

Abstract
Production of pathogenic Abs contributes to disease progression in many autoimmune disorders. The immunosuppressant agent mycophenolic acid (MPA) has shown clinical efficacy for patients with autoimmunity. The goal of these studies was to elucidate the mechanisms of action of MPA on B cells isolated from healthy individuals and autoimmune patients. In this study, we show that MPA significantly inhibited both proliferation and differentiation of primary human B cells stimulated under various conditions. Importantly, MPA did not globally suppress B cell responsiveness or simply induce cell death, but rather selectively inhibited early activation events and arrested cells in the G0/G1 phase of the cell cycle. Furthermore, MPA blocked expansion of both naive and memory B cells and prevented plasma cell (PC) differentiation and Ab production from healthy controls and individuals with rheumatoid arthritis. Finally, whereas MPA potently suppressed Ig secretion from activated primary B cells, terminally differentiated PCs were not susceptible to inhibition by MPA. The target of MPA, IMPDH2, was found to be downregulated in PCs, likely explaining the resistance of these cells to MPA. These results suggest that MPA provides benefit in settings of autoimmunity by directly preventing activation and PC differentiation of B cells; however, MPA is unlikely to impact autoantibody production by preexisting, long-lived PCs.
Mycophenolic acid (MPA; CellCept) is an immunosuppressant agent used primarily in transplantation to prevent allograft rejection and as an alternative therapy for several autoimmune disorders. MPA selectively inhibits inosine 5′-monophosphate dehydrogenase (IMPDH), the rate-limiting enzyme in the de novo pathway of purine synthesis. Two isoforms of IMPDH have been described. Whereas IMPDH1 is widely expressed, IMPDH2 is more abundant in activated lymphocytes and is five times more susceptible than IMPDH1 to inhibition by MPA (1–5). Inhibition of IMPDH2 by MPA leads to depletion of guanosine nucleotides, which blocks DNA synthesis and cell division (1, 6). Lymphocytes rely exclusively on the de novo pathway to generate GTP, whereas other cell types use additional, alternative pathways; therefore, MPA primarily targets lymphocytes.
MPA significantly blunts the proliferative and cytokine response of human lymphocytes. In T cells, MPA inhibits expansion to the mitogen PHA (7, 8). MPA also reduces adhesion of both CD4+ and CD8+ T cells to endothelial cells by impairing upregulation of VCAM-1, E-selectin, and P-selectin (9). Additionally, in a murine model of colitis, splenic T cells from mice pretreated with MPA have reduced expression of TNF-α, IFN-γ, and IL-4 after in vitro restimulation with anti-CD3 and anti-CD28 (10). Similarly, MPA targets B cells as MPA blocks proliferation and Ab secretion from both B cell hybridomas (11) and primary B cells (12). Ab formation in rats immunized with sheep RBCs is also inhibited by MPA (7).
Given its potent ability to inhibit lymphocyte division and effector functions, MPA is used clinically to treat a number of autoimmune disorders. In mouse models of lupus, MPA prolongs survival and reduces kidney disease and autoantibody levels in both MRL-lpr/lpr (13) and NZB×W mice (14, 15). Improved disease in MRL lpr/lpr mice after treatment with MPA is associated with decreased numbers of both splenic B cells and CD4+ T cells. Mechanism of action studies demonstrate that ex vivo treatment of splenocytes from MRL-lpr/lpr mice with MPA leads to a reduction of proliferation, cytokine secretion, and autoantibody production (16). In humans, clinical trials have shown that MPA has efficacy comparable with that of cyclophosphamide in treating lupus nephritis (17). In addition, treatment with MPA has benefit in patients with rheumatoid arthritis (RA), psoriasis, and Crohn’s disease (18–20). Individuals with a polymorphism in IMPDH2 are less responsive to MPA therapy. This polymorphism is present in 19% of healthy individuals and may explain differences in MPA efficacy among patients (21).
A hallmark of autoimmunity is the production of pathogenic autoantibodies. As such, it is important to determine whether current treatment options effectively target and inhibit Ab-secreting cells. One example is rituximab (Rituxan), an anti-CD20 mAb used therapeutically to deplete CD20+ B cells. Plasma cells (PCs), however, do not express CD20 and thus are not targeted by rituximab. Notably, reductions in circulating autoantibodies levels, which play an important role in autoimmune disease, are not consistently achieved after treatment with rituximab (22).
CellCept is currently being used as the standard of care for the treatment of lupus nephritis. Gaining a better understanding of the mechanistic action of MPA would provide a critical step toward improving the study design of future clinical trials. Specifically, several important aspects of the impact of MPA on B cell function are unknown. In particular, a number of key questions remain regarding the effect of MPA on Ab production. The primary goal of this study was to examine the effect of MPA on expansion, PC differentiation, and Ig secretion of primary human B cells from healthy individuals and autoimmune patients as well as to determine its direct effect on terminally differentiated PCs.
Materials and Methods
Cells
Human blood was collected from healthy donors after receiving informed consent. RA blood samples were obtained from the Warren G. Magnuson Clinical Center Blood Bank (Bethesda, MD) as approved by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health. All studies have been approved by either the MedImmune Human Biological Sample Review Committee or the Warren G. Magnuson Clinical Center Institutional Review Board. PBMCs were isolated from CPT tubes (BD Biosciences) after centrifugation. For most studies, B cells and CD4+ T cells were negatively selected using MACS cell separation reagents (Miltenyi Biotec). For isolation of B cells, the following kits were used from Miltenyi Biotec: total B cells, B cell Isolation Kit II, human (130-091-151); naive B cells, Naive B cell Isolation Kit II, human (130-091-150); preswitched memory B cells, IgM+ Memory B cell Isolation Kit, human (130-093-619); and postswitched memory B cells, IgG+ Memory B cell Isolation Kit, human (130-094-350). For total peripheral blood CD4+ T cells for the T/B cell coculture assays, total CD4+ T cells were negatively selected using the CD4+ T cell Isolation Kit II (130-091-155; Miltenyi Biotec). Routine purity for total B cells and CD4+ T cells was >97% and for the specific B cell subsets was >95%. The degree of purity and initial B cell phenotype were determined by flow cytometry for all experiments on day 0. Of note, total CD4+ T cells contained all subpopulations of CD4+ T cells, and total B cell preparations isolated using this method contain populations of both naive and memory B cells, but PCs were excluded due to expression of CD43, which is targeted by the Ab selection mixture of the MACS kit.
For Th17 cell differentiation assays, PBMCs were isolated from peripheral blood using CPT tubes (BD Biosciences). Total CD4+ T cells were isolated from the PBMCs using the EasySep CD4+ T cell Negative Selection Kit (cat. no. 19052; StemCell Technologies, Vancouver, BC, Canada) and the negative selection program on a RoboSep automated cell separator (StemCell Technologies). Total CCR6+ CD4+ T cells were then isolated using bispecific anti-CCR6 tetrameric Ab complexes generated using the EasySep Do-It-Yourself kit (cat. no. 18099; StemCell Technologies) and unconjugated anti-CCR6 (clone 11A9; Becton Dickinson). The separations were performed using a RoboSep (StemCell Technologies) and the positive selection program, collecting the positive (CD4+CCR6+) fraction. Routine purity for total CD4+ T cells was 98% and for CD4+ CCR6+ cells was 97%.
Tonsillar B cell subtypes were isolated by cell sorting performed on a BD FACSAria II high-speed cell sorter (BD Biosciences). Naive tonsillar B cells were defined as CD19+CD27−CD38int; memory B cells as CD19+ CD27+CD38int; and germinal center (GC) B cells as CD19+CD27+ CD38hi. Greater than 90% purity was achieved for all cell populations.
Culture conditions: B cells and activated T cells
T cell/B cell cocultures have been previously described (23). Briefly, 96-well flat-bottom wells were coated overnight at 37°C with anti-CD3 diluted in PBS (10 μg/ml; clone UCHT1; BD Biosciences) and washed before addition of cells. Alternatively, cells were stimulated with anti-CD3/anti-CD28–coated beads (Invitrogen), in a 5:1 T cell to bead ratio. Both ways of activating T cells gave comparable results. CD4+ T cells were treated with mitomycin C (30 μg/ml; Sigma Aldrich) for 30 min at 37°C then washed and rested in complete media at 37°C for an additional 30 min. Then, 1.0 × 105 mitomycin C-treated CD4+ T cells and 0.5 × 105 purified B cells (per well) were cocultured in a final volume of 200 μl on anti-CD3–coated plates or with anti-CD3/anti-CD28 beads. For determination of CD40L expression and other activation markers in the absence of B cells, T cells were stimulated alone as described earlier. Culture medium for all experiments was RPMI 1640 (Invitrogen) supplemented with 10% FCS, penicillin–streptomycin (100 U/ml penicillin, 100 μg/ml streptomycin), 2-mercaptoethanol (55 μM), L-glutamine (2 mM), and HEPES (5 mM). MPA (Sigma Aldrich) reconstituted in DMSO (<0.001% of the final volume; Sigma Aldrich) or DMSO-only control was added to cultures at various concentrations (0.1–3.0 μM).
