

Abstract
Autophagy, the process by which proteins and organelles are sequestered in double-membrane structures called autophagosomes and delivered to lysosomes for degradation, is critical in diseases such as cancer and neurodegeneration1,2. Much of our understanding of this process has emerged from analysis of bulk cytoplasmic autophagy, but our understanding of how specific cargo including organelles, proteins, or intracellular pathogens are targeted for selective autophagy is limited3. We employed quantitative proteomics to identify a cohort of novel and known autophagosome-enriched proteins, including cargo receptors. Like known cargo receptors, NCOA4 was highly enriched in autophagosomes, and associated with ATG8 proteins that recruit cargo-receptor complexes into autophagosomes. Unbiased identification of NCOA4-associated proteins revealed ferritin heavy and light chains, components of an iron-filled cage structure that protects cells from reactive iron species4 but is degraded via autophagy to release iron5,6 through an unknown mechanism. We found that delivery of ferritin to lysosomes required NCOA4, and an inability of NCOA4-deficient cells to degrade ferritin leads to decreased bioavailable intracellular iron. This work identifies NCOA4 as a selective cargo receptor for autophagic turnover of ferritin (ferritinophagy) critical for iron homeostasis and provides a resource for further dissection of autophagosomal cargo-receptor connectivity.
Autophagosomes are decorated by a family of ubiquitin-like adaptor ATG8 proteins that are conjugated to phosphatidylethanolamine through the action of an autophagy-specific E1-E2-E3 cascade. While ATG8 proteins are known to recruit a small number of cargo receptors to insipient autophagosomes, the full repertoire of selective autophagic cargo and their cognate receptor proteins remain poorly defined3. Selective autophagy may be particularly important for the survival or growth of particular cancer cell types7,8 but in other contexts may act as a tumor suppressor to maintain normal cellular homeostasis and constrain tumor initiation9,10. Thus, a more comprehensive understanding of autophagy cargo-receptor pairs is required for understanding autophagic mechanisms that contribute to proteostasis.
Three previous studies described the use of mass spectrometry to identify proteins in autophagosomal preparations, but the low overlap in the proteins identified between these studies and limitations of the approaches used led us to catalog resident autophagosomal proteins using quantitative proteomics (Extended Data Fig. 1a)11-13. We combined stable isotopic labeling by amino acids in cell culture (SILAC) with an established density gradient separation protocol14,15 to quantitatively identify proteins enriched in autophagosome preparations. This analysis was performed using two pancreatic cancer cell lines (PANC1 and 8988T) that require autophagy for growth, as well as the MCF7 breast cancer cell line, which is less reliant on autophagy for growth7. Given the high basal autophagy of PANC1 and 8898T cells, light cells were briefly treated with the PI3 kinase inhibitor Wortmannin to suppress autophagosome formation, while heavy cells were treated with the lysosomal inhibitor Chloroquine (CQ) to maximize the number of autophagosomes (Fig. 1a, Extended Data Fig. 1b). This approach allows for robust identification of proteins intimately associated with autophagosome-enriched samples as opposed to proteins that simply co-migrate with these vesicles during gradient centrifugation. As expected, the autophagosome-enriched fraction was enriched for the ATG8 protein MAP1LC3B (LC3B) as assayed by immunoblotting or immunofluorescence and contained characteristic double-membrane vesicles by electron microscopy (Extended Data Fig. 1c-h, k-m). These autophagosomes were intact as assessed by LC3B and p62/SQSTM1 release upon detergent treatment (Extended Data Fig. 1i). We also note, that autophagosomes and autophagolysosomes are heterogeneous in nature, as they form via a dynamic interplay between other membrane-rich organelles, each containing their own specific complement of proteins.
Figure 1. Quantitative proteomics for identification of autophagosome-associated proteins.

(a) Autophagosome enrichment workflow. (b) Log2(H:L) plot for autophagosome proteins from PANC1 cells (Ex. 3, Table S3) and scheme for identification of candidate autophagosome proteins. (c) Autophagosome candidate overlap from biologic replicate experiments for PANC1 and MCF7 cells, as well as overlap between PANC1 and MCF7 datasets. (d) Pearson correlation plot for overlapping candidates from PANC1 experiments (86 proteins, comparing Ex. 2 vs. Ex. 3). (e) Log2(H:L) heat map of Class 1A candidates from PANC1 and MCF7 cells.
Single-label (heavy Lys) profiling of the autophagosomal fraction from PANC1 after 4 or 16 h of CQ treatment, as well as double-label (heavy Lys and Arg) profiling of PANC1 and MCF7 derived autophagosomal preparations at 16 h of CQ treatment resulted in the quantification of >2000 proteins (Supplementary Tables 1-4, see Methods)16,17. Proteins were selected based on significantly increased log2 (heavy:light) ratios and the presence of 2 or more peptides, and subsequently filtered against the relative abundance of the proteome measured independently by LC-MS (Fig. 1b, see Methods), thereby removing abundant proteins that may be non-specifically captured by bulk autophagy. We identified 86 proteins with log2(H:L)>1.5 in all 3 PANC1 replicates (Pearson correlation of 0.92 for a representative pair), and 102 proteins with log2(H:L)>1.0 in both MCF7 replicates (Pearson correlation of 0.89) (Fig. 1c, d, Extended Data fig. 1j, Supplementary Tables 3-5). We will refer to the union of these two high stringency datasets as Class 1 autophagosome-enriched proteins, and a high priority subset of these proteins based on their presence in ≥3 data sets or known involvement in autophagy as Class 1A (Fig. 1e, Extended data Fig. 2a, Supplementary Table 5). We also identified non-Class 1 proteins with a log2(H:L)>2.0 in any 2 of the 5 PANC1 or MCF7 experiments (16 h in CQ), and will refer to this lower stringency dataset as Class 2 autophagosome-enriched proteins (Supplementary Table 5). As expected, PANC1 cells treated with CQ for 16 h showed a greater accumulation of candidate proteins than PANC1 or 8988T cells treated for 4 h (Fig. 1e, Extended Data Fig. 2).
Within the Class 1A proteins, we identified 2 ATG8 paralogs (GABARAPL2, LC3B), 4 known autophagy cargo receptors (SQSTM1/p62, CALCOCO2/NDP52, OPTN, NBR1) and 4 proteins that were previously reported to associate with ATG8 family members and/or cargo receptors, or to be involved in autophagosomal membrane fusion (KEAP1, TMEM59, FYCO1, STX17) (Fig. 1e, Extended Data Fig. 2a). Moreover, we identified 2 proteins reported as selective autophagy cargo (APP, NRP1), and 7 proteins identified as high-confidence interacting proteins in our previously published Autophagy Interaction Network18 (Fig. 1e, Extended Data Fig. 2a). Furthermore, the autophagy pathway proteins RB1CC1, ATG9A, TBC1D15, and the OPTN-binding protein TBK1 were identified in the Class 2 dataset (Supplementary Table 5). The presence of several plasma membrane and endocytosis-related proteins (Fig. 1e, Extended Data Fig. 2a) is consistent with intermixing of these membrane sources during autophagosome maturation or lysosomal fusion. Co-localization with LC3B-positive puncta was observed for 7 of 11 Class 1A proteins tested (Extended Data Fig 2a, 3).
Figure 3. NCOA4 mediates autophagic delivery of ferritin to lysosomes to control iron homeostasis.

(a) NCOA4 depletion rescues ferritin degradation upon 9 h iron chelation in 8988T. Relative FTH1 levels (n=3, biologic triplicate) for each chelator are quantified. Bars and error bars represent mean values and s.d., respectively: ** (p<0.01) and * (p<0.02) comparing FTH1 levels between different cell lines (one-sided t-test). (b) DFO chelation time course in U2OS cells, DFO added at time 0, two NCOA4 antibodies are used for immunoblotting (top panel Sigma antibody, 2nd panel Bethyl antibody). (c) GFP-NCOA4 (green) co-localizes with endogenous LC3 (red) and endogenous ferritin (blue) in U2OS cells subjected to DFO chelation in the presence of lysosomal protease inhibitors for 6 h. Scale bar, 20 μm. (d) Immunostaining of 8988T cells subjected to DFO chelation in the presence of lysosomal protease inhibitors for 9 h. Scale bar, 10 μm. Punctate ferritin fraction was quantified from >100 cells per cell line from 2 independent experiments (biologic duplicate). Bars and error bars represent mean values and s.d., respectively: ***denotes p<0.001 using a one-sided t-test. (e) Lysates from 8988T cells as in panel A were analyzed using indicated antibodies (lanes 1-3, untreated), with quantification based on at least 3 independent experiments (biologic triplicate). Bars and error bars represent mean values and s.d., respectively: * (p<0.05) using a one-sided t-test. (f) 8988T cells stably expressing shGFP or shNCOA4-1 were treated with H2O2 and cell viability was measured at 72 h. Bars and error bars represent mean values and s.d., respectively of technical triplicates: *** (p<0.001) using a two-sided t-test.
