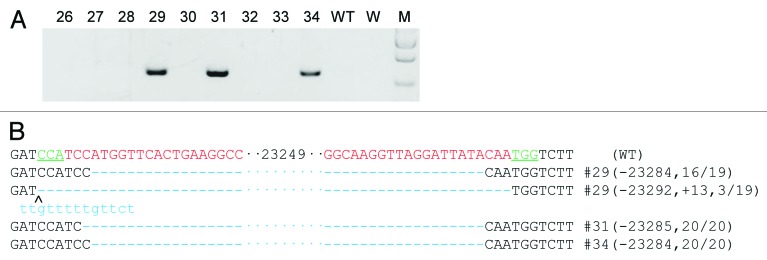
Figure 2. Four sgRNA:Cas9-mediated Rian gene modification (A) Demonstration of 23kb deletions in the Rian gene in founder animals from co-injection of Cas9 and four sgRNAs. 23kb deletions were present in three of the nine founders. Primers used were Rian-F1 and R2. WT, wild-type control. W, water. M, DNA marker. (B) Sequences of mutant alleles present in the 3 mutant founder animals. The PAM sites are underlined and highlighted in green; the target sequences are in red; the mutations are in blue, lower case; deletions (-), insertions (+). WT, wild-type control.
Heritability of large fragment deletions
We successfully generated 7 founder animals with 23kb genomic deletions. Four of them (#17, #20, #22, #34) were males and the other 3 (#10, #29, #31) were females. To study whether the deletions were heritable, the 7 founders were mated with wild-type C57BL6/J. Litters from F1 animals were genotyped (Fig. 3A). The results showed that 4 of them (#22, #29, #31, #34) passed the mutation to their offspring, while the other 3 (#10, #17, #20) did not (Table 2; Table S2), demonstrating the large fragment deletions are inheritable. Noticeably, all 3 founders with no transmission of the mutation were products of injections with dual sgRNAs. In contrast, all 3 mutant founders derived from co-injection of Cas9 and four sgRNAs transmitted the deletion to their offspring (Table 2; Table S2), which suggests the use of multiple sgRNAs could allow gene targeting events to take place more efficiently after injection, which would produce more mutant cells, therefore improve the chance of mutant cells contributing to the development of the genital ridge, thus increasing the efficiency of transmitting the mutations from the founders to the F1 offspring accordingly.

Figure 3. The inheritance of Rian mutations and the reduction of expression of Rian transcript variants from the maternally inherited mutant allele. (A) Transmission of deletion alleles to offspring of founder animals. PCR results showed that four (#22, #29, #31, and #34) of the 7 founders transmitted mutations to their progeny, as also shown in Table 2 and Table S2. Primers used were Rian-F1 and R2 (Table S1). M, DNA marker. PC, positive founder genome control. WT, wild-type control. (B) Q-PCR analysis of the expression of Rian transcript variants in three organs from litters with mutant Rian alleles of either maternal or paternal origin. The founder mice were mated with wild-type C57BL/6J. Heterozygote Rian mutant offspring from the crosses between male wild-type C57BL/6J and female founders (#29 and #31) are named Rian+/−(ma) while mutant offspring from female wild-type C57BL/6J and male founders (#22 and #34) are named Rian+/−(pa). The three paired primer pairs (Pr-1, Pr-2 and Pr-3) used detected different Rian transcript variants. The results showed that transcripts from the Rian locus were reduced significantly in Rian+/−(ma) mice, while no reduction was observed in Rian+/−(pa) mice. Pr, primers. WT, wild-type. Rian+/−(ma), heterozygous Rian mutant mice with a mutant allele of maternal origin. Rian+/−(pa), heterozygous Rian mutant mice with a mutant allele of paternal origin.