Culture conditions: purified B cells
B cells (0.5 × 105 to 1.0 × 105) were cultured in 96-well round-bottom plates in a final volume of 150–200 μl complete medium. At initiation of culture, B cells were stimulated with either CpG-B (1 μg/ml; Invivogen) or a combination of IL-21 (33 ng/ml; Peprotech), anti-CD40 (0.1 μg/ml, goat IgG; R&D Systems), and anti-IgM F(ab′)2 (5.0 μg/ml; Jackson Immuno Research Laboratories). B cells were cultured for up to 21 d. For some experiments, B cells were CFSE-labeled prior to culture. Briefly, B cells were incubated with CFSE (10 μM; Invitrogen) diluted in serum-free media for 4 min with gentle rocking at room temperature. Cells were quenched with FBS and were washed extensively before culture (24). In most experiments, MPA or DMSO-only control was added to cultures at various concentrations (0.1–3.0 μM) at the initiation of culture. However, to determine the effect of MPA on established PCs, MPA was added after 7 d of B cell stimulation, and supernatants were removed after several days of cultures to assess Ig content as described later.
Culture conditions: purified T cells
T cell differentiation and staining of intracellular cytokines
Isolated CCR6+CD4+ cells were activated with anti-CD3/anti-CD28–coated beads (Invitrogen) for 3 d in X-VIVO-15 serum-free medium (Lonza, Walkersville, MD) supplemented with 2-mercaptoethanol, L-glutamine, and antibiotics. For expansion of Th17 cells from CD4+CCR6+ precursors, the following soluble mixture was used: IL-1β, IL-23, TGF-β, and anti–IFN-γ. Cytokines and Abs were used at the following concentrations: IL-1β (20 ng/ml; R&D Systems, Minneapolis, MN), IL-23 (50 ng/ml; R&D Systems), TGF-β (5 ng/ml; R&D Systems), anti–IFN-γ (clone AHC4032, 10 μg/ml; Invitrogen). After 3 d, MPA was added, and the cultures were continued overnight. The following day, the cells were restimulated with leukocyte activation cocktail (Becton Dickinson) for an additional 4–6 h. After fixation and permeabilization, cells were stained for intracellular Ags using the following Abs: anti–IL-2 (clone MQ1-17H12), anti–IL-17A (clone eBio64DEC17), and anti–IL-21 (clone 3A3-N2) (all from eBioscience). Data were acquired using a FACSCalibur (Becton Dickinson) and analyzed with FlowJo software (Tree Star).
Cytokine production
Supernatants from T cell/B cell cocultures activated with anti-CD3/anti-CD28 microbeads were harvested after 48 h, and cytokine production (IL-2, IL-4, IL-5, IL-10, IL-12, IL-13, and IFN-γ) was quantified using a multiplex cytokine assay (Meso Scale Discovery) according to the manufacturer’s instructions. IL-2, IL-17, and IL-21 expression were measured by flow cytometry as described earlier.
B cell expansion
B cell expansion was determined in cultures of purified B cells or in T cell/ B cell cocultures stimulated as described earlier. B cell expansion was quantified by measuring ATP levels on day 3–4 of culture using the Cell Titer-Glo Luminescent Assay (Promega) according to the manufacturer’s instructions. As CD4+ T cells were mitomycin C treated, increases in ATP units in coculture experiments reflected B cell expansion exclusively.
Flow cytometry
At a variety of time points, cells were harvested from culture wells and stained for 30 min at 4°C. The following combinations of Abs were used: from BD Pharmingen: anti-CD19 (PerCP–Cy5.5); anti-IgD (FITC or PE); anti-CD38 (PE or allophycocyanin, clone HB7); anti-CD27 (PE); from Invitrogen: anti-CD4 (FITC or PE); anti-CD69 (allophycocyanin); and anti-CD25 (PE, Beckman Coulter), anti-CD40 (PE, eBioscience), anti-CD40L (PE, BioLegend). 7-Aminoactinomycin D (7-AAD; BD Pharmingen) was used to determine cell viability. Cell cycle analysis was performed on ethanol-fixed cells using propidium iodide (50 μg/ml). Cells were analyzed on an LSR II flow cytometer (BD Biosciences) using FACSDiva software. All conditions were collected for the same time, thus the number of events displayed is reflective of relative cell number. However, total cell numbers were determined using AccuCount Particles (Spherotech).
Ig production
Secreted Ig was quantified by ELISA after stimulation of B cells as described previously (24). Briefly, 96-well flat-bottom plates were coated overnight at 4°C with either 5 μg/ml goat anti-human IgM or IgG diluted in PBS (Bethyl Laboratories). Plates were washed and blocked with 0.2% BSA in PBS. Supernatants were diluted and incubated on plates overnight. Bound Ig was detected with goat anti-human IgM− or goat anti-human IgG–alkaline phosphatase (0.2 μg/ml; Bethyl Laboratories) all diluted in blocking buffer. Plates were developed with SigmaFast p-Nitrophenyl phosphate Tablets (Sigma Aldrich), and specific absorbance was measured at 405 nm using a SpectraMax plate reader (Molecular Devices).
Apoptosis assay
Apoptosis of B cells stimulated either with CpG-B or with a combination of IL-21/anti-CD40/anti-IgM was assessed after various days of culture using the Caspase Glo 3/7 Assay (Promega).
IMPDH expression/real-time PCR
For analysis of predifferentiated PCs, 1 × 106 purified B cells were cultured with IL-21/anti-CD40/anti-IgM at the concentrations indicated above in 24-well plates in a final volume of 1.5 ml culture medium. RNA was isolated from total unfractionated B cells on days 0–3 of culture and from sort-purified PCs after 7–8 d of culture using an RNeasy Plus Mini Kit (Qiagen). Predifferentiated day 7–8 PCs were sorted based on high expression of CD27 and CD38; >95% purity was achieved. For RNA isolation of purified B cell subpopulations from the bone marrow, cells were isolated by cell sorting on a BD FACSAria II high-speed cell sorter (BD Biosciences). Total B cells from the bone marrow were defined as CD19+ CD20+ cells and PCs as CD19+CD27hiCD38hiCD138+. RNA was directly isolated as described above. All RNA was reverse transcribed with SuperScript III Reverse Transcriptase (Invitrogen) and random primers (Invitrogen). cDNA was analyzed for expression of IMPDH1, IMPDH2, and syndecan-1 by TaqMan PCR assay using a CFX96 Real-time PCR detection system (Bio-Rad). Probes used were as follows: IMPDH1, Assay ID Hs00265302_m1; IMPDH2, Assay ID Hs00168418_m1; CD138, Assay ID Hs00896423_m1 (Applied Biosystems).
Statistics
Mean of triplicate wells for each condition is shown. SD or SEM was calculated for each experiment as indicated in the legends to figures that accompany this article. Statistics were performed using a one-tailed, two-sample t test. Significant differences relative to vehicle-alone (DMSO) controls are noted by p value: *p < 0.05, **p < 0.01, ***p < 0.005.
Results
MPA inhibits T cell-induced B cell activation
Activation of B cells in vivo can be driven by interactions with activated T cells that express costimulatory molecules such as CD154 and produce B cell tropic cytokines. To mimic this process in vitro and to elucidate the specific mechanism of action of MPA, B cells were cocultured with anti-CD3/anti-CD28–stimulated, mitomycin C-treated CD4+ T cells. First, the effect of MPA on early activation events was determined. Primary (day 0) T cells and B cells were small resting cells, which expressed low densities of the activation markers CD25 and CD69. By day 3, anti-CD3/ anti-CD28 stimulation induced CD4+ T cells to increase in cell size and upregulate expression of CD25 and CD69 (Fig. 1A). Concomitant with T cell activation, B cells enlarged and upregulated CD25 and CD69 as well as expression of CD19 (Fig. 1B). MPA inhibited T cell blasting, especially at higher doses. Whereas an intermediate concentration of MPA (0.3 μM) had little effect on activation markers, higher concentrations (1.0 and 3.0 μM) inhibited upregulation of CD25 on CD4+ T cells (Fig. 1A). In B cells, MPA also reduced cell size as well as CD25 and CD19 expression in a dose-dependent manner (Fig. 1B). MPA more profoundly reduced cell size than CD25 expression on both T cells and B cells. In contrast to its effect on cell size and CD25 expression, MPA did not inhibit upregulation of CD69 in either cell population. CD69 upregulation in B cells, however, was impaired through blockade of the CD40–CD40L pathway with TRAM1 anti-CD40L mAb (data not shown and Ref. 23). MPA also did not significantly hamper CD40 upregulation in B cells even at high concentrations of 3.0 μM (Fig. 1C, 1D). The effect of MPA on CD25 and CD69 expression was similar for purified activated T cells in the absence of B cells (data not shown). Additionally, CD154 expression increased after activation by activated T cells and was slightly further increased in the presence of low concentrations of MPA; however, at higher concentrations, CD40L expression was reduced by ~40% (Fig. 1C, 1D).
FIGURE 1.