As further validation, 8988T pancreatic cancer cells were subjected to both SILAC-based autophagosomal profiling using density gradient purification and a semi-quantitative proteomic approach using an immunopurification-based autophagosome enrichment scheme by immunoprecipitation of tagged GFP-LC3B autophagosomes12 (Extended Data Fig. 2b-d and Extended Data Fig. 4, Supplementary Tables 6-7). In total, 40 proteins were identified in common between the 50 Class 1A autophagosomal proteins and those identified as enriched in autophagosomes purified from 8988T cells. Comparative analysis of our MCF7 candidate proteins with a previously reported autophagosomal quantitative proteomics analysis13 that used the same cell line revealed 2 overlapping proteins from their final combined data set (SQSTM1 and GABARAPL2) (Extended Data Fig. 5, Supplementary Table 8). Expanding the analysis to also include our PANC1 datasets only increased the overlap to a total of 4 proteins from our Class 1 and 2 list. This low rate of overlap appears to reflect the fact that a large fraction of proteins previously reported to reside in autophagosomes would have been removed upon filtering at high stringency for abundance in the total proteome (see Methods, Extended Data Fig. 5, Supplementary Table 8), indicating that these likely are present in autophagosomes due to non-selective bulk degradation of cytosolic contents, or are co-purifying contaminants. Likewise there is minimal overlap between our Class 1 and 2 proteins with two additional proteomics efforts11,12. While understandable that there would be variation between datasets depending on cell type, autophagy stimulus, and purification technique, our dataset represents the most robust autophagosome proteomics effort to date given the number of bona fide autophagy related proteins identified among our Class 1 and 2 candidates.
Among the most highly and consistently enriched autophagosomal proteins was NCOA4 (nuclear receptor coactivator 4) (Fig. 1e, Supplementary Tables 2-4, 6-7), which was also enriched in an independent autophagosome proteomics study, although not included in their final list of autophagosomal proteins due to the method of analysis13 (Extended Data Fig. 5 and Methods). NCOA4 was originally identified as a protein that interacts with the androgen receptor (AR), and its overexpression was reported to activate transcription of AR-regulated genes19. However, not all studies have supported a role for NCOA4 in AR function20, and the data described below reveals a previously unrecognized role for NCOA4 as an autophagy cargo receptor.
We initially examined the localization of NCOA4. GFP-NCOA4 was diffusely localized in the cytoplasm in U2OS cells that have a low level of basal autophagy but accumulated in cytoplasmic puncta that are largely co-incident with LC3B-positive puncta in response to CQ in U2OS cells as well as 8988T cells, consistent with localization of GFP-NCOA4 in autophagosomes (Fig. 2a, Extended Data Fig. 6a). In addition, NCOA4 showed significant co-localization with GABARAPL2-positive puncta (Extended Data Fig. 6b-c), which is consistent with it being amongst the strongest interactors in a GST-ATG8 binding assays performed in vitro using cell extracts (Fig. 2b). Consistent with our proteomic data, NCOA4 is enriched in purified autophagosomes by immunoblotting and its levels are markedly increased in response to blockade of autophagosome degradation by CQ or Bafilomycin A1 (BAF) (Fig. 2c-d), Moreover, NCOA4 does not co-localize with the late endosome marker Mannose 6-Phosphate Receptor (Extended Data Fig. 6d). Together this data supports the predominantly autophagosomal localization of NCOA4 puncta. We were unable to identify a canonical LIR (LC3-interacting region) motif in NCOA4, although the existence of non-canonical ATG8 binding motifs21 may suggest that NCOA4 employs such an alternative motif for interaction.
Figure 2. NCOA4 associates with and co-localizes with ferritin.

(a) GFP-NCOA4 (green) co-localizes with endogenous LC3 (red) in CQ-treated cells. Scale bar, 20 μm. (b) GST-pull-down assay of NCOA4-HA-FLAG from stable 293T cells using GST-ATG8 proteins. HA immunoblot for NCOA4-HA-FLAG. (c) Lysates from 8988T cells treated with CQ or BAF (8h) were immunoblotted for NCOA4, LC3B, and ACTB as a loading control. *, cross-reactive band (see Extended Data Fig. 7d-e). (d) 8988T autophagosome purification fractions were analyzed using antibodies to NCOA4 and LC3B. TAX1BP1, a newly identified autophagy receptor, was included as a positive control. LD is gradient load, A1 is autophagosome fraction, A2 is the autophagolysosome fraction, L is the lysosome fraction, M is the mitochondrial fraction. (e) NCOA4 interaction network from cells expressing NCOA4-HA-FLAG or FTH1-HA-FLAG (Table S9). Black lines (this study) depict directionality of interaction observed with line thickness weighted by WDN-score (293T dataset). Dotted lines, STRING database. Numbers in parentheses indicate log2(H:L) ratio of NCOA4, FTH1, and FTL from MCF7 Ex. 1 dataset (Table S4). (f,g) Extracts from 293T cells stably expressing the indicated proteins were immunoprecipitated with α-FLAG and immunoblotted with the indicated antibodies. α-ACTB, loading control. (h) Representative confocal images of GFP-NCOA4 (green) and ferritin (red) after no treatment or FAC treatment (24 h). Scale bar, 10 μm.
To begin to understand potential roles for NCOA4 in autophagy, we performed affinity purification-mass spectrometry (AP-MS) of stably expressed NCOA4-HA-FLAG and used the Comparative Proteomics Software Analysis Suite (CompPASS) to identify high confidence candidate interacting proteins (HCIPs)22. AP-MS of NCOA4-HA-FLAG from PANC1, 8988T, and 293T cells revealed a number of HCIPs including both the ferritin heavy chain (FTH1) and ferritin light chain (FTL), as well as HERC2 and NEURL4, which are known to associate with each other (Fig. 2e, Supplementary Table 9)23. Interaction of NCOA4-HA-FLAG with selected endogenous HCIPs was verified by anti-FLAG immunoprecipitation followed by immunoblotting (Fig. 2f). FTH1-HA-FLAG reciprocally associated with endogenous NCOA4 as determined by AP-MS (Fig. 2e, Supplementary Table 9) and also associated with co-expressed MYC-NCOA4 (Fig. 2g). The absence of HERC2-NEURL4 in ferritin immune complexes (Supplementary Table 9) suggests that NCOA4 makes distinct complexes with ferritin and HERC2-NEURL4. Consistent with this, neither HERC2 nor NEURL4 were enriched in autophagosomal fractions (Supplementary Tables 2-7) and HERC2 does not colocalize with autophagosomes (Extended Data Fig. 6e).
FTH1 and FTL form a 24-subunit macromolecular iron-storage complex critical for iron homeostasis4. Early electron microscopy studies identified iron-laden ferritin in lysosomes and more recently, it was shown that in cells subjected to iron chelation, ferritin is delivered to the lysosome for degradation via autophagy, presumably to promote iron availability5,6,24. Interestingly, FTH1 and FTL were enriched in autophagosomal fractions from MCF7 and 8988T cells based on mass spectrometry (Fig. 2e, Extended Data Fig. 2, Supplementary Tables 4, 6). Furthermore, ferritin and NCOA4 demonstrated extensive co-localization in puncta in several cell lines upon stimulation of ferritin expression with ferric ammonium citrate (FAC), reflecting the high level of ferritin undergoing autophagic targeting and lysosomal degradation (Fig. 2h). We refer to this process as “ferritinophagy”.
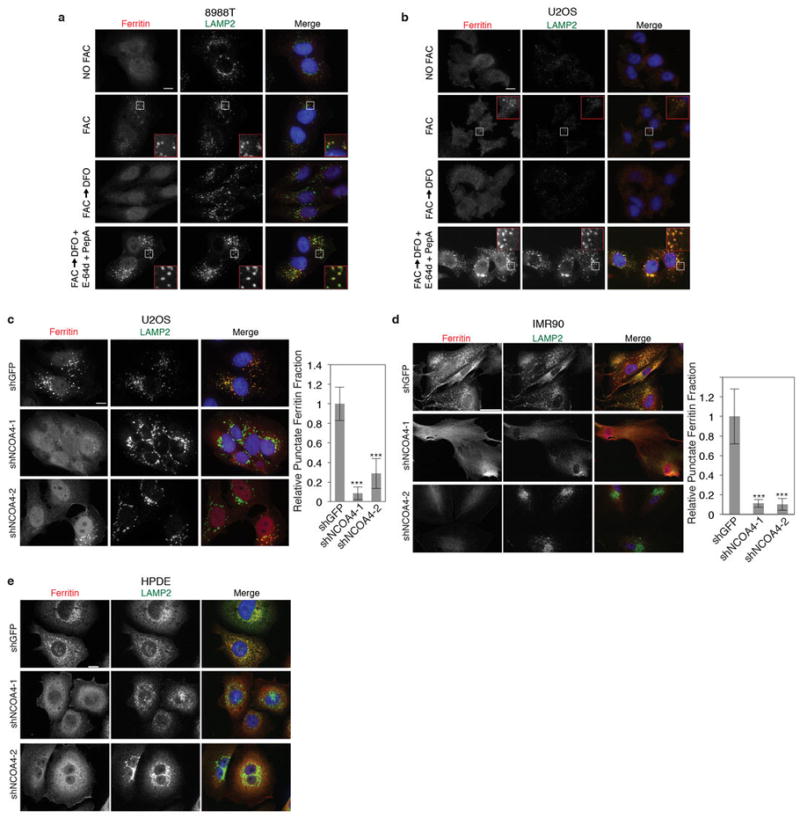
Given the interaction between NCOA4 and ferritin, and their localization in autophagosomes, we tested the hypothesis that NCOA4 acts as an autophagy receptor for ferritinophagy. In response to low intracellular iron levels, ferritin is degraded to release its iron stores. This can be stimulated experimentally by chelation of iron5. While some reports have suggested that ferritin is degraded via the proteasome25, we validated that in the cell lines we examined, ferritin is primarily degraded by the lysosome in response to multiple distinct chelators (Extended Data Fig. 7a-b), as observed previously5. In addition, genetic inhibition of autophagy using RNAi against ATG5 reduced ferritin degradation in response to iron chelation (Extended Data Fig. 7c). Importantly, suppression of NCOA4 expression with multiple shRNAs followed by iron chelation abrogated ferritin degradation in multiple cell lines and with chemically distinct chelators (Fig. 3a-b, Extended Data Fig. 7d-g). Consistent with the above results, ferritin localized to lysosomes and accumulated to a significant degree upon lysosomal protease inhibition (Extended Data Fig. 8a-b). Furthermore, ferritin simultaneously colocalizes with NCOA4 and LC3B positive puncta representing autophagosomes (Fig. 3c). If NCOA4 functions as an essential receptor for ferritinophagy, depletion of NCOA4 would be predicted to block ferritin localization with lysosomes. Indeed, depletion of NCOA4 blocked ferritin co-localization with lysosomes and led to a diffuse localization pattern (Fig. 3d, Extended Data Fig. 8c). NCOA4 is also critical in mediating the targeting of ferritin to autophagosomes in non-transformed IMR90 and HPDE cells, indicating that this is a general cellular mechanism for the degradation of ferritin by autophagy (Extended Data Fig. 8d-e). As a control for the specificity of the shRNAs, a non-degradable murine NCOA4 cDNA was able to significantly rescue the ferritin lysosomal localization (Extended Data Fig. 9a-c). These results were confirmed with two additional siRNAs against NCOA4 (Extended Data Fig. 10a). Again, consistent with a distinct role for the HERC2/NCOA4 complex, HERC2 knockdown had no effect on lysosomal delivery of ferritin (Extended Data Fig. 10b-c).