Since previous studies have identified the potential to generate off-target mutations of the CRISPR/Cas9 system,19,20 we set out to look for off-target effects of our strategy. We first identified candidate off-target sites for each of the 4 sgRNAs used in this study using the Optimized CRISPR Design web tool (http://crispr.mit.edu/). The top 5 potential off-target sites for each sgRNA were selected for T7EN1 assays (Tables S1 and S3). The three founders (#29, #31 and #34) with mutations generated by multiple sgRNAs were analyzed as this approach is more likely to induce off-target effects compared with dual sgRNA-induced targeting.21 We did not find off-target mutations at any of the sites tested (Fig. S3).
Decrease in Rian gene expression specifically in Rian+/−(ma) mice
After successfully generating a Rian knockout mouse model, we proceeded to validate the effect of the deletion on gene expression and its relevance in modeling imprinting. As Rian is a maternally expressed gene, the effect on Rian expression will depend on the parental origin of the mutant allele inherited by offspring.22 The heterozygous offspring from wild-type C57BL/6J and founder mice with a mutant allele of maternal origin (#29, #31) were denoted as Rian+/− (ma), while those with knockout alleles of paternal origin (#22, #34) were designated as Rian+/− (pa). Having already shown that Rian is highly expressed in brain, ovary and heart (Fig. S1B), we measured expression in these three tissues from mutant mice. Q-PCR results showed the expression level of Rian was minimal in Rian+/− (ma) mice, while in Rian+/− (pa) mice, the expression level was similar to that of wild-type mice (Fig. 3B). This result indicated that the CRISPR/Cas9-mediated genomic deletion does not interfere with expression of the non-imprinted allele. Notably, the deletion not only affected expression of the Rian transcripts from exons covered or partially covered by the deletion, but also affected transcriptional activity of the sequence upstream of the deleted fragment. Amplicons detected by Rian-Pr-3 do not lie within the deleted region (Fig. S1A), however their expression level decreased as well (Fig. 3B). We designed primers to measure the expression of the other four Rian variants (Rian-002, 003, 011, 012) not covered by the deleted region (Table S1). Interestingly, their expression levels were also significantly decreased (Fig. S4), while the expression of three miRNAs in the N-terminal of region of the Rian gene did not change in Rian+/− (ma) mice (Fig. S5).
Increased expression of genes adjacent to Rian in Rian+/−(ma) mice
One of the known functions of lncRNAs is to regulate the transcription of target genes in the genome,23 but the molecular mechanisms involved are often diverse and complex. LncRNAs can regulate transcription of nearby genes either in cis or in trans. Some lncRNAs act through epigenetic pathways, while others interact directly with RNA polymerases or transcription factors.24 To understand the effect of deleting the Rian locus on neighboring genes, we examined the transcription of 6 nearby genes (Wdr25, Begain, Dlk1, Meg3, Mirg and Dio3) in both Rian+/− (ma) and Rian+/− (pa) mice by Q-PCR (Fig. 4A; Table S1). In Rian+/− (ma) mice we found that expression levels of Dlk1 and Mirg were increased in brain, and expression levels of Mirg, Meg3 and Dio3 were increased in ovary (Fig. 4B). We did not observe changes in expression of neighboring genes in heart (data not shown). In contrast, the expression of these genes which flank Rian did not change in both brain and ovary in Rian+/−(pa) mice, similar to our observation that expression of Rian remains unchanged in Rian+/−(pa) mice (Fig. 3B). These results suggest Rian selectively regulates nearby genes in different tissues. The selective regulation of nearby genes in different tissues may indicate a tissue-specific function of Rian. It would be interesting to study the molecular mechanism for Rian-mediated regulation of neighboring genes and its role in the development of different somatic tissues.

Figure 4. Mutation of the Rian locus affects expression of nearby genes specifically in Rian+/−(ma) mice. (A) Relative locations of six genes around Rian on mouse chromosome 12. The sharp corner shows the direction of transcription. The scale bar represents 100 kb. (B) The expressions of six genes flanking Rian was assayed by Q-PCR in brain and ovary from heterozygous mice. In Rian+/−(ma) mice, expression of Dlk1 and Mirg increased in brain and Meg3, Mirg, and Dio3 increased in ovary. In Rian+/−(pa) mice, expression of genes adjacent to Rian showed minimal changes.