MPA selectively inhibits early activation events. A–D, B cells were cocultured with mitomycin C-treated, anti-CD3/anti-CD28–activated CD4+ T cells in the presence of DMSO control or a range of MPA concentrations as indicated. A and B, Cell size and surface expression of activation markers expressed by (A) CD4+ T cells or (B) CD19+ B cells were determined 72 h after coculture. Mean flourescence intensity is indicated in each histogram. Data are representative of one of six independent donors tested. C and D, CD154 (CD40L) expression was determined in purified mitomycin C-treated CD4+ T cell cultures after 48 h of stimulation with anti-CD3/anti-CD28 as described. CD40 expression was quantified on B cells after 48 h of coculture with activated T cells. C, Data from one representative donor shown as percentage of DMSO control. D, Representative histograms of CD40L and CD40 expression in the presence of 3 μm MPA (red histogram). **p < 0.01.
We next assessed the impact of MPA on early cytokine production in the T cell/B cell cocultures. Within 48 h of activation, IL-2, IL-5, IL-10, IL-13, IFN-γ, and to a lesser extent IL-4 could be detected in culture supernatant (Fig. 2). Addition of MPA significantly reduced production of IFN-γ and IL-10 as well as IL-5 and IL-13 (Fig. 2). Surprisingly, however, levels of IL-2 and IL-4 were unaffected by the presence of MPA (Fig. 2). These results demonstrate that MPA does not globally prevent cell activation but rather selectively exerts its effect on specific activation events.
FIGURE 2.

MPA differentially affects cytokine production. B cells were activated with anti-CD3/anti-CD28–stimulated T cells as described in Fig. 1, and DMSO control or MPA was added at the start of culture at the concentrations indicated. Cytokine levels in the supernatant were measured after 48 h of T cell/B cell coculture. Data shown represent the mean (±SD) of triplicate wells from one of three unique donors. *p < 0.05, **p < 0.01, ***p < 0.005.
The impact of MPA on T cell-driven B cell expansion and PC differentiation at later time points was determined. By day 4 of culture, activated T cells had induced B cells to greatly expand (Fig. 3A). However, as expected, mitomycin C treatment prevented the majority of T cell expansion (Fig. 3A). Moreover, in the T/B cell assay, very few T cells were found in day 7 cultures indicating, as expected, that the mitomycin C-treated T cells had not expanded in the presence of B cells (data not shown). In day 4 cultures, MPA significantly blunted the T cell-dependent B cell proliferative response in a dose-dependent manner (Fig. 3A).
FIGURE 3.
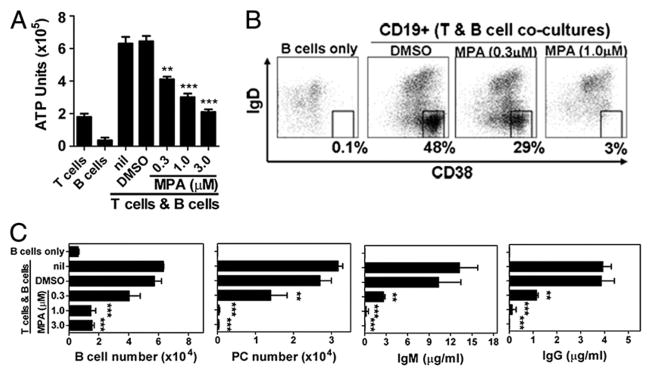
MPA inhibits T cell-dependent B cell expansion and PC differentiation. B cells were activated with anti-CD3/anti-CD28–stimulated T cells as described in Fig. 1, and DMSO control or MPA was added at the start of culture at the concentrations indicated. A, Cell expansion quantified by ATP units was determined on day 4. B, On day 7, B cell phenotype was defined by CD19+CD4− cells with percentage PCs indicated as CD19loIgD−CD38hi cells in black box. C, Total CD19+ B cell number, total CD19loIgD−CD38hi PC number, IgM, and IgG levels were all determined after 7 d of coculture. Data in A and C are mean (±SD) of triplicate T cell/B cell cocultures isolated from one of four independent donors; representative results are shown. **p < 0.01, ***p < 0.005.
By day 7, CD19loIgD−CD38hi PCs were noted in the cultures (Fig. 3B, 3C), and IgM and IgG were detected in culture supernatants (Fig. 3C). Addition of MPA blocked T cell-induced B cell expansion and PC differentiation as noted by significantly fewer total B cells and a loss of PCs in the presence of 1.0 μM MPA (Fig. 3B, 3C). Inhibition of PC differentiation by MPA was further evidenced by a dose-dependent loss of IgM and IgG (Fig. 3C). Maximal inhibition of PC differentiation and Ig production was reached with 1 μM MPA. However, in contrast to the minimal effect that 0.3 μM MPA had on cell size and CD25 upregulation, this concentration of MPA reduced Ig production by >70%.
IL-21 is known to be a potent stimulator of plasma cell differentiation and Ab production (25). Although IL-21 is present in these T/B cell cocultures and IL-21R–Fc consistently inhibits T cell-induced PC differentiation (23), the amount of IL-21 is limiting, and thus the effect of MPA on its expression is difficult to determine. To address if loss of PC differentiation in the presence of MPA is secondary to loss of IL-21 production, recombinant IL-21 (33 ng/ml) was added at the initiation of the T/B cell cocultures. Exogenous addition of IL-21 was not found to overcome the inhibition of PC differentiation induced by MPA (data not shown). To discern directly the effect of MPA on IL-21 production, primary CD4+ T cells were isolated from the blood and differentiated into Th17 cells. As shown in Supplemental Fig. 1, IL-21 and IL-17 were found to be greatly upregulated 4–6 h after restimulation of Th17 cells. Addition of MPA 24 h prior to the restimulation resulted in a dose-dependent inhibition of IL-21 expression in the activated Th17 cells. Although MPA significantly inhibited IL-21 even at low concentrations, it had a minimal effect at inhibiting IL-17 production (Supplemental Fig. 1). Consistent with data observed in the T/B cell cocultures, MPA also had little effect on IL-2 expression in Th17-restimulated cells (Supplemental Fig. 1). Importantly, these data further emphasize the differential impact of MPA on specific cytokines and its selective inhibition on both T and B cell activation.
MPA directly inhibits B cell expansion and PC differentiation
The ability of MPA specifically to target highly purified peripheral blood B cells was evaluated next. B cells were stimulated either with IL-21 and anti-CD40 mAb to mimic T cell-dependent responses (24) with BCR cross-linking or with the TLR ligand CpG-B to mimic T cell-independent responses. B cells costimulated with IL-21/anti-CD40/anti-IgM maximally activated the B cells with peak upregulation of CD19, CD25, and CD69 observed on day 2 of culture, and maximal cell blasting was detected on day 3 (Supplemental Fig. 2A and data not shown). Although similar in trend, CpG-B stimulation of B cells induced a more modest upregulation of CD19 and CD25 (Supplemental Fig. 2B). Importantly, under both stimulation conditions, MPA significantly blocked cell blasting as well as upregulation of CD19 and CD25 (Supplemental Fig. 2). Consistent with observations of T cell/ B cell cocultures, MPA did not affect upregulation of CD69 (Supplemental Fig. 2, right panel). In addition, these stimulation conditions also induced upregulation of IL-21R (day 3), which was greatly inhibited by MPA in a dose-dependent manner, suggesting a contributing mechanism by which MPA may downregulate B cells responses to IL-21 costimulation (data not shown). These data support a direct role for MPA as a selective inhibitor of early B cell activation events independent of T cell influences.
Stimulation of purified B cells with IL-21/anti-CD40/anti-IgM or CpG-B induced a quantitatively similar degree of B cell expansion as measured by ATP after 4 d of culture (Fig. 4A, 4B). However, as shown by CFSE dilution, B cells had a qualitatively different proliferative response to these two stimuli (Fig. 4C, 4D). By day 7, in the presence of IL-21/anti-CD40/anti-IgM, most cells underwent at least one division (86.33 ± 0.39% diluted CFSE) compared with CpG-B–stimulated cells, of which only about half were induced to divide (55 ± 0.57%). Further, in response to CpG-B stimulation, although a reduced percentage of B cells divided, those that did divide underwent a greater number of cell divisions (CpG-B stimulation: 19.7 ± 0.21% underwent more than three cell divisions; IL-21/anti-CD40/anti-IgM stimulation: 6.04 ± 0.39% underwent more than three cell divisions). Importantly, however, under both stimulation conditions, MPA significantly blunted the B cell proliferative response, as noted by both a dose-dependent reduction in the number of ATP units and a lack of CFSE dilution (Fig. 4A–D).
FIGURE 4.

Direct inhibition of primary human B cells by MPA. Peripheral blood B cells were labeled with CFSE and stimulated with either (A, C, E) IL-21, anti-CD40, and anti-IgM or (B, D, F) CpG-B in the presence of vehicle control alone (DMSO) or MPA at various concentrations as indicated. A and B, On day 4 of culture, B cell expansion was quantified by ATP. C and D, On day 7 after activation, CFSE dilution and CD38 expression were determined. CD19lo PCs are indicated in black box as CFSEdimCD38hi cells. Number indicates percentage PCs of total CD19+ B cell population. E and F, IgG and IgM content were also determined after 7 d. Data show mean (±SD) of one of three representative experiments. *p < 0.05, ***p < 0.005.