Iron plays an essential role in multiple cellular processes and as such iron metabolism is a tightly regulated process controlled by a network of iron-dependent proteins4. Ferritin forms an intracellular iron storage protein complex capable of chelating up to 4500 atoms of iron, thus protecting the cell from free iron participating in the generation of free radicals via Fenton-like reactions26. Iron stored in ferritin is utilized during periods of low iron levels and recent evidence suggests that iron can be liberated from ferritin upon degradation of ferritin in the lysosome5. As our evidence suggested that NCOA4 mediates the transport of ferritin to the lysosome via the autophagosome, we were interested in how NCOA4 knockdown affects the network of iron regulatory proteins. First, NCOA4 depletion increased basal ferritin levels relative to control cells (Fig. 3a lane 1 versus lanes 6 and 11, Extended Data Fig. 7f, lane 1 versus lanes 5 and 9). This suggests that NCOA4-deficient cells would display reduced iron bioavailability. Therefore, we examined the levels of the iron-response protein 2 (IRP2), an RNA-binding protein that binds to iron response elements (IRE) on a set of iron-regulated mRNAs to control their translation4. Cellular iron levels control IRE-binding activity of IRP2, with high and low iron levels promoting IRP2 turnover and stabilization, respectively4. We found that NCOA4 depletion increased IRP2 abundance to an extent comparable to control cells subjected to iron chelation (Fig. 3e lane 7 versus lane 6). A higher IRP2 level in NCOA4-depleted cells would be anticipated to stabilize transferrin receptor mRNA and increase translation of the transferrin receptor to increase intracellular iron. Indeed, the abundance of the transferrin receptor is increased in NCOA4-depleted 8988T cells (Fig. 3e lane 1 versus lanes 2-3). These results were confirmed with two independent siRNAs to NCOA4 in 8988T, U2OS, and IMR90 cell lines (Extended Data Fig. 10d). Conversely, ectopic expression of NCOA4 in 8988T cells attenuates the increase of ferritin seen in FAC treated control cells implying that the increased NCOA4 is promoting ferritin turnover (Extended Data Fig. 10e). We also note that the abundance of NCOA4 was altered by iron loading or chelation, although no canonical iron response element was identified (Fig. 3b, Extended Data Fig. 10e)4.
We next examined the biological consequences of reduced iron availability and lysosomal ferritin degradation in response to NCOA4 depletion. Lysosomal iron has been shown to react with reactive oxygen species (ROS) leading to free radical formation via Fenton-like reactions, which results in lysosomal bursting and cell death26. Therefore, NCOA4 knockdown should provide protection from cell death after ROS challenge. As shown in Figure 3f, control cells were more sensitive to hydrogen peroxide challenge than NCOA4-depleted cells.
Selective autophagy is increasingly recognized as a regulated process through which specific cellular proteins, complexes, and organelles are degraded in the lysosome in response to diverse stimuli. Previous work suggested that delivery of the ferritin complex to lysosomes occurs via autophagy and regulates iron bioavailability5,6. Our identification of NCOA4 as a specific cargo receptor for ferritin provides the first mechanistic understanding of how the ferritin complex is selectively delivered to autophagosomes. Flux through the pathway is regulated by iron availability, and in turn, disruption of the pathway through modulation of NCOA4 levels leads to alterations in the activity of IRP2, and altered sensitivity of cells to ROS. Moreover, NCOA4 mRNA is induced in red blood cells during erythropoiesis, and its expression correlates with genes involved in heme-biosynthesis27, raising the possibility that NCOA4 function is important for both cellular remodeling and iron availability during differentiation28. While our data provides compelling evidence for the role of NCOA4 as a ferritinophagy cargo receptor, it does not rule out that it may have other roles in specific cellular contexts. However, given our findings in normal and tumor cells of diverse tissue origin, our work suggests that the targeting of ferritin to autophagosomes by NCOA4 is a general cellular mechanism for regulating bioavailable iron. While further functional studies are required to elucidate whether selective autophagy of particular cargo underlies the strong reliance of pancreatic and other cancer cells on autophagy for growth, our work reveals the potential of quantitative autophagy proteomics to uncover receptor-cargo relationships and to further elucidate the mechanisms underlying both macro and selective autophagy.
Methods Summary
Cells were grown in Lys(K)-free and Arg(R)-free DMEM/dialyzed fetal bovine serum, with light (K0, R0) or heavy (K8/R0 or K8/R10) Lys/Arg, and treated with CQ (25 μM) for 4 or 16 hours. Mixed cells were lysed and autophagosomes were purified as described14,15, prior to SDS-PAGE and in-gel digestion with trypsin or Lys-C, and LC-MS/MS (see METHODS). Candidate autophagosomal proteins were identified by employing a multi-step filtering process including a log2(heavy:light) ratio cut-off and enrichment in autophagosomes versus whole proteome. Interaction proteomics employed 293T, 8988T, or PANC1 cells stably expressing NCOA4 or FTH1 HA-FLAG-tagged constructs and CompPASS to identify HCIPs22. Chelation assays were performed similarly to those previously described with slight modifications5. Cells stably expressing shRNAs to NCOA4 were cultured for 24 hours in iron-enriched media (supplemental Ferric Ammonium Citrate added) and subsequently subjected to iron chelation with multiple different chelators prior to immunoblotting or immunofluorescence.
Full Methods
Cell culture and reagents
PANC1, PATU-8988T (8988T), MCF7, U2OS, IMR90, and 293T cell lines were obtained from the American Type Culture Collection or the German Collection of Microorganisms and Cell Cultures. HPDE cells were cultured as previously described7. All cell lines were tested routinely for mycoplasma contamination.
Antibodies
The following antibodies were used in these studies. Lamp 2 (Abcam Ab25631; Western 1:1000; IF 1:100); HA(Covance MMS 101P; Western 1:2000; IF 1:100); MAP1LC3B (LC3B) (Cell Signaling 2775; Western 1:2000); MAP1LC3B (LC3B) (Cell Signaling 3868; IF 1:200); MAP1LC3B (LC3B) (nanoTools 0231-100/LC3-5F10; IF 1:100); VDAC1 (Abcam Ab28777; Western 1:1000); Ferritin (Rockland 200-401-090-0100; IF 1:400); FTH1 (Cell Signaling 3998; Western 1:1000); NCOA4 (ARA70) (Bethyl Laboratories A302-272A; Western 1:1000); NCOA4 (Sigma SAB1404569; Western 1:1000); SQSTM1 (Abnova H00008878-M01; Western 1:5000); TAX1BP1 (Cell Signaling 8182; Western 1:1000); ATG5 (Cell Signaling 2630; Western 1:1000); HERC2 (BD Transduction Laboratories 612366; Western 1:1000); FTL (Abnova Ab69090; Western 1:1000); ACTB (Sigma A2066; Western 1:5000); MYC (Santa Cruz Sc-40; Western 1:1000); IRP2 (Santa Cruz Sc-33682; Western 1:500); TFRC (BD Transduction Laboratories 612124; Western 1:2000); Mannose 6-Phosphate Receptor (Abcam ab2733; IF 1:100); GABARAPL2 (gift from Millennium Pharmaceuticals, IF 1:100). The following secondary antibodies were used: Anti-Rabbit IgG (H+L) HRP Conjugate (Western Secondary 1:7500); Anti-Mouse IgG (H+L) HRP Conjugate (Western Secondary 1:7500); Alexa Fluor® 488 anti-Mouse IgG (H+L) (IF Secondary 1:1000); Alexa Fluor® 594 anti-Rabbit IgG (H+L) (IF Secondary 1:1000), Alexa Flour® 633 anti-Rabbit IgG (H+L) (IF Secondary 1:1000).