Taken together, our work establishes an efficient approach for the generation of precisely defined large genomic deletions with the CRISPR/Cas9 system. We demonstrate that the use of multiple sgRNA pairs increases the targeting efficiency. Our study provides researchers with a robust and cost-effective tool to study the function of ncRNAs, especially imprinted lncRNAs, in vivo. The method described in this work may also be applicable to other species.
Materials and Methods
DNA constructs
The pST1374-Cas9-N-NLS-flag-linker plasmid(Addgene ID 44758) used to express Cas9 protein, was described previously.25 The Rian gene sequence was downloaded from the UCSC Genome Browser website (http://genome.ucsc.edu/) (Mouse July 2007 (NCBI37/mm9) Assembly). SgRNA oligos were synthesized and annealed to the pUC57-sgRNA construct as described.15
In vitro transcription
In vitro transcription was performed as described previously.15 For 2 sgRNA and 4 sgRNA co-injection experiments, 2 μg of each vector were mixed and digested together following the procedures as described.15
Cas9/sgRNA co-injection of one-cell embryos
The Cas9/sgRNA co-injection method was described previously.15,25 CBA and C57BL6/J mice were mated to produce the hybrid strain B6CBAF1. Zygotes obtained by mating B6CBAF1 males with superovulated B6CBAF1 females were injected with the Cas9 mRNA and sgRNA mixtures described in Table 1 and then transferred to pseudopregnant B6CBAF1 females.
T7EN1 Cleavage and Sequencing
Mouse tail tips (2–3mm) were digested in lysis buffer (10mM TRIS-HCl, 0.4 M NaCl, 2mM EDTA, 1% SDS, and 100μg/ml Proteinase K) as described previously.25 Lysates were treated with phenol-chloroform and supernatants were mixed with 2 volumes of ethanol to extract genomic DNA. PrimerSTAR HS DNA polymerase (Takara, DR010A) was used to amplify target sites. PCR products were denatured and annealed in NEB buffer 2 before addition of T7 endonuclease 1. The T7EN1-digested products were separated on a 2.5% agarose gel.
PCR products from mutant founders were purified using a PCR cleanup kit (Axygen, APPCR-50) and cloned into a T vector (Takara, D103A). For each sample, at least 17 clones were picked and sequenced using the M13–47 primer. Primers are listed in Table S1.
Real-time and quantitative PCR
Mouse tissues were disrupted in TRIzol (Life Tech, 15596018) with a homogenizer. RNA was extracted following the manufacturer’s instructions (Life Tech). cDNAs were made with the RevertAid First Strand cDNA Synthesis Kit (Thermo, K1622) with Oligo(dT)18 and random primer. Quantitative PCR was performed with SYBR Premix Ex Taq (Takara, DDR420A).
A stem-loop strategy was used to detect the expression of miRNAs.26 The stem-loop RT primers specific to each miRNA were used for reverse transcription. The cDNA products were used for Q-PCR with miRNA specific forward primers and a universal reverse (UR) primer which binds to the 3′ portion of the stem-loop RT primer. Primers are listed in Table S1.
Off-target assay
The potential off-target sites for each sgRNA were analyzed by Optimized CRISPR Design (http://crispr.mit.edu/). The top 5 potential off-target sites for each sgRNA were selected for T7EN1 assay. PCR primers are shown in Table S1.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Ethics Statement
All mice used in this project were housed in an AAALAC (Assessment and Accreditation of Laboratory Animal Care)-accredited SPF animal facility at the Model Animal Research Center, Nanjing University, China. The animal protocols are approved by the IACUC (Institutional Animal Care and Use Committee) of the Model Animal Research Center.