Although the proliferative responses were qualitatively different, both stimuli induced a similar degree of PC differentiation as noted by the percentage of CD38hi cells that had completely diluted CFSE (Fig. 4C, 4D). Moreover, by day 7, CpG-B induced robust production of IgM (Fig. 4F) and smaller amounts of IgA and IgG (~0.4–1 μg/ml, respectively, data not shown), whereas the combination of IL-21/anti-CD40/anti-IgM promoted class switching and secretion of IgG Abs (Fig. 4E) and ~1 μg/ml IgA (data not shown). IgM levels could not be measured under these conditions because of the addition of exogenous anti-IgM in the culture. Notably, MPA completely blocked PC differentiation as indicated by an absence of CD19loCFSEdimCD38hi PCs under both stimulation conditions (Fig. 4C, 4D, right panels). Additionally, IgM, IgG, and IgA production were completely inhibited in the presence of MPA in a dose-dependent manner (Fig. 4E, 4F and data not shown).
Peripheral blood and tonsillar B cells contain several B cell subsets including naive, preswitched and postswitched memory B cells and GC B cells (present in tonsil). These B cell sub-populations were isolated as described in Materials and Methods and activated with either CpG or a combination of IL-21/anti-CD40/anti-IgM either alone or cultured in the presence of increasing concentrations of MPA. Notably, all subpopulations of peripheral blood (Fig. 5A) and tonsillar (Fig. 5B) B cells examined were sensitive to inhibition by MPA as shown by a significant dose-dependent decrease of B cell expansion as measured by ATP units 3 d after activation. MPA inhibited GC B cell activation as well, although, as expected, the GC B cells died readily in culture even after stimulation (Fig. 5B). In data not shown, MPA also inhibited PC differentiation of the various B cell subsets examined in a dose-dependent manner at later time points.
FIGURE 5.
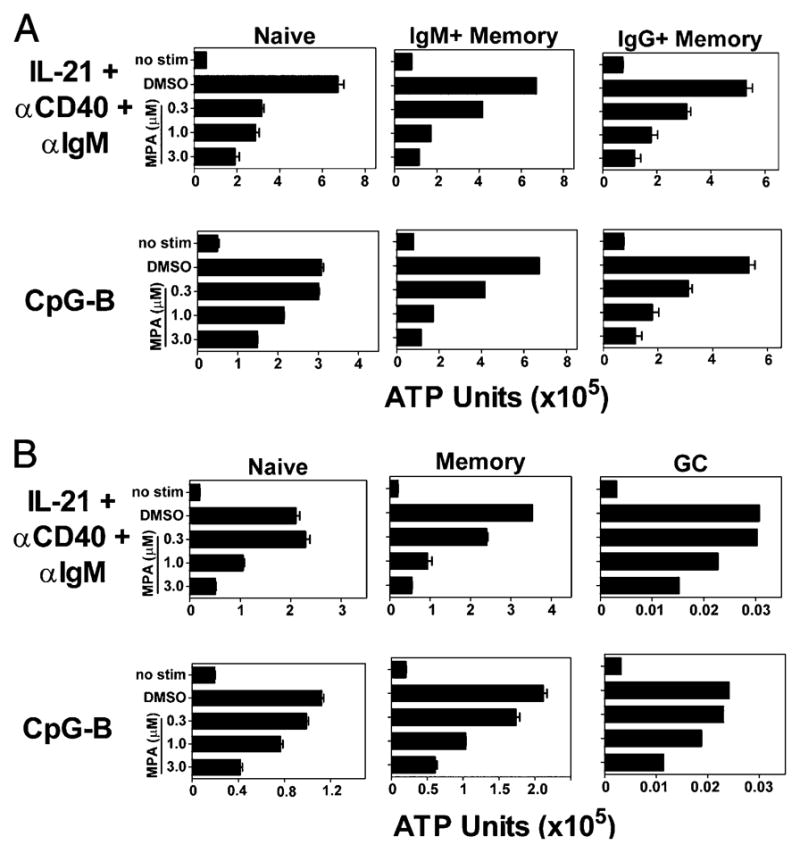
MPA inhibited all non-PC subpopulations of B cells isolated from both the peripheral blood and tonsil. A, Naive and preswitched and postswitched memory B cells were isolated from peripheral blood. Data represent three independent experiments. B, Naive, memory, and GC B cells were isolated from the tonsil. Cells were stimulated with either IL-21, anti-CD40, and anti-IgM or CpG-B in the presence of DMSO control or MPA at the indicated concentrations. ATP units were quantified on day 3. Data represent three independent experiments. Mean and SD of triplicate wells is shown where possible. For IgM+ memory B cells (A) and GC B cells (B), the average of duplicate wells is shown.
Inhibition of B cell activation and differentiation is not due to induction of cell death by MPA
To determine if the inhibitory effect of MPA on B cells was due to induction of cell death, cell viability in the presence of MPA was assessed. At early time points (24 h), before cell division was noted (data not shown), stimulation with IL-21/anti-CD40/anti-IgM induced a modest increase of apoptosis as measured by caspase 3/7 activity (Fig. 6A). Addition of MPA did not increase apoptosis in these cultures. Conversely, compared with unstimulated cells, stimulation of B cells with CpG-B led to a slight reduction of apoptosis that was unchanged in the presence of vehicle (DMSO) alone or low concentrations of MPA (0.3, 1.0 μM). At higher concentrations (3.0 μM), caspase activity in these cultures was comparable with what is observed with unstimulated cells (Fig. 6A). Cumulatively, these data demonstrate that MPA does not induce early apoptosis of primary B cells stimulated under these culture conditions.
FIGURE 6.
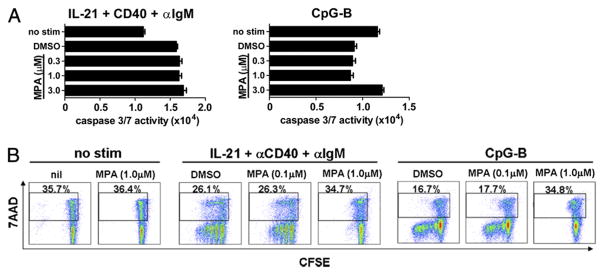
MPA treatment does not induce cell death. Purified B cells were stimulated with IL-21, anti-CD40, and anti-IgM or CpG-B as indicated in the presence of DMSO control or increasing amounts of MPA. A, Apoptosis was quantified by caspase 3/7 activity after 24 h of culture. Mean (±SD) is shown for one of three independent donors. B, Purified B cells were CFSE labeled and stimulated as indicated. CFSE dilution as well as cell death of resting (no stim) and stimulated cells was determined with the vital dye 7-AAD on day 4. Percentage of dead (7-AAD+) cells is indicated. Data represent two independent experiments with three unique donors tested.
To examine further the effect of MPA on cell survival, cell viability and proliferation were assessed at later time points by flow cytometry. A direct comparison of cell division as determined by CFSE dilution and cell death (as measured by incorporation of the vital dye 7-AAD) was performed (Fig. 6B). By day 4, in the absence of stimulation, approximately one-third of all B cells had undergone cell death (7-AAD+, Fig. 6B). Addition of MPA did not affect cell viability of unstimulated B cells (35.7% versus 36.4%). After 4 d of activation with either IL-21/anti-CD40/anti-IgM or with CpG-B, B cells were induced to undergo robust cell division (Fig. 6B). Importantly, addition of 1.0 μM MPA to stimulated cultures completely inhibited cell division and resulted in cell viability comparable with that observed in unstimulated cells (Fig. 6B). These data suggest that at the concentrations used, MPA did not directly or indirectly induce cell death, but instead prevented B cell activation and division.
G0/G1 cell cycle arrest in the presence of MPA
To define further the effect of MPA on cell cycle progression, purified B cells were activated with various stimuli, and DNA content was measured by propidium iodide staining. Compared with unstimulated cells, activation of B cells with either IL-21/anti-CD40/anti-IgM or CpG-B drove an increased percentage of B cells into the S and G2/M phases of the cell cycle (Fig. 7). Cell cycle progression into S phase peaked by day 3 and then started to decline after day 4 of culture (data not shown). On day 3, at the peak of cell cycle progression, consistent with CFSE dilution, fewer B cells stimulated with CpG-B entered S phase compared with those stimulated with IL-21 costimulation (Fig. 7). Under both stimulation conditions, however, in the presence of MPA there was a dose-dependent decrease in the percentage of cells that entered S phase and a corresponding increase in the percentage of cells that remained in G0/G1. At higher concentrations of MPA (1.0 or 3.0 μM), the cell cycle profile of B cells was comparable with that observed in the absence of stimulation (Fig. 7), indicating that MPA induced a G0/G1 cell cycle arrest.
FIGURE 7.