RNAi
ATG5 siRNA (Gene ID: 9474, NM_004849.3) was purchased from Invitrogen as previously published7. siRNAs were transfected using a reverse transfection protocol and RNAiMax (Invitrogen). siControl was a siRNA designed against luciferase. Lentiviral shRNA plasmid clones (pLKO.1) were obtained from the RNAi Consortium collection: shNCOA4-1: 5′ CCCAGGAAGTATTACTTAATT 3′ (TRCN0000019724), shNCOA4-2: 5′ GCTGGCAAACAGAAGTTTAAA 3′ (TRCN0000019726) and shGFP: 5′ GCAAGCTGACCCTGAAGTTCAT 3′ (Addgene plasmid #30323). NCOA4 Gene ID: 8031, NM_001145263.1. Lentivirus was produced as described previously7. Additional siRNAs used in this work were purchased from Invitrogen against NCOA4 (Gene ID: 8031, NM_001145263.1), siNCOA4-1: 5′ ACAAAGAUCUAGCCAAUCA 3′ and siNCOA4-2: 5′ GACCUUAUUUAUCAGCUUA 3′ and against HERC2 (Gene ID: 8924, NM_004667.5), siHERC2-1: 5′ GCACAGAGUAUCACAGGUA 3′ and siHERC2-2: 5′ CGAUGAAGGUUUGGUAUUU 3′.
Chemicals
Ferric ammonium citrate (FAC; Fisher Scientific, 36 to 180 μM titrated for each cell line based on level of FTH1 translation at 24h post-addition of FAC), deferoxamine mesylate (DFO; BioVision; 100 μM), bathophenanthroline disulfonate (BPS; Sigma; 300 μM), Deferiprone (DFP, Sigma, 100 μM), Deferasirox (DFX; Selleckchem; 30 μM), E64-d (Sigma; 10 μg/mL), PepstatinA (CalBiochem; 10 μg/mL), Bortezomib (BTZ; gift from Millenium Pharmaceuticals; 1 μM), Chloroquine (CQ; Sigma; 10 or 25 μM), Bafilomycin A1 (BAF; Sigma; 50 nM), hydrogen peroxide (H2O2; Sigma), CellTiter-Glo® (Promega).
SILAC-based Density Gradient Centrifugation Autophagosome Enrichment
PANC1, 8988T, and MCF7 cells were grown in lysine and arginine free DMEM supplemented with 10% dialyzed FBS (Gibco), 2 mM L-glutamine, penicillin-streptomycin, and light (K0) lysine (50 μg/mL) and light (R0) arginine (85 μg/mL). Heavy media was the same except the light lysine was replaced with K8-lysine (Cambridge Isotopes) and the light arginine was replaced with R10-arginine at the same concentrations (PANC1 and 8988T cell lines were also grown in K8-only heavy media (K8, R0)). Where indicated, cells (108) were treated with Wortmannin (200 nM) or CQ (25 μM) for the times indicated. After the indicated treatments, heavy and light cells were mixed 1:1 by cell number. Autophagosome purification was performed as described previously14,15 with slight modifications. All steps were carried out at 4°C. Briefly, cells were washed three times with PBS and resuspended in Buffer A (250 mM Sucrose, 10 mM HEPES, pH 7.4, 1 mM EDTA, protease inhibitors (EDTA-free, Roche)). Cells were lysed by nitrogen cavitation and homogenized using a potter-elvehjem homogenizer with a teflon pestle. Lysates were centrifuged at 2000×g, the supernatant was centrifuged at 17,000×g and resuspended in 0.95 mL Buffer A. Lysate was diluted with 1.45 mL 85.6% Nycodenz (Sigma-Aldrich) solution. A discontinuous Nycodenz gradient (26%, 24%, 20%, 15%) was layered on top of the lysate and spun at 24,700 rpm in a SW41 rotor (Beckman). Fractions were collected as indicated, pelleted at 24,000×g, and used for downstream applications (immunoblotting, electron microscopy, mass spectrometry).
Autophagosome Immunoisolation
8988T cells stably expressing GFP-LC3B were treated as above. Lysis and clarification centrifugation steps are as described above. GFP-based immunoisolation was performed as previously described12. Briefly, lysates were incubated with μMACS™ microbeads (magnetic beads coated with anti-GFP, MACS Miltenyi Biotec) for 1 hour at 4°C with mixing. Lysate-bead mixture was applied to a LS Column in a MidiMACS™ Separator, washed, eluted, pelleted and prepared for downstream analysis (mass spectrometry).
Mass spectrometry analysis of Autophagosomes
Enriched autophagosomes were prepared for mass spectrometry analysis as described17. Briefly, pelleted autophagosomes were resuspended in 2% SDS, 50 mM Tris, pH 7.5, 2 mM EDTA, boiled for 10 minutes, and centrifuged at 16,100×g for 5 minutes at room temperature. Supernatants were subjected to SDS-PAGE followed by in-gel digestion with trypsin (K8, R10 heavy samples and 8988T K8 sample) or Lys-C (PANC1 K8 heavy samples). For generation of comparison whole cell lysate datatsets, untreated heavy and light labeled cells were mixed in a 1:1 ratio, lysed as described above, and subjected to SDS-PAGE followed by in-gel digestion. Approximately 10 μg of total whole cell lysate was used for this analysis, however, multiple dilutions of whole cell lysate extract were separated by SDS-PAGE and a lane with equal intensity of overall coommassie staining to the autophagosome separations was chosen for processing. Peptides were subjected to the C18 stage-tip method and resuspended in 5% formic acid, 5% acetonitrile prior to mass spec analysis.
Peptides were separated on 100 μm × 25 cm C18 reversed phase (Maccel C18 3μm 200Å, The Nest Group, Inc.) with a 90 min gradient of 6% to 27% acetonitrile in 0.125% formic acid. The twenty-two most intense peaks from each full MS scan acquired in the Orbitrap Velos Pro (Thermo) were selected for MS/MS (RAW files available upon request). Sequest-based identification using a Human UNIPROT database followed by a target decoy-based linear discriminant analysis was used for peptide and protein identification as described16. Several experiments were processed in tandem using a protein sieve and protein assembler in-house processing tool, including PANC1 CQ 4 hr with PANC1 Ex. 1 and PANC1 Ex. 2, PANC1 Ex. 3 and PANC1 WCL together. MCF7 Ex. 1, MCF7 Ex. 2, and MCF7 WCL were processed together. However, all datasets were processed independently for calculation of Pearson correlation of log2(H:L) ratios between datasets for Fig. 1d and Extended Data Fig. 1j. Other parameters used for database searching include: 50 ppm precursor mass tolerance; 1.0 Da product ion mass tolerance; tryptic or Lys-C digestion with up to three missed cleavages; and variable oxidation of Met (+15.994946). A protein level false-discovery rate of <1% was used as a threshold for protein identifications using the target decoy strategy. Quantification of each protein was determined using the peak heights for light and heavy forms for that protein. The criterion for protein quantification was a summed signal-to-noise ratio of >10. Quantification of protein level was by calculating the median value of the ratios of light to heavy. Processed mass spectrometry data including SILAC profiling information is available in Supplementary Tables 2-4, 6-7, with a guide to the experimental conditions for each experiment in Supplementary Table 1. Of note, contaminants including Keratin, Lys-C, or Trypsin were removed from the data in order to avoid interference with data filtering as below. In addition, peptides identified for MAP1LC3B (Gene ID: 81631, NP_073729.1) are shared with a protein from an additional gene coding region with a gene symbol MAP1LC3B2 (Gene ID 643246, NP_001078950.1). The protein assembler program reported MAP1LC3B2 as the protein identified. However there were no peptides to discriminate the two proteins. There is one amino acid difference between MAP1LC3B and MAP1LC3B2 (C113Y). But no peptides were identified within the region of C113Y (identified peptides are as follows: 31IPVIIER37 and 52FLVPDHVNMSELIK65). We therefore report the log2(H:L) ratios for MAP1LC3B in our figures and Extended Data Fig. 2 but leave the protein identifier as MAP1LC3B2 in our supplementary tables. The processed proteomics data reported in the paper are available in the Supplementary Information; raw data files are available upon request.
Bioinformatic analysis
Candidate autophagosomal proteins were identified by employing a multi-step filtering process beginning with an enrichment cut-off including proteins with log2(heavy:light) ratios greater than 1.0 for MCF7 datasets, 1.5 for PANC1 16 h datasets and 0.5 for PANC1 and 8988T 4 h CQ datasets (0.5 was used as a cut-off in the 4 h CQ datasets given the overall relatively lower level of maximum log2(heavy:light) ratios). To be included, proteins had to be represented by 2 or more peptides. At this point in the analysis, approximately 600 proteins remained per dataset as potential candidates. In order to remove abundant proteins that may be non-specifically captured by bulk autophagy, candidates were filtered against the relative abundance of the proteome measured independently by LC-MS by directly comparing number of peptides identified per protein. Of note, a whole cell lysate proteome dataset was prepared from MCF7, PANC1, and 8988T cells as detailed above and was used for comparison within cell lines. In the datasets not processed together (PANC1 4h CQ and PANC1 Ex. 1 versus PANC1 WCL) comparisons were done based on pairing of gene symbols that did not distinguish between isoforms within the same gene symbol. Finally, a two-sided Student's t-test was used as a measure of statistical confidence of the observed log2(heavy:light) ratio taking into account the standard deviation of the log2(heavy:light) ratio and number of peptides measured per protein. A p-value of less that 0.05 was used for inclusion as a candidate (except for the PANC1 Ex. 1 dataset that used Lys-C for digestion where a p-value of less than 0.1 was used). For each dataset, this typically left approximately 150 candidates. We subsequently determined the overlap between the three PANC1 16 h CQ biological replicate datasets, identifying 86 proteins in common. The overlap between two MCF7 datasets consisted of 102 proteins. Finally, the overlap between the PANC1 and MCF7 datasets was determined (33 proteins in common, 122 proteins specific to either PANC1 or MCF7 datasets). This set of 155 proteins is referred to as Class 1 candidate autophagosomal proteins. A subset of the top 50 candidate proteins, termed Class 1A candidates, was developed based on presence in typically 3 or more independent experiments and those with known or potential links with autophagy. Finally, we also identified non-Class 1 proteins with a log2(H:L)>2.0 in any 2 of the 5 independent PANC1 (PANC1 Ex. 1-3) or MCF7 (Ex. 1-2) profiling experiments (16 h in CQ), and will refer to this dataset as Class 2 autophagosome-enriched proteins (Supplementary Table 5).