Author Contributions
J.H., J.Z., X.H., and W.Z. designed the experiments and analyzed the data, J.H., J.Z., L.C., B.S., J.Z., B.H., and Y.D. performed experiments, W.Z. and J.H. wrote the manuscript. W.Z., X.H., and P.H.T. revised the manuscript. X.H. and W.Z. supervised the project.
Acknowledgments
We thank all members of Huang Lab for technical advice and helpful discussions. This work was supported by the National Natural Science Foundation of China (31171377).
Glossary
Abbreviations:
- CRISPR
clustered regularly interspaced short palindromic repeats
- Cas9
CRISPR associated 9
- lncRNA
long non-coding RNA
- miRNA
microRNA
- ncRNA
non-coding RNA
- Q-PCR
quantitative reverse transcription polymerase chain reaction
- sgRNA
single guide RNA
- siRNA
small interfering RNA
- TALEN
transcription activator-like effector nuclease
- piRNA
Piwi-interacting RNA
- T7EN1
T7 endonuclease I
References
- 1.Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14:699–712. doi: 10.1038/nrm3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152:1308–23. doi: 10.1016/j.cell.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 3.Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–41. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brockdorff N, Ashworth A, Kay GF, Cooper P, Smith S, McCabe VM, Norris DP, Penny GD, Patel D, Rastan S. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature. 1991;351:329–31. doi: 10.1038/351329a0. [DOI] [PubMed] [Google Scholar]
- 7.Sado T, Brockdorff N. Advances in understanding chromosome silencing by the long non-coding RNA Xist. Philos Trans R Soc Lond B Biol Sci. 2013;368:20110325. doi: 10.1098/rstb.2011.0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Luo D, Zhao H, Zhu Z, Hu W, Cheng CH. Inheritable and precise large genomic deletions of non-coding RNA genes in zebrafish using TALENs. PLoS One. 2013;8:e76387. doi: 10.1371/journal.pone.0076387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–8. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 10.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–9. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Auer TO, Duroure K, De Cian A, Concordet JP, Del Bene F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 2014;24:142–53. doi: 10.1101/gr.161638.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–8. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou J, Shen B, Zhang W, Wang J, Yang J, Chen L, Zhang N, Zhu K, Xu J, Hu B, et al. One-step generation of different immunodeficient mice with multiple gene modifications by CRISPR/Cas9 mediated genome engineering. Int J Biochem Cell Biol. 2014;46:49–55. doi: 10.1016/j.biocel.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 16.Fujii W, Kawasaki K, Sugiura K, Naito K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 2013;41:e187. doi: 10.1093/nar/gkt772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu T, He H, Han Z, Zeng T, Huang Z, Liu Q, Gu N, Chen Y, Sugimoto K, Jiang H, et al. Expression of macro non-coding RNAs Meg8 and Irm in mouse embryonic development. Acta Histochem. 2012;114:392–9. doi: 10.1016/j.acthis.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 18.Gu T, He H, Xing Y, Liu Q, Gu N, Kenkichi S, Jiang H, Wu Q. Expression of non-coding RNA AB063319 derived from Rian gene during mouse development. J Mol Histol. 2011;42:105–12. doi: 10.1007/s10735-011-9312-z. [DOI] [PubMed] [Google Scholar]
- 19.Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–43. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–41. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou J, Wang J, Shen B, Chen L, Su Y, Yang J, Zhang W, Tian X, Huang X. Dual sgRNAs facilitate CRISPR/Cas9-mediated mouse genome targeting. FEBS J. 2014;281:1717–25. doi: 10.1111/febs.12735. [DOI] [PubMed] [Google Scholar]
- 22.Lawson HA, Cheverud JM, Wolf JB. Genomic imprinting and parent-of-origin effects on complex traits. Nat Rev Genet. 2013;14:609–17. doi: 10.1038/nrg3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell. 2011;145:178–81. doi: 10.1016/j.cell.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 24.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–46. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen B, Zhang J, Wu H, Wang J, Ma K, Li Z, Zhang X, Zhang P, Huang X. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013;23:720–3. doi: 10.1038/cr.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.