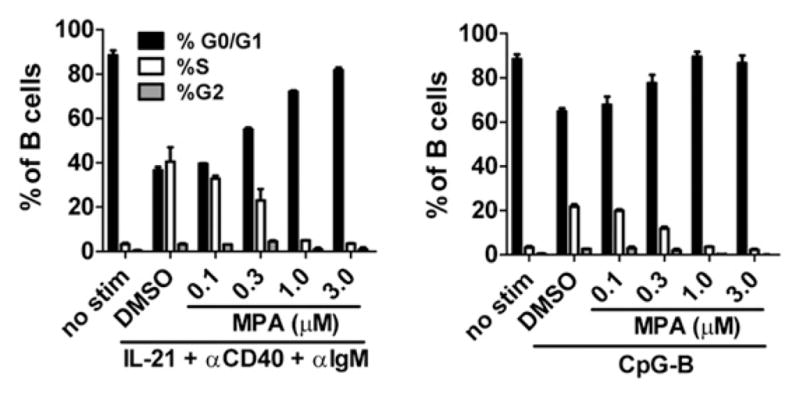
MPA induces G0/G1 cell cycle arrest. Purified B cells were stimulated with IL-21, anti-CD40, and anti-IgM or CpG-B as indicated. After 3 d of culture, cell cycle analysis was performed by propidium iodide staining. Percentage of cells in G0/G1, S, and G2/M phases of the cell cycle is indicated. Data represent the mean (±SEM) of three donors tested.
Terminally differentiated PCs are not susceptible to inhibition by MPA
Abs are generated in vivo by both newly activated/differentiated B cells as well as by long-lived Ig-secreting PCs. Thus, the effect of MPA on both primary and pre-established, terminally differentiated PCs was examined. To assess the ability of MPA to impact Ig production of pre-established PCs, purified B cells were stimulated as described earlier with either IL-21/anti-CD40/anti-IgM or CpG-B, and MPA (or control) was added on day 7, after a population of CD19loIgD−CD38hi Ab-secreting PCs had clearly been established by both stimulation conditions (Fig. 8A, insets). As noted above, when MPA was added at the initiation of culture, a complete block of IgM, IgG, and IgA production induced by CpG-B or IL-21 costimulation was noted (Fig. 8A, open squares and data not shown). Moreover, bortezomib (Velcade), a proteasome inhibitor that has been shown to deplete PCs in murine models of lupus (26), when added to pre-established PCs completely prevented further secretion of Ig as noted by a plateau of IgM, IgG, and IgA levels immediately after addition of the drug on day 7 (Fig. 8A, open circles and data not shown). Strikingly, however, addition of MPA to B cell cultures stimulated for 7 d with either CpG-B or IL-21/anti-CD40/anti-IgM had no impact on the ability of pre-established PCs to continue to secrete IgM, IgG, or IgA (Fig. 8A, triangles, and data not shown). Notably, when concentrations of MPA that were equivalent or higher than those achieved in clinical settings [up to 100 μM MPA (27–30)] were added to the day 7 cultures of pre-established PCs, no impact on Ig production was noted throughout the 21-d culture period (data not shown).
FIGURE 8.

Terminally differentiated PCs have reduced expression of IMPDH2 and are not susceptible to inhibition by MPA. A, Purified B cells were cultured and stimulated as indicated. DMSO control alone, MPA (1.0 μM), or bortezomib (Velcade, 100 ng/ml) was either added at initiation of culture (day 0) or after 7 d of culture. Supernatants were harvested on days 7, 10, 14, and 21, and Ig content was determined by ELISA. Inset indicates the percentage of CD19loIgD−CD38hi PCs in culture on day 7 at the time when DMSO, MPA, or bortezomib was added late. Mean (±SD) of one of three representative experiments is shown. B and C, Purified B cells were stimulated with IL-21, anti-CD40, and anti-IgM and cultured for the days (d) indicated. mRNA was isolated either from unfractionated B cell cultures on days 0, 1, 2, and 3 or from sorted PCs (CD27hiCD38hi) on day 7. B, Pre-sort and post-sort PC fractions on day 7 are shown. C, IMPDH1, IMPDH2, and syndecan-1 levels were quantified by RT-PCR. Fold change shown is relative to resting B cells (day 0). Data represent the mean ± SEM of three donors tested. D, Total B cells and PCs were sort purified from human bone marrow, and expression of IMPDH2, IMPDH1, and syndecan-1 was quantified by RT-PCR. Four unique donors are shown. **p < 0.01, ***p < 0.005.
To address the impact of MPA on primary PCs, CD27hiCD38hi PCs were directly sorted from human bone marrow. As PCs die very rapidly in vitro, the isolated PCs were directly sorted into ELISPOT plates that were incubated overnight in the presence of increasing concentrations of MPA (up to 100 μM). These sorted PCs produced IgM, IgG, and IgA in the 18-h assay. Consistent with the earlier findings from predifferentiated PCs, MPA had no impact on the ability of the primary bone marrow PCs to secrete Ig even in the presence of very high concentration of MPA (data not shown). Importantly, in concurrent (or parallel) overnight cultures, the same dilutions of MPA were able to inhibit B cell activation as noted by a reduced upregulation of CD19 and CD25 expression on total bone marrow CD19+CD20+ B cells activated with IL-21/anti-CD40/anti-IgM (data not shown). These data demonstrate that MPA is able to exert its inhibitory effect on primary bone marrow B cells, but not primary terminally differentiated PCs in short overnight cultures.
Terminally differentiated PCs express reduced levels of IMPDH2
The effect of MPA is mediated through direct inhibition of the enzyme IMPDH2. Thus, peripheral blood B cells were activated with a combination of IL-21/anti-CD40/anti-IgM, and the expression level of IMPDH was examined by either total B cells at various time points after activation or in the resultant PC population that emerged from these cultures after 7 or 8 d of stimulation (Fig. 8B). Stimulation with IL-21/anti-CD40/anti-IgM resulted in an approximate 10-fold increase in IMPDH2 mRNA expression in total B cells within 24 h compared with freshly isolated, non-stimulated B cells (Fig. 8C). IMPDH2 mRNA levels were further upregulated in total B cells by 48 h (25-fold increase), and began to decline by 72 h after stimulation. We noted a similar pattern of expression of IMPDH1; however, the induction of IMPDH2 mRNA was significantly greater than the upregulation of IMPDH1 mRNA (Fig. 8C). By day 7, a population of PCs emerged that were defined as CD19loIgD−CD27hiCD38hi, a portion of which expressed the PC Ag CD138 (syndecan-1) (data not shown and Ref. 23). These PCs were purified by cell sorting based on CD27 and CD38 expression (Fig. 8B), and mRNA from these cells was compared with that from resting primary B cells. As expected, differentiated PCs highly upregulated expression of CD138 mRNA (Fig. 8C). In contrast, IMPHD2 levels in sorted predifferentiated PCs were downregulated 2- to 4-fold below that noted in freshly isolated primary B cells and 50-fold below that of day 2-stimulated cells, suggesting that these PCs are not simply quiescent cells, but rather that IMPDH2 mRNA has actively been switched off in these cells (Fig. 8C). Importantly, IMPHD2 mRNA was also found to be expressed significantly less in primary terminally differentiated bone marrow CD19loCD20−CD27hiCD38hi CD138+ PCs compared with that in total bone marrow CD19+ CD20+ B cells, which do not contain the PC population (Fig. 8D). Although IMPDH1 was found to be reduced in primary bone marrow PCs compared with that in total bone marrow B cells, the differences were not as significant (Fig. 8D). These results suggest that through downregulation of IMPDH2, terminally differentiated PCs become refractory to inhibition by MPA.
MPA inhibits B cells isolated from RA patients
MPA has shown promise as an immunosuppressive agent for the treatment of autoimmune disorders such as systemic lupus erythematosus (SLE) and RA (17, 19). As such, it is important to determine if B cells isolated from patients with autoimmunity are directly susceptible to inhibition by MPA. Comparable with healthy controls, B cells isolated from patients with RA expanded robustly in response to either IL-21 costimulation (Fig. 9A) or CpG-B (Fig. 9B) as noted by a 7- to 10-fold increase in ATP units compared with unstimulated B cells on day 4. Importantly, MPA blocked the expansion of B cells from both healthy controls and patients with RA to a comparable degree. PC differentiation and Ig secretion were quantified after 7 d of culture with the above stimuli. Approximately equivalent numbers of CD19loIgD− CD38hi PCs emerged from B cells isolated from normal controls or patients with RA after stimulation with IL-21/anti-CD40/anti-IgM or CpG-B (Fig. 9), and production of Ig was also similar. Notably, PC differentiation and Ab production of B cells isolated from several normal donors or patients with RA were completely blocked in the presence of MPA (Fig. 9).
FIGURE 9.