For analysis of the semi-quantitative 8988T GFP immunoisolation experiment, data was sorted by comparing log2 ratios of peptide numbers of proteins identified in autophagosomes purified from chloroquine versus wortmannin treated cells and log2 ratios of peptide numbers of proteins identified in autophagosomes purified from chloroquine treated cells versus peptide numbers from a whole cell lysate sample (Supplementary Table 7). Zero value denominators were systematically replaced with a value of 0.5 in order to generate a log2 ratio. Candidates were qualified as enriched if both log2 ratios were greater than 0.5.
For preparation of Extended Data Figure 5, the overlap between the MCF7 candidate proteins identified in this work and the stimulus independent autophagosomal candidates identified in Dengjel et al. was determined (Table 1 from Reference 13)13. Only two proteins overlapped, namely p62/SQSTM1 and GABARAPL2. To understand the lack of overlap between the datasets, we analyzed the proteins from the Dengjel et al. list using the data from our MCF7 experiments. Of note, the Dengjel et al. experiments also used the MCF7 cell line. We curated the Dengjel datasets to ensure the gene symbols published in the Dengjel study matched the updated version of the database used in our proteomics analyses (Supplementary Table 8). For the 96 Dengjel candidate proteins, we first determined the log2(H:L) ratio observed in both MCF7 Ex. 1 and 2 autophagosome enrichments (Supplementary Tables 4, 8). A majority of the proteins had a log2(H:L) ratio greater than 1.0 (77 out of 96). However, of the 77 proteins with a log2(H:L) ratio greater than 1.0, only 2 proteins passed the whole proteome abundance filter used in our analysis to remove abundant proteins that may be non-specifically captured by bulk autophagy (Extended Data Fig. 5a, Supplementary Table 8). We subsequently included the 3, 16 h PANC1 autophagosome proteomics datasets used to create the final Class 1 and 2 list in order to obtain as much coverage and comparison of the proteins identified in the Dengjel datasets as possible. The overlap with the Dengjel dataset is not significantly improved even when we expanded the data to include our PANC1 datasets (Extended Data Fig. 5b, Supplementary 8) now with an overlap of 4 proteins (SQSTM1, GABARAPL2, VPS35, MAP1LC3B). As the Dengjel authors derived their final autophagosomal candidate proteins from the overlap of lists from 3 different experiments, we also analyzed the 3 experiments independently using the same approach as above. Similarly, while a large proportion of the ‘Cluster A’ proteins from each experiment were enriched by log2(H:L) ratio in our datasets, only a small number of these proteins passed the whole proteome abundance filter and were found in our Class 1 and 2 dataset (Extended Data Fig. 5c-e). By relaxing the log2(H:L) ratio cutoff and redundancy stringency (reduced stringency column in the Supplementary Table 8 accompanying Extended Data Fig. 5b-e) to include non-class 1 and 2 proteins in the analysis, the additional overlap only ranges from 5-15% depending on the individual dataset. Of note, the Class 1-2 candidates that were also identified in the Dengjel Concanamycin A ‘Cluster A’ proteins included NCOA4. The likely reason for the large number of proteins with a log2(H:L) ratio greater than 1.0 but that did not pass the whole proteome abundance filter stems from the protein correlation profiling methodology used by Dengjel et al. Specifically, the authors relied on an ‘autophagosomal’ enrichment profile that required identification and quantification of candidate proteins in all 6 iodixanol fractions for creation of an evaluable profile. This likely biased their identification towards proteins with a high abundance in the whole proteome that would be more likely to be identified in all 6 fractions. As above, abundant proteins are more likely to be captured by non-selective bulk autophagy. Finally, by using the overlap of the 3 different experiments, they further biased their candidate list towards abundant proteins captured by autophagy under all 3 conditions tested such as proteasome subunits and heat-shock proteins.
Electron Microscopy
Pelleted autophagosome fractions (A1) were fixed in 2.5% Glutaraldehyde, 1.25% Paraformaldehyde, and 0.03% picric acid in 0.1 M sodium cacodylate buffer (pH 7.4) for 1 hour at room temperature, washed in 0.1 M Sodium Cacodylate buffer (pH 7.4), and post fixed for 30 minutes in 1% Osmium tetroxide (OsO4)/1.5% Potassium ferrocyanide (KFeCN6). Autophagosome pellets were washed in water 3 times and incubated in 1% aqueous uranyl acetate for 30 minutes followed by 2 washes in water and subsequent dehydration in grades of alcohol (5 minutes each; 50%, 70%, 95%, 2 times 100%). Autophagosome pellets were infiltrated overnight in a 1:1 mixture of propylene oxide and TAAB Epon (Marivac Canada Inc. St. Laurent, Canada). The samples were subsequently embedded in TAAB Epon and polymerized at 60°C for 48 hours. Ultrathin sections (about 60 nm) were cut on a Reichert Ultracut-S microtome, picked up on to copper grids stained with lead citrate and examined in a TecnaiG2 Spirit BioTWIN transmission electron microscope and images were recorded with an AMT 2k CCD camera.
Interaction Proteomics
Interaction proteomics was performed essentially as described previously, but with small modifications22. Briefly, 293T, PANC1, or 8988T cells were transduced with a lentiviral vector expressing NCOA4-HA-FLAG (NP_001138735.1) or FTH1-HA-FLAG (293T only, NP_002023.2) and stable cell lines were selected in puromycin. Cells from 4 × 15 cm dishes at 80% confluence were harvested and lysed in 3 ml of 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% Nonidet P40, 2 mM DTT and protease inhibitors. Cleared lysates were filtered through 0.45 μm spin filters (Millipore Ultrafree-CL) and immunoprecipitated with 30 μl anti-HA resin (Sigma). Complexes were washed 4 times with lysis buffer, exchanged into PBS for a further 3 washes, eluted with HA peptide and precipitated with 10% TCA. TCA-precipitated proteins were trypsinized, purified with Empore C18 extraction media (3 M), and analyzed via LC-MS/MS with a LTQ-Velos linear ion trap mass spectrometer (Thermo) with an 18 cm3 125 μm (ID) C18 column and a 50 minute 8%–26% acetonitrile gradient. All AP-MS experiments in 293T cells were performed in biological duplicate and for each biological experiment, complexes were analyzed twice by LC-MS/MS to generate technical duplicates. AP-MS experiments in PANC1 and 8988T cells were performed on a single AP but with technical duplicates. Spectra were searched with Sequest against a target-decoy human tryptic UNIPROT-based peptide database, and these results were loaded into the Comparative Proteomics Analysis Software Suite (CompPASS) to identify high confidence candidate interacting proteins (HCIPs)22. Here a statistics table, derived from analogous AP-MS data for 172 unrelated proteins in 293T cells was employed to determine weighted and normalized D-scores (WDN-score) as well as Z-scores based on spectral counts. The PANC1 data was analyzed using a PANC1 specific statistics table with 12 unrelated AP-MS PANC1 experiments and the 8988T AP-MS experiment was analyzed using the 293T statistics table given no statistics table was available for the 8988T cell line. The D-score measures the reproducibility, abundance, and frequency of individual proteins detected in each individual analysis. To identify NCOA4 associated proteins, we filtered proteins at a 2% false discovery rate for those with a WDN-score ≥ 1.0, Z and average assembled peptide spectral matches (APSMs) ≥ 2 in both biological duplicates. Data presented in Fig. 2e are derived from the 293T AP-MS experiments and the figure was made using Cytoscape29. STRING database data are represented as previously22. FTH1 and FTL interactors were confirmed in both PANC1 and 8988T AP-MS experiments and HERC2 in the 8988T AP-MS. NEURL4 was included in the interaction network given identification in the 293T AP-MS experiment and previously published data revealing a HERC2-NEURL4 interaction23.
Immunological Methods and Microscopy
To assess enrichment of autophagosome associated proteins in autophagosome purifications, the gradient load and autophagosome fractions were extracted in 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P40, 0.1% SDS, 2 mM EDTA, 2 mM DTT, protease inhibitors and subjected to immunoblotting with the indicated antibodies. To assess autophagosome integrity, purified autophagosome fractions (A1) were incubated in buffer A for 1 hour at 37°C plus or minus 0.5% Triton X-100, centrifuged at 16,000×g for 10 minutes, and the supernatant and pellet were subjected to immunoblotting with the indicated antibodies. To validate interactions between NCOA4 and candidate interacting proteins, 293T cells stably expressing NCOA4-HA-FLAG were harvested at 80% confluency. Extracts (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% Nonidet P40, 2 mM DTT, and protease inhibitors) from cells were subjected to immunoprecipitation with anti-FLAG resin (M2 agarose; Sigma), and washed complexes subjected to immunoblotting with the indicated antibodies. Likewise, 293T cells stably expressing FTH1-HA-FLAG with transient expression of MYC-NCOA4 were harvested at 80% confluency, lysed as above, subjected to immunoprecipitation as above, and immunoblotted with the indicated antibodies.