B cells isolated from patients with RA are susceptible to inhibition by MPA. B cells were purified from healthy control donors (black bars) or from patients with RA (white bars). B cells were stimulated either with (A) IL-21, anti-CD40, and anti-IgM or (B) CpG-B in the presence of DMSO-vehicle alone or increasing concentrations of MPA. B cell expansion was quantified on day 4 by ATP. On day 7, PC number was quantified from CD19loIgD−CD38hi cells, and Ab content was measured by ELISA. All data are shown as mean (±SD) from one of three representative control donors, and one of four RA donors whose B cells were treated with IL-21, anti-CD40, and anti-IgM, and one of four unique RA donors treated with CpG.
Discussion
MPA has potent anti-inflammatory properties in various disease settings. Previous reports have suggested a number of distinct mechanisms by which MPA inhibits inflammatory responses. These mechanisms include inhibition of lymphocyte proliferation, reduced cytokine production, and diminished mononuclear cell recruitment to sites of inflammation (8–12, 31, 32). MPA also affects proliferation of nonlymphoid cells (33–37). Data presented in the current study extend previous findings and further define the actions of MPA on primary human B cells and terminally differentiated PCs. More specifically, we show that MPA selectively inhibited B cell activation and potently blocked PC differentiation of primary B cells isolated from both healthy donors and patients with RA. Terminally differentiated PCs, however, were found to express significantly reduced IMPDH2 mRNA levels compared with those of resting, noncycling B cells, thus suggesting that IMPDH2 may be actively downregulated, allowing plasma cells to escape inhibition by MPA.
Previous studies demonstrate that MPA affects lymphocyte activation in vitro. In unfractionated splenocyte cultures, MPA differentially inhibits early activation markers on B cells and T cells (8). More specifically, whereas MPA inhibits both CD25 and HLA-DR expression on B cells, it inhibits CD25 but not HLA-DR expression on T cells (8). MPA has also been shown to inhibit selectively some aspects of dendritic cell maturation and function in that MPA-treated dendritic cells fail to stimulate T cells (38). Moreover, differential effects of MPA on CD83 upregulation were noted on stimulated monocytes depending on when during the dendritic cell maturation process MPA was added. MPA, however, did downmodulate expression of CD205, CD80, and CD86 regardless of when MPA was added to the cultures and had no effect on HLA-DR or CD1a expression (38). Our studies greatly extend these findings and further demonstrate that MPA specifically inhibited some but not all early activation events on both CD4+ T cells and B cells. These data demonstrate that MPA does not globally blunt lymphocyte activation, but rather selectively modulates specific early activation events.
Another mechanism by which MPA downmodulates the immune response is through alteration of cytokine production. Human monocytes treated with MPA in vitro produce reduced levels of the proinflammatory cytokine IL-1β and increased levels of the anti-inflammatory IL-1R antagonist, IL-1Rα (37). In renal allograft models, treatment with mycophenolate mofetil (MMF), the pro-drug of MPA, reduced expression of a variety of cytokines (36). In a murine model of inflammatory bowel disease, splenic T from animals treated with MMF had reduced expression of TNF-α, IFN-γ, IL-4, and IL-10 (10). Our data expand on these findings and demonstrate that MPA potently inhibits expression of an array of cytokines. More specifically, MPA inhibited expression of IL-10, IFN-γ, as well as IL-21 and to a lesser extent IL-5, IL-13, and IL-17, without altering production of IL-2 or IL-4. These results are somewhat surprising given publications that demonstrate co-regulation of some of these cytokines, such as IL-4, IL-5, and IL-13 (39). However, reports have also established that IL-13 and IL-4 can be differentially regulated, as IL-13, but not IL-4, production by human T cells is susceptible to inhibition by IFN-α (40). Differential regulation of IL-4 and IL-13 in this model is thought to be transcriptionally regulated. These data suggest that MPA exerts its effects on lymphocyte activation in part by selectively inhibiting an array of specific cytokines known to be important to both T and B cell immunoactivation/regulation.
In addition to an effect on cytokine production, MPA may also influence sensitivity to local cytokines by altering the expression level of cytokine receptors. Indeed, we observe both a reduction in IL-21 production by Th17 cells as well as reduced upregulation of IL-21R expression on activated B cells in the presence of MPA. Both of these mechanisms may contribute to the downstream effect of MPA on B cell expansion and PC differentiation.
In regard to proliferation, MPA inhibits cell division, as well as cytokine production, and Ig secretion by the B cell hybridoma MAR 18.5 (11). Studies of MPA on primary human B cells demonstrated that MPA was almost as potent as rapamycin at inhibiting B cell proliferation and Ig production in response to a variety of stimuli (12). However, in these studies, MPA induced substantial apoptosis in the majority of responding B cells (12). Other studies have shown that high concentrations of MPA (50 μg/ml or 156 μM) inhibit memory B cell formation and PC differentiation and reduce HLA-specific ASC when MPA is added to stimulated total PBLs (38). In the current study, we show that concentrations of MPA that did not induce apoptosis had a direct effect on B cells isolated from both healthy controls as well as from patients with autoimmune disease. MPA dramatically and directly inhibited the proliferative response and PC differentiation of B cells from control and RA donors. Additionally, purified naive, memory, and GC B cell populations isolated from either blood or tonsil were all susceptible to inhibition by MPA. Notably, PC differentiation and Ig production were substantially blocked at intermediate concentrations of MPA (0.3 μM), whereas this concentration had little or no effect on many early activation events. These data suggest that although MPA affects many aspects of B cell function, later-stage events, such as class switch recombination and differentiation into Ab-secreting PCs, are more sensitive to MPA-mediated inhibition.
Whereas the inhibitory effect of MPA on proliferative responses has been well demonstrated, its effects on cell death are less clear. Studies of several cell lines suggest that MPA induces apoptosis at high concentrations (11, 33, 41). Additionally, reports suggest that MPA induces apoptosis of primary human B cells (12). However, conflicting studies report that MPA protects against p53-mediated apoptosis (42). Finally, a recent investigation suggests that MPA does not trigger apoptosis, but rather induces a nonclassical caspase-independent necrotic signal involving Rho-GTPase Cdc42 as well as actin polymerization (43). These varied results on cell death are likely due to the various cell types or concentrations of MPA used. Data reported in this study show that, at concentrations sufficient to inhibit B cell proliferation and PC differentiation, MPA did not induce apoptosis either in non-activated, resting primary B cells or in activated B cells at early time points. Further, at later time points, overall cell death in B cells activated in the presence of MPA was comparable with what is observed in unstimulated B cells. These results do not rule out the possibility that MPA may be toxic at high concentrations; however, our data demonstrate that at lower concentrations, MPA inhibits B cell proliferation and differentiation independently of an effect on cell viability.
MPA has been shown to induce arrest in cell cycle progression at the G0/G1 phase in primary T cells and cell lines (8, 44–46). In an osteosarcoma cell line, cell cycle arrest mediated by MPA is dependent on p53 activation and requires expression of the ribosomal stress-sensing proteins L5 and L11 (47). Our data complement these findings and show that MPA induces primary human B cells to arrest in the G0/G1 phase of the cell cycle and prevents entry into S phase after activation with several stimuli. These results are consistent with the observation that MPA does not kill primary B cells directly, but rather selectively inhibits cell activation and completely blocks cell division.
Previously, we have shown that IL-21 and anti-CD40 induce the generation of terminally differentiated, noncycling PCs that are resistant to hyaluronic acid-induced cell death (which only targets cycling cells) (24). Importantly, we now demonstrate that in vitro-generated terminally differentiated PCs are also not susceptible to inhibition by MPA. Heidt and colleagues (12) demonstrated that the inhibitory effect of MPA is maintained when added to cultures of activated B cells after 48 h. However, MPA is likely to mediate its effect primarily through inhibition of the proliferative response at this early time point. Moreover, in studies of B cell hybridomas (11), the ability of MPA to inhibit Ig production is likely secondary to inhibition of cell division of immortalized hybridomas and does not necessarily translate to the ability of MPA to act on terminally differentiated, nondividing PCs. Whereas, our studies directly demonstrate that when MPA is added to primary B cells that have been predifferentiated into PCs in which division has largely ceased, MPA does not impact Ig production. In data not shown, MPA remains active in culture for >5 d, indicating that the inability of MPA to target PCs over time in culture is not due to instability of the drug at 37°C. These data demonstrate that although MPA inhibited proliferation and PC differentiation of freshly isolated primary B cells, it did not target nondividing, terminally differentiated PCs under these assay conditions.
In the clinic, the steady-state Cmax for MPA in serum of patients with lupus nephritis has been reported to be 21 μg/ml (66 μM) (29), the Cmax of MMF in children with autoimmunity, including SLE, is reported to be in the same range, 18.5 μg/ml (28), and the Cmax of MMF for patients with SLE was found to be 16.6 μg/ml (30). A similar range for transplant patients was reported (27). Whereas the concentrations of MPA routinely used (0.3–3 μM) in the current study are well below those reported in the circulation of patients, when comparable or higher concentrations of MPA found in circulation (66–100 μM) were used, there was still no impact of MPA on Ig production from either pre-established or primary plasma cells. Thus, not only does MPA have minimal impact on PC function in ex vivo settings, but also more than likely it will not target long-lived, resident bone marrow PCs in vivo. In clinical trials, MPA has been shown to have a variable effect on humoral immune responses, with statistically significant reductions in anti-DNA Ab titers achieved in some but not all patient subgroups after treatment with MMF (48, 49). In one study, Bijl and colleagues (50) described a significant reduction in anti-dsDNA Ab titers in SLE patients after 6 mo of treatment with MMF. Importantly, in this study, patients only began with MMF therapy after a rise (of at least 25%) in anti-dsDNA titers. It was reported that MMF treatment reduced anti-DNA titers 2-fold and that this reduction was associated with a decreased activation state of B cells in circulation (50). These data suggest that in settings of clinical flares, MPA is likely effective at preventing production of autoantibodies from newly or chronically activated B cells.