To examine localization of high-priority candidate proteins, full-length clones of the indicated candidate proteins were stably expressed as HA-FLAG fusions in 8988T and U2OS cells. Cells were plated on glass coverslips, treated with vehicle or CQ (10 μM, 4h), and fixed with 4% paraformaldehyde prior to immunofluorescence using anti-HA to detect candidate proteins and anti-LC3B to detect autophagosomes. All images were collected with a Yokogawa CSU-X1 spinning disk confocal with Borealis modification on a Nikon Ti-E inverted microscope equipped with 100× Plan Apo NA 1.4 objective lens. HA-candidate protein fluorescence was excited with the 488nm line (selected with an AOTF) from Spectral Applied Precision LMM-7 solid state laser launch. Emission was collected with a quad band pass polychroic mirror (Semrock) and a Chroma ET525/50m emissions filter. LC3B fluorescence was excited with the 561 nm line from the LMM-7 launch, and emission collected with the Semrock polychroic and a Chroma ET620/60m emission filter. For triple colocalization (Fig. 3c), ferritin fluorescence was excited with the 642 nm line from the LMM-7 launch, and emission collected with the Semrock polychroic and a Chroma ET700/75m emission filter. Images were acquired with a Hamamatsu ORCA-AG cooled CCD camera controlled with MetaMorph 7 software. Z-series optical sections were collected with a step size of 0.2 microns, using the internal Nikon Ti-E focus motor. Co-localization was determined based on examination of single z slices using MetaMorph 7 software (results are tabulated in Extended Data Fig. 2a).
To examine NCOA4 protein levels while blocking autophagic flux, 8988T cells were treated with vehicle (PBS or DMSO), CQ (10 μM, 8 hours), or Bafilomycin A1 (50 nM, 8 hours), extracted in 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P40, 0.1% SDS, 2 mM EDTA, 2 mM DTT, protease inhibitors and subjected to immunoblotting with the indicated antibodies.
To examine co-localization of NCOA4 with LC3B and ferritin, a N-terminal GFP fusion of NCOA4 was used. U2OS and 8988T cells stably expressing GFP-NCOA4 were treated with vehicle (PBS or DMSO), CQ (10 μM, 4 hours), Bafilomycin A1 (50 nM, 4 hours), or FAC (ferric ammonium citrate, 36 μM or 180 μM, 24 hours) and fixed with 4% paraformaldehyde prior to immunofluorescence using anti-LC3B to label autophagosomes or anti-ferritin in FAC loaded cells. Images were collected as described above with 6-10 z-series optical sections collected with a step size of 0.2 microns, using the internal Nikon Ti-E focus motor. Z-series are displayed as maximum z-projections.
GST Pull-down
The six human paralog ATG8 proteins were produced as N-terminal GST fusions (GST-ATG8) and transformed in E. coli BL21 (DE3) cells. Protein expression was induced with 1 mM IPTG for 3 hours at 37°C. Cells were collected by centrifugation, re-suspended in 25 mM Tris–HCl (pH 7.4), 10% (w/v) sucrose and lysed with a single freeze–thaw cycle followed by sonication. After centrifugation, lysates were incubated with 10μl of a 50% (v/v) slurry of glutathione Sepharose 4B beads for 30 minutes at 4°C. Beads were washed thoroughly with 150 mM NaCl, 20 mM Tris (pH 7.4), 2 mM DTT, 0.5% (w/v) Nonidet P-40, leaving 50–100μg of GST-fusion protein bound to the beads. Beads were then mixed with 300μg of lysate from a NCOA4-HA-FLAG stably expressing 293T cell line. The assay mix was incubated for 30 minutes at 4°C, and beads were washed four times with 1 ml wash buffer. Proteins were eluted with SDS sample buffer and analyzed by 4–20% gradient SDS–PAGE and immunoblotting with HA antibody. Pounceau stain was used to visualize GST-ATG8 bands.
Chelation assays
Chelation assays were performed similarly to those previously described with slight modifications5. Briefly, U2OS and 8988T cells stably expressing shRNAs as described above (shGFP, shNCOA4-1, shNCOA4-2, selected with puromycin) were plated (2×105 cells/well) and cultured for 24 hours in FAC (36 μM or 180 μM). Cells were washed three times with PBS and subjected to iron chelation with the described chelators minus or plus lysosomal protease inhibitors (E-64d and PepstatinA) or the proteasomal inhibitor, Bortezomib. Cells were harvested at the indicated time points, washed with PBS, and lysed in 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P40, 0.1% SDS, 2 mM EDTA, 2 mM DTT, protease inhibitors and analyzed by 4-20% gradient SDS-PAGE followed by immunoblotting with the indicated antibodies. Experiments were performed at a minimum in biological triplicate. Quantitation of western blots was performed using ImageJ30. Cells were treated similarly for immunofluorescence experiments (including IMR90 and HPDE cells). Cells plated on glass coverslips were treated as described and fixed with 4% paraformaldehyde prior to immunofluorescence using anti-ferritin and anti-LAMP2. Data were collected as described above. For quantitation of ferritin localization, at least 5, 40× fields were collected for biological replicate experiments. The fraction of punctate ferritin was quantitated by measuring the percentage of punctate ferritin immunofluorescence divided by total cellular ferritin immunofluorescence using MetaMorph software. At least 5 fields were quantitated from biological duplicate experiments. For Fig. 3d, the number of 8988T cells quantitated expressing shGFP was 155, for 8988T cells expressing shNCOA4-1: 113, and shNCOA4-2: 138. For Extended data fig. 8c, the number of U2OS cells quantitated is as follows: shGFP: 133 cells, shNCOA4-1: 103 cells, shNCOA4-2: 79 cells. For Extended data fig. 8d, the number of IMR90 cells quantified is as follows: shGFP: 29 cells, shNCOA4-1: 26 cells, shNCOA4-2: 31 cells. For Extended data fig. 9a, the number of 8988T cells quantified is as follows: 8988T-control MSCV-shGFP: 100 cells, 8988T-control-MSCV-shNCOA4-1: 125 cells, 8988T-control-MSCV-shNCOA4-2: 132 cells, 8988T-mouse-NCOA4-shGFP: 151 cells, 8988T-mouse-NCOA4-shNCOA4-1: 153 cells, 8988T-mouse-NCOA4-shNCOA4-2: 172 cells. Exposure times were held constant between experiments. Rescue experiments were performed using retroviral (MSCV blasticidin) based expression of the murine homolog of NCOA4 (NP_001029160.1) with a N-terminal HA tag.
H2O2 assay
Experiments were performed as described previously with slight modifications26. 8988T cells stably expressing shGFP or shNCOA4-1 were plated on a 96-well plate and treated with increasing concentrations of H2O2 as indicated for 30 minutes followed by 72 hours of culturing in complete media. Relative cell viability was measured using CellTiter-Glo®. Experiments were performed at least three times, each time in technical triplicate.
Extended Data
Extended Data Figure 1. Autophagosome enrichment protocol validation.
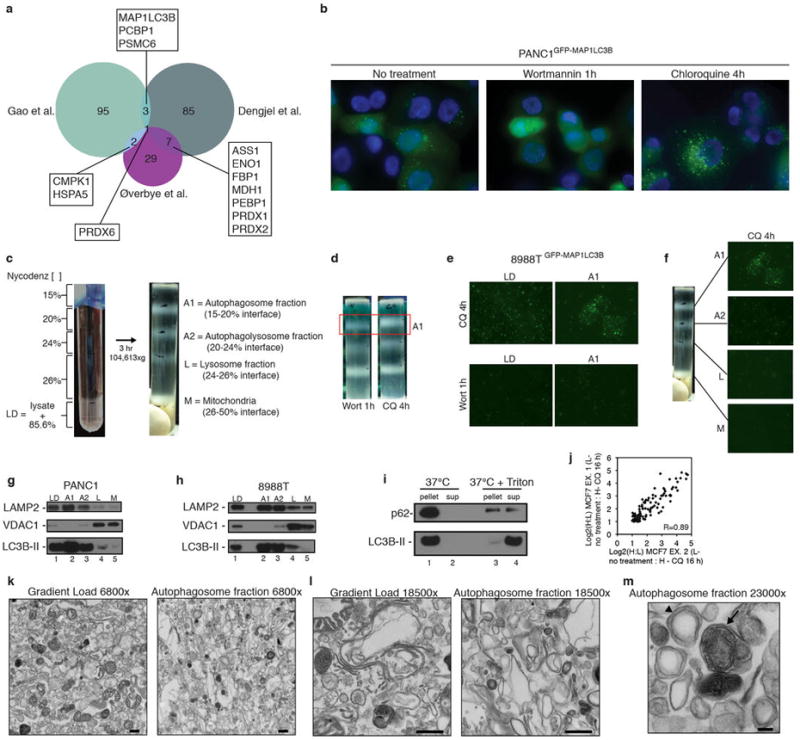
a, Overlap of proteins identified in three prior autophagosome proteomics studies11-13 as evaluated by area-proportional Venn diagram. Proteins overlapping between datasets are noted. b, PANC1 cells stably expressing GFP-MAP1LC3B have a high level of basal autophagy (left panel). 1h Wortmannin (200 nM) treatment blocks autophagosome formation (middle panel) and 4h chloroquine treatment (25 μM) causes accumulation of autophagosomes (right panel). c, Lysed 8988T cells mixed with Nycodenz and placed at the bottom of a discontinuous density gradient with Nycodenz layers at the indicated concentrations (left). After 3h centrifugation at indicated speed, 4 bands appear at the indicated interfaces with enrichment of the indicated organelles in each interface (right). d, 8988T cells treated with either Wortmannin (1h, 200 nM) or Chloroquine (4h, 25 μM) subjected to gradient centrifugation. A decreased amount of material is recovered from the A1 (autophagosome) interface due to the effect of Wortmannin on autophagosome formation. e, Fluorescence microscopy of gradient load (LD) and autophagosome fraction (A1) from 8988T cells stably expressing GFP-MAP1LC3B after either chloroquine or wortmannin treatment. f, Fluorescence microscopy of indicated fractions from density gradient of 8988T cells stably expressing GFP-MAP1LC3B treated with chloroquine (A1 fraction image is also presented in panel e and gradient picture is also presented in panel c). g, PANC1 autophagosome fractions analyzed by immunoblotting using antibodies to LAMP2, VDAC1, and LC3B. LD is gradient load, A1 is autophagosome fraction from 15-20% nycodenz interface, A2 is the autophagolysosome fraction from the 20-24% nycodenz interface, L is the lysosome fraction from the 24-26% nycodenz interface, M is the mitochondrial fraction from the 26%-50% nycodenz interface. h, 8988T autophagosome fractions analyzed as in g. i, 8988T autophagosomes (A1 fraction) were incubated at 37°C for 1 hour -/+ Triton X-100 and centrifuged at high speed. The resulting pellet was resuspended in equal volume to supernatant and assayed by immunoblotting with antibodies to p62 and LC3B. j, Pearson correlation plot for overlapping candidates from MCF7 experiments (102 proteins, comparing Ex. 1 vs. Ex. 2). k, Electron micrographs of 8988T gradient load (LD, left panel) and 8988T autophagosome fraction (A1, right panel) at 6800× magnification, scale bar 500 nm. l, 8988T gradient load (LD, left panel) and 8988T autophagosome fraction (A1, right panel) at 18500× magnification, scale bar 500 nm. m, 8988T autophagosome fraction (A1) at 23000× magnification, scale bar 100 nm. Arrowhead: double-membrane autophagosome, Arrow: fused autophagolysosome.