The ability of MPA to target activated lymphocytes correlates with high expression of the molecular target of MPA, IMPDH2, in these cells (3). Indeed, after stimulation with IL-21 costimulation, there was a >25-fold increase in IMPDH2 mRNA expression in activated B cells compared with that in resting B cells. Expression of IMPDH1 mRNA was significantly lower than that of IMPDH2. These observations are consistent with previous reports that IMPDH2 is the predominant isoform expressed in activated lymphocytes (5). Importantly, IMPDH2 levels were greatly reduced in both in vitro-differentiated and primary human bone marrow PCs compared with those of resting primary B cells. These results suggest that reduced IMPDH2 expression is the main mechanism by which PCs evade inhibition by MPA. Moreover, in addition to our previous data that IL-21–generated PCs are nondividing, terminally differentiated cells (24), these current data further confirm our previous findings that these in vitro-generated PCs are terminally differentiated, as cells that both lack IMPDH2 expression and are immune to MPA suppression would not be expected to have the capacity to divide.
Overall, our data show a potential therapeutic benefit accruing from the use of MPA in autoimmune individuals is likely derived from its ability to block B cell activation, proliferation, and PC differentiation. These studies show, however, that MPA is not likely to impact Ig production from terminally differentiated, nondividing PCs in vivo. This may be beneficial in situations where protective humoral immunity has been established. However, it may also be a limitation in disease settings where long-lived PCs contribute to pathogenic autoantibody production.
Supplementary Material
Abbreviations used in this article
- 7-AAD
7-aminoactinomycin D
- GC
germinal center
- IMPDH
inosine 5′-monophosphate dehydrogenase
- MMF
mycophenolate mofetil
- MPA
mycophenolic acid
- PC
plasma cell
- RA
rheumatoid arthritis
- SLE
systemic lupus erythematosus
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology. 2000;47:85–118. doi: 10.1016/s0162-3109(00)00188-0. [DOI] [PubMed] [Google Scholar]
- 2.Carr SF, Papp E, Wu JC, Natsumeda Y. Characterization of human type I and type II IMP dehydrogenases. J Biol Chem. 1993;268:27286–27290. [PubMed] [Google Scholar]
- 3.Dayton JS, Lindsten T, Thompson CB, Mitchell BS. Effects of human T lymphocyte activation on inosine monophosphate dehydrogenase expression. J Immunol. 1994;152:984–991. [PubMed] [Google Scholar]
- 4.Jain J, Almquist SJ, Ford PJ, Shlyakhter D, Wang Y, Nimmesgern E, Germann UA. Regulation of inosine monophosphate dehydrogenase type I and type II isoforms in human lymphocytes. Biochem Pharmacol. 2004;67:767–776. doi: 10.1016/j.bcp.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 5.Nagai M, Natsumeda Y, Weber G. Proliferation-linked regulation of type II IMP dehydrogenase gene in human normal lymphocytes and HL-60 leukemic cells. Cancer Res. 1992;52:258–261. [PubMed] [Google Scholar]
- 6.Allison AC, Eugui EM. The design and development of an immunosuppressive drug, mycophenolate mofetil. Springer Semin Immunopathol. 1993;14:353–380. doi: 10.1007/BF00192309. [DOI] [PubMed] [Google Scholar]
- 7.Allison AC. Mechanisms of action of mycophenolate mofetil. Lupus. 2005;14(Suppl 1):s2–s8. doi: 10.1191/0961203305lu2109oa. [DOI] [PubMed] [Google Scholar]
- 8.Heinschink A, Raab M, Daxecker H, Griesmacher A, Müller MM. In vitro effects of mycophenolic acid on cell cycle and activation of human lymphocytes. Clin Chim Acta. 2000;300:23–28. doi: 10.1016/s0009-8981(00)00297-7. [DOI] [PubMed] [Google Scholar]
- 9.Blaheta RA, Leckel K, Wittig B, Zenker D, Oppermann E, Harder S, Scholz M, Weber S, Encke A, Markus BH. Mycophenolate mofetil impairs transendothelial migration of allogeneic CD4 and CD8 T-cells. Transplant Proc. 1999;31:1250–1252. doi: 10.1016/s0041-1345(98)01984-8. [DOI] [PubMed] [Google Scholar]
- 10.Lee J, Kim MS, Kim EY, Park HJ, Chang CY, Park KS, Jung DY, Kwon CH, Joh JW, Kim SJ. Mycophenolate mofetil promotes down-regulation of expanded B cells and production of TNF-alpha in an experimental murine model of colitis. Cytokine. 2008;44:49–56. doi: 10.1016/j.cyto.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Jonsson CA, Carlsten H. Mycophenolic acid inhibits inosine 5′-monophosphate dehydrogenase and suppresses immunoglobulin and cytokine production of B cells. Int Immunopharmacol. 2003;3:31–37. doi: 10.1016/s1567-5769(02)00210-2. [DOI] [PubMed] [Google Scholar]
- 12.Heidt S, Roelen DL, Eijsink C, van Kooten C, Claas FH, Mulder A. Effects of immunosuppressive drugs on purified human B cells: evidence supporting the use of MMF and rapamycin. Transplantation. 2008;86:1292–1300. doi: 10.1097/TP.0b013e3181874a36. [DOI] [PubMed] [Google Scholar]
- 13.Jonsson CA, Erlandsson M, Svensson L, Mölne J, Carlsten H. Mycophenolate mofetil ameliorates perivascular T lymphocyte inflammation and reduces the double-negative T cell population in SLE-prone MRLlpr/lpr mice. Cell Immunol. 1999;197:136–144. doi: 10.1006/cimm.1999.1570. [DOI] [PubMed] [Google Scholar]
- 14.Corna D, Morigi M, Facchinetti D, Bertani T, Zoja C, Remuzzi G. Mycophenolate mofetil limits renal damage and prolongs life in murine lupus autoimmune disease. Kidney Int. 1997;51:1583–1589. doi: 10.1038/ki.1997.217. [DOI] [PubMed] [Google Scholar]
- 15.Yung S, Zhang Q, Zhang CZ, Chan KW, Lui SL, Chan TM. Anti-DNA antibody induction of protein kinase C phosphorylation and fibronectin synthesis in human and murine lupus and the effect of mycophenolic acid. Arthritis Rheum. 2009;60:2071–2082. doi: 10.1002/art.24573. [DOI] [PubMed] [Google Scholar]
- 16.Jonsson CA, Carlsten H. Inosine monophosphate dehydrogenase (IMPDH) inhibition in vitro suppresses lymphocyte proliferation and the production of immunoglobulins, autoantibodies and cytokines in splenocytes from MRLlpr/lpr mice. Clin Exp Immunol. 2001;124:486–491. doi: 10.1046/j.1365-2249.2001.01545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones RB, Walsh M, Smith KG. What is the value of mycophenolate mofetil as induction and maintenance therapy in lupus nephritis? Curr Opin Rheumatol. 2009;21:256–261. doi: 10.1097/bor.0b013e32832a0698. [DOI] [PubMed] [Google Scholar]
- 18.Epinette WW, Parker CM, Jones EL, Greist MC. Mycophenolic acid for psoriasis. A review of pharmacology, long-term efficacy, and safety. J Am Acad Dermatol. 1987;17:962–971. doi: 10.1016/s0190-9622(87)70285-0. [DOI] [PubMed] [Google Scholar]
- 19.Goldblum R. Therapy of rheumatoid arthritis with mycophenolate mofetil. Clin Exp Rheumatol. 1993;11(Suppl 8):S117–S119. [PubMed] [Google Scholar]
- 20.Neurath MF, Wanitschke R, Peters M, Krummenauer F, Meyerzum Büschenfelde KH, Schlaak JF. Randomised trial of mycophenolate mofetil versus azathioprine for treatment of chronic active Crohn’s disease. Gut. 1999;44:625–628. doi: 10.1136/gut.44.5.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winnicki W, Weigel G, Sunder-Plassmann G, Bajari T, Winter B, Herkner H, Sengoelge G. An inosine 5′-monophosphate dehydrogenase 2 single-nucleotide polymorphism impairs the effect of mycophenolic acid. Pharmacogenomics J. 2010;10:70–76. doi: 10.1038/tpj.2009.43. [DOI] [PubMed] [Google Scholar]