Extended Data Figure 2. Autophagosome proteomics candidate list and validation in 8988T cells.
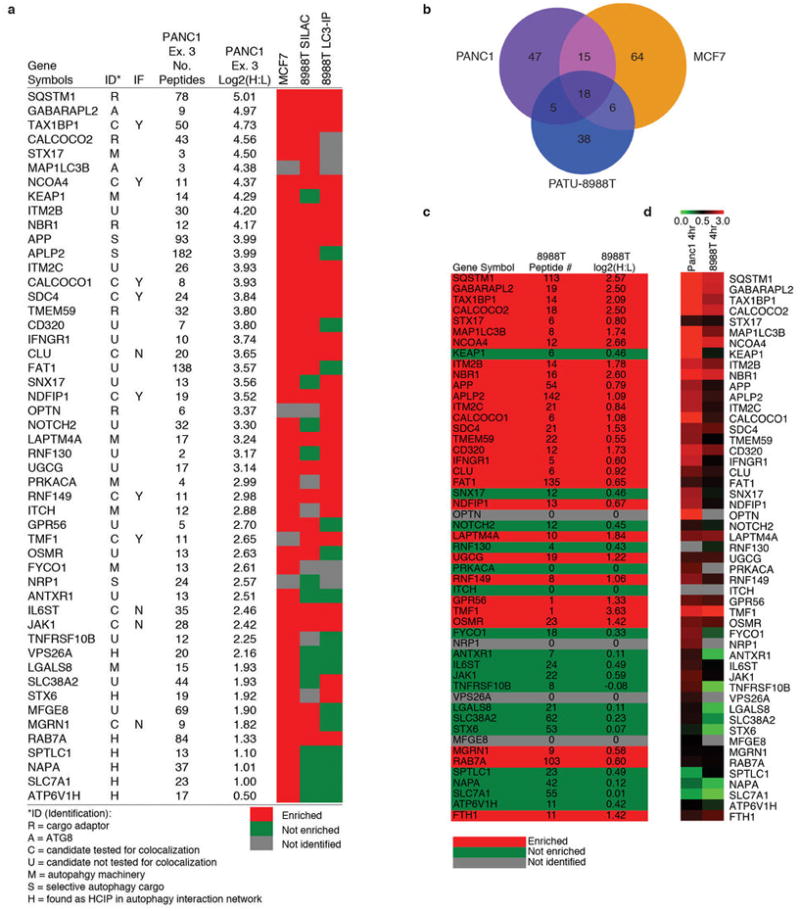
a, Autophagosome proteomics class 1A candidate list b, Overlap between Class 1 candidates (MCF7 and PANC1) and candidates from 4 hr CQ SILAC 8988T gradient autophagosome purification experiment. c, Data from 8988T SILAC gradient autophagosome for Class 1A candidates (and FTH1, see also Table S6). d, Heat map of Class 1A candidates (and FTH1) comparing PANC1 4h CQ and 8988T 4h CQ experiments.
Extended Data Figure 3. Candidates from autophagosome proteomics colocalize in autophagosomes.
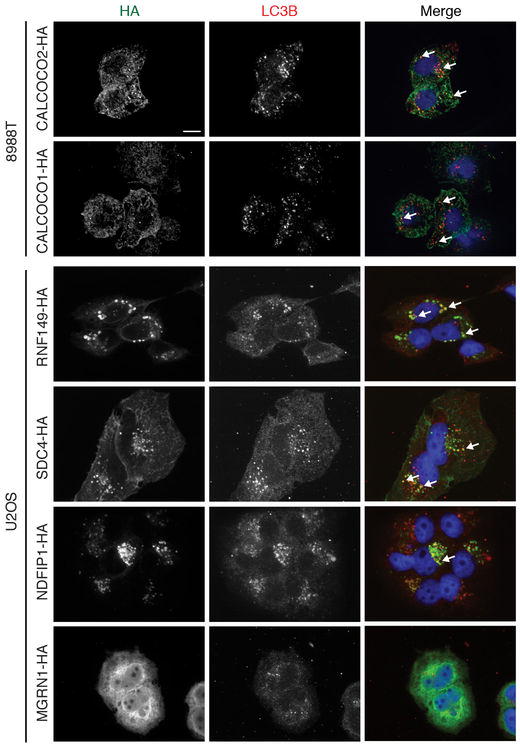
Representative confocal images of 8988T and U2OS cells expressing HA-tagged candidates after CQ treatment. Colocalization of HA-tagged candidates (green) with endogenous LC3B (red). Representative colocalization marked by white arrows. Scale bar, 10 μm. MGRN1 is included as an example of one of the candidates that did not show colocalization.
Extended Data Figure 4. Immunopurification-based autophagosome proteomics.
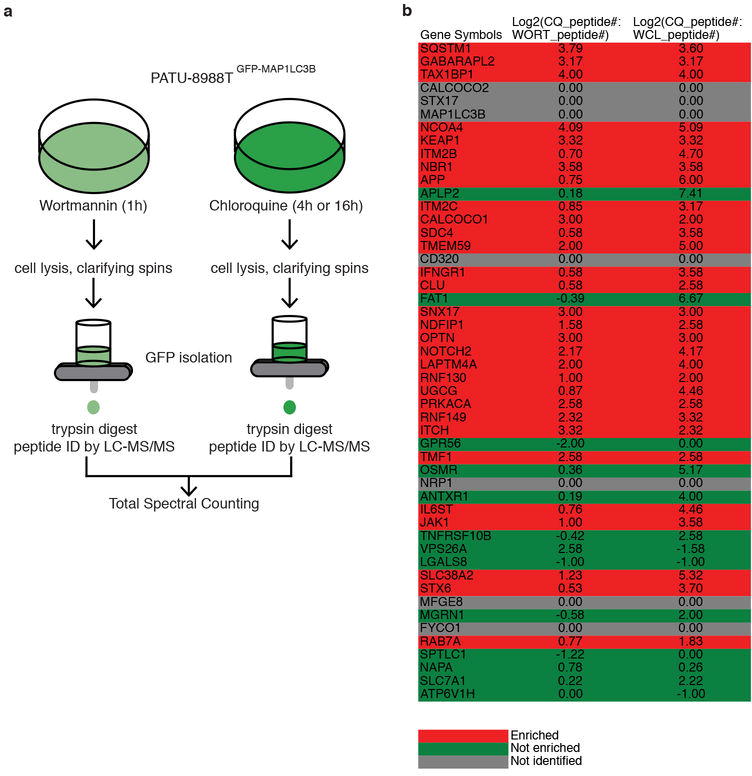
a, Schematic for GFP-immunoisolation of GFP-LC3B labeled autophagosomes from 8988T cells. b, Data from GFP-immunoisolation for Class 1A candidates (see also Table S7).
Extended Data Figure 5. Comparative analysis of Class 1 and 2 proteins with Dengjel et al. autophagosome proteomics.