- 22.Favas C, Isenberg DA. B-cell-depletion therapy in SLE—what are the current prospects for its acceptance? Nat Rev Rheumatol. 2009;5:711–716. doi: 10.1038/nrrheum.2009.218. [DOI] [PubMed] [Google Scholar]
- 23.Kuchen S, Robbins R, Sims GP, Sheng C, Phillips TM, Lipsky PE, Ettinger R. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. J Immunol. 2007;179:5886–5896. doi: 10.4049/jimmunol.179.9.5886. [DOI] [PubMed] [Google Scholar]
- 24.Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Spolski R, Leonard WJ, Lipsky PE. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol. 2005;175:7867–7879. doi: 10.4049/jimmunol.175.12.7867. [DOI] [PubMed] [Google Scholar]
- 25.Ettinger R, Kuchen S, Lipsky PE. The role of IL-21 in regulating B-cell function in health and disease. Immunol Rev. 2008;223:60–86. doi: 10.1111/j.1600-065X.2008.00631.x. [DOI] [PubMed] [Google Scholar]
- 26.Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, Wiethe C, Winkler TH, Kalden JR, Manz RA, Voll RE. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. 2008;14:748–755. doi: 10.1038/nm1763. [DOI] [PubMed] [Google Scholar]
- 27.Bullingham RE, Nicholls AJ, Kamm BR. Clinical pharmaco-kinetics of mycophenolate mofetil. Clin Pharmacokinet. 1998;34:429–455. doi: 10.2165/00003088-199834060-00002. [DOI] [PubMed] [Google Scholar]
- 28.Filler G, Hansen M, LeBlanc C, Lepage N, Franke D, Mai I, Feber J. Pharmacokinetics of mycophenolate mofetil for autoimmune disease in children. Pediatr Nephrol. 2003;18:445–449. doi: 10.1007/s00467-003-1133-1. [DOI] [PubMed] [Google Scholar]
- 29.Joy MS, Hilliard T, Hu Y, Hogan SL, Dooley MA, Falk RJ, Smith PC. Pharmacokinetics of mycophenolic acid in patients with lupus nephritis. Pharmacotherapy. 2009;29:7–16. doi: 10.1592/phco.29.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roland M, Barbet C, Paintaud G, Magdelaine-Beuzelin C, Diot E, Halimi JM, Lebranchu Y, Nivet H, Büchler M. Mycophenolate mofetil in patients with systemic lupus erythematosus: a prospective pharmacokinetic study. Lupus. 2009;18:441–447. doi: 10.1177/0961203308098631. [DOI] [PubMed] [Google Scholar]
- 31.Allison AC, Eugui EM. Mechanisms of action of mycophenolate mofetil in preventing acute and chronic allograft rejection. Transplantation. 2005;80(2 Suppl):S181–S190. doi: 10.1097/01.tp.0000186390.10150.66. [DOI] [PubMed] [Google Scholar]
- 32.Heemann U, Azuma H, Hamar P, Schmid C, Tilney N, Philipp T. Mycophenolate mofetil inhibits lymphocyte binding and the upregulation of adhesion molecules in acute rejection of rat kidney allografts. Transpl Immunol. 1996;4:64–67. doi: 10.1016/s0966-3274(96)80039-6. [DOI] [PubMed] [Google Scholar]
- 33.Gu JJ, Santiago L, Mitchell BS. Synergy between imatinib and mycophenolic acid in inducing apoptosis in cell lines expressing Bcr-Abl. Blood. 2005;105:3270–3277. doi: 10.1182/blood-2004-10-3864. [DOI] [PubMed] [Google Scholar]
- 34.Laurent AF, Dumont S, Poindron P, Muller CD. Mycophenolic acid suppresses protein N-linked glycosylation in human monocytes and their adhesion to endothelial cells and to some substrates. Exp Hematol. 1996;24:59–67. [PubMed] [Google Scholar]
- 35.Morath C, Zeier M. Review of the antiproliferative properties of mycophenolate mofetil in non-immune cells. Int J Clin Pharmacol Ther. 2003;41:465–469. doi: 10.5414/cpp41465. [DOI] [PubMed] [Google Scholar]
- 36.Nadeau KC, Azuma H, Tilney NL. Sequential cytokine expression in renal allografts in rats immunosuppressed with maintenance cyclosporine or mycophenolate mofetil. Transplantation. 1996;62:1363–1366. doi: 10.1097/00007890-199611150-00034. [DOI] [PubMed] [Google Scholar]
- 37.Waters RV, Webster D, Allison AC. Mycophenolic acid and some antioxidants induce differentiation of monocytic lineage cells and augment production of the IL-1 receptor antagonist. Ann N Y Acad Sci. 1993;696:185–196. doi: 10.1111/j.1749-6632.1993.tb17151.x. [DOI] [PubMed] [Google Scholar]
- 38.Wadia PP, Herrera ND, Abecassis MM, Tambur AR. Mycophenolic acid inhibits maturation and function of human dendritic cells and B cells. Hum Immunol. 2009;70:692–700. doi: 10.1016/j.humimm.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 39.Kelly BL, Locksley RM. Coordinate regulation of the IL-4, IL-13, and IL-5 cytokine cluster in Th2 clones revealed by allelic expression patterns. J Immunol. 2000;165:2982–2986. doi: 10.4049/jimmunol.165.6.2982. [DOI] [PubMed] [Google Scholar]
- 40.Essayan DM, Krishnaswamy G, Oriente A, Lichtenstein LM, Huang SK. Differential regulation of antigen-induced IL-4 and IL-13 generation from T lymphocytes by IFN-alpha. J Allergy Clin Immunol. 1999;103:451–457. doi: 10.1016/s0091-6749(99)70470-7. [DOI] [PubMed] [Google Scholar]
- 41.Takebe N, Cheng X, Fandy TE, Srivastava RK, Wu S, Shankar S, Bauer K, Shaughnessy J, Tricot G. IMP dehydrogenase inhibitor mycophenolate mofetil induces caspase-dependent apoptosis and cell cycle inhibition in multiple myeloma cells. Mol Cancer Ther. 2006;5:457–466. doi: 10.1158/1535-7163.MCT-05-0340. [DOI] [PubMed] [Google Scholar]
- 42.Boldt A, Barten MJ, Sagner A, Mohr FW, Adams V, Dhein S, Gummert JF. The influence of immunosuppressive drugs on T- and B-cell apoptosis via p53-mediated pathway in vitro and in vivo. Transplantation. 2006;82:422–427. doi: 10.1097/01.tp.0000229036.75483.15. [DOI] [PubMed] [Google Scholar]
- 43.Chaigne-Delalande B, Guidicelli G, Couzi L, Merville P, Mahfouf W, Bouchet S, Molimard M, Pinson B, Moreau JF, Legembre P. The immunosuppressor mycophenolic acid kills activated lymphocytes by inducing a nonclassical actin-dependent necrotic signal. J Immunol. 2008;181:7630–7638. doi: 10.4049/jimmunol.181.11.7630. [DOI] [PubMed] [Google Scholar]
- 44.Laliberté J, Yee A, Xiong Y, Mitchell BS. Effects of guanine nucleotide depletion on cell cycle progression in human T lymphocytes. Blood. 1998;91:2896–2904. [PubMed] [Google Scholar]
- 45.Linke SP, Clarkin KC, Di Leonardo A, Tsou A, Wahl GM. A reversible, p53-dependent G0/G1 cell cycle arrest induced by ribonucleotide depletion in the absence of detectable DNA damage. Genes Dev. 1996;10:934–947. doi: 10.1101/gad.10.8.934. [DOI] [PubMed] [Google Scholar]
- 46.Quéméneur L, Gerland LM, Flacher M, Ffrench M, Revillard JP, Genestier L. Differential control of cell cycle, proliferation, and survival of primary T lymphocytes by purine and pyrimidine nucleotides. J Immunol. 2003;170:4986–4995. doi: 10.4049/jimmunol.170.10.4986. [DOI] [PubMed] [Google Scholar]
- 47.Sun XX, Dai MS, Lu H. Mycophenolic acid activation of p53 requires ribosomal proteins L5 and L11. J Biol Chem. 2008;283:12387–12392. doi: 10.1074/jbc.M801387200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan TM, Li FK, Tang CS, Wong RW, Fang GX, Ji YL, Lau CS, Wong AK, Tong MK, Chan KW, Lai KN Hong Kong-Guangzhou Nephrology Study Group. Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. N Engl J Med. 2000;343:1156–1162. doi: 10.1056/NEJM200010193431604. [DOI] [PubMed] [Google Scholar]
- 49.Pisoni CN, Sanchez FJ, Karim Y, Cuadrado MJ, D’Cruz DP, Abbs IC, Khamasta MA, Hughes GR. Mycophenolate mofetil in systemic lupus erythematosus: efficacy and tolerability in 86 patients. J Rheumatol. 2005;32:1047–1052. [PubMed] [Google Scholar]
- 50.Bijl M, Horst G, Bootsma H, Limburg PC, Kallenberg CG. Mycophenolate mofetil prevents a clinical relapse in patients with systemic lupus erythematosus at risk. Ann Rheum Dis. 2003;62:534–539. doi: 10.1136/ard.62.6.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.