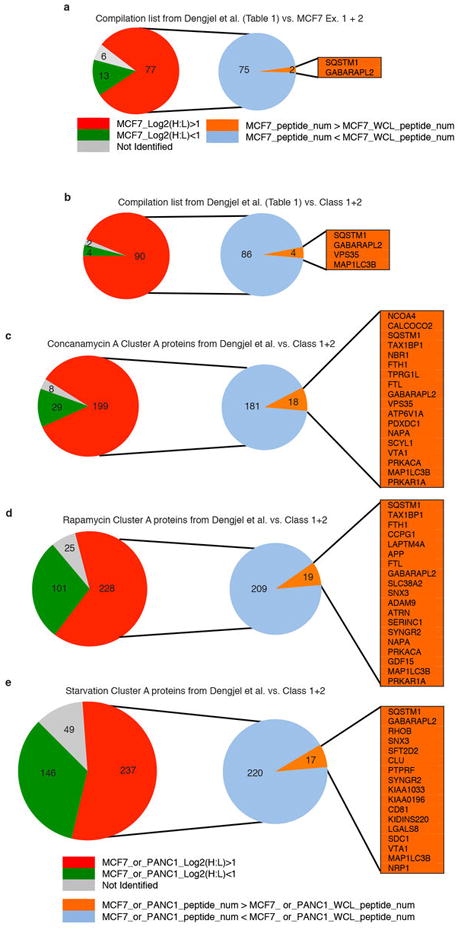
a, Analysis and comparison of Dengjel et al13 data and candidate list with data derived from MCF7 autophagosome proteomics experiments as detailed in methods section (see also Table S8). Orange shading of gene symbols denotes proteins identified as MCF7 candidates. The key for panel a only is below the pie charts. b-e, Analysis and comparison of Dengjel et al data and candidate list with data derived from both MCF7 and PANC1 autophagosome proteomics experiments (Class 1 and 2 proteins) as detailed in methods section. The key for panels b-e is below panel e. Orange shading of gene symbols denotes proteins identified as Class 1 or 2 candidates. (see also Table S8).
Extended Data Figure 6. NCOA4 colocalizes with LC3B and GABARAPL2 in autophagosomes.
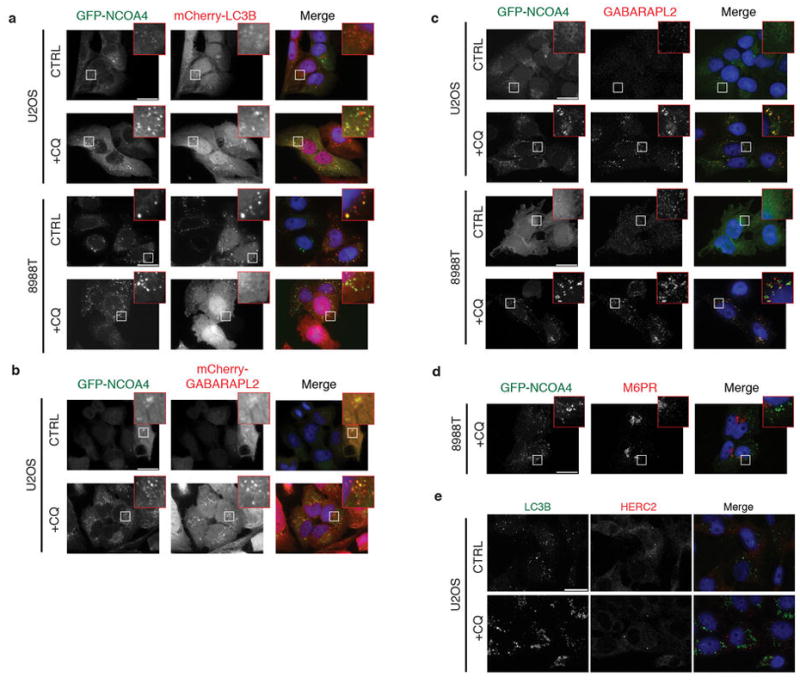
a, GFP-NCOA4 (green) co-localizes with mCherry-LC3B (red) in CQ-treated cells. Scale bar, 20 μm. b, GFP-NCOA4 (green) co-localizes with mCherry-GABARAPL2 (red) in CQ-treated cells. Scale bar, 20 μm. c, GFP-NCOA4 (green) co-localizes with endogenous GABARAPL2 (red) in CQ-treated cells. Scale bar, 20 μm. d, GFP-NCOA4 (green) does not colocalize with endogenous Mannose 6-Phosphate Receptor (M6PR) (red) in CQ-treated cells. Scale bar, 20 μm. e, HERC2 does not co-localize in autophagosomes. Immunostaining of U2OS cells subjected to CQ treatment, endogenous LC3 (green) and endogenous HERC2 (red), scale bar, 20 μm.
Extended Data Figure 7. Ferritin undergoes primarily lysosomal mediated degradation.
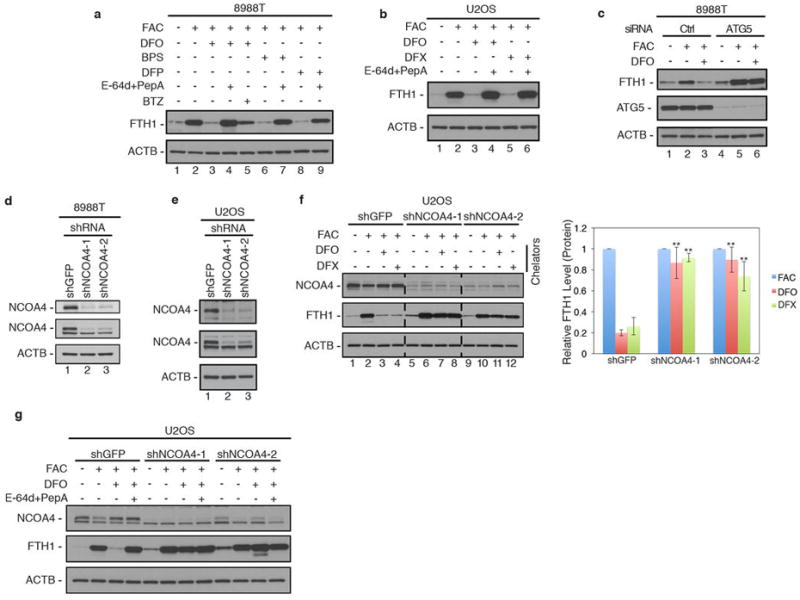
a, 8988T cells were cultured with FAC for 24 hours, washed, followed by chelation with 3 chelators (DFO, BPS, DFP) -/+ lysosomal protease inhibitors (E-64d and PepstatinA) or proteasomal inhibitor, Bortezomib (lane 5) for 8 hours. Lysates were immunoblotted using antibodies to ACTB and FTH1. b, U2OS cells were cultured with FAC for 24 hours, washed, followed by chelation with 2 chelators (DFO, DFX) -/+ lysosomal protease inhibitors (E-64d and PepstatinA) and analyzed as in panel A. c, 8988T cells transfected with luciferase control siRNA or validated siRNA to ATG5 were cultured with FAC, washed, and subjected to DFO chelation for 9 hours. Lysates were immunoblotted using antibodies to FTH1, ATG5, and ACTB. d, RNAi-mediated knockdown of NCOA4 in 8988T cells. 8988T cells stably expressing a control shRNA (shGFP) and two independent shRNAs to NCOA4 (shNCOA4-1 and shNCOA4-2) were lysed and analyzed by immunoblotting with two different antibodies to NCOA4 and ACTB as a loading control. Middle panel shows immunoblot probed with NCOA4 antibody from Bethyl Laboratories (#A302-272A). A non-specific band migrates just below the NCOA4 specific band. Top panel shows immunoblot probed with NCOA4 antibody from Sigma (SAB1404569). e, U2OS cells stably expressing shGFP, shNCOA4-1, or shNCOA4-2 were analyzed by immunoblotting as in panel d. f, NCOA4 depletion rescues ferritin degradation upon 9 hours iron chelation in U2OS. Relative FTH1 levels (n=3) for each chelator are quantified. Bars and error bars represent mean values and s.d., respectively: ** (p<0.01) and * (p<0.02) comparing FTH1 levels between different cell lines to shGFP control (one-sided t-test). g, U2OS cells stably expressing shGFP, shNCOA4-1, or shNCOA4-2 were cultured with FAC for 24 hours, washed, and subjected to DFO chelation -/+ lysosomal protease inhibitors. Lysates were immunoblotted using antibodies to NCOA4, FTH1, and ACTB.
Extended Data Figure 8. NCOA4 mediates autophagic delivery of ferritin to lysosomes.
a, 8988T cells were cultured in the presence or absence of FAC for 24 hours, washed, and subjected to DFO chelation -/+ lysosomal protease inhibitors (E-64d and PepstatinA). Cells were fixed and immunostained using antibodies to ferritin (red) and LAMP2 (green). Higher magnification views of the boxed areas are shown in the insets. Scale bar, 10 μm. b, U2OS cells treated and analyzed as in panel a. c, Immunostaining of U2OS cells expressing a control shRNA (shGFP) and two independent shRNAs to NCOA4 (shNCOA4-1 and shNCOA4-2) subjected to DFO chelation in the presence of lysosomal protease inhibitors for 9 hours. Scale bar, 10 μm. Punctate ferritin fraction was quantified from >75 cells per cell line from 2 independent experiments (number of U2OS cells quantitated is as follows: shGFP: 133 cells, shNCOA4-1: 103 cells, shNCOA4-2: 79 cells). Bars and error bars represent mean values and s.d., respectively: ***denotes p<0.001 using a one-sided t-test. d, IMR90 cells as in panel c were treated and immunostained as in panel c. Scale bar, 50 μm. Punctate ferritin fraction was quantified from >25 cells per cell line in two independent experiments and from more than 10 microscopy fields (number of IMR90 cells quantified is as follows: shGFP: 29 cells, shNCOA4-1: 26 cells, shNCOA4-2: 31 cells). Bars and error bars represent mean values and s.d., respectively: ***denotes p<0.001 using a one-sided t-test. e, HPDE cells as in panel c were treated and immunostained as in panel c. Scale bar, 10 μm. Quantitation was not possible due to the high background signal in shGFP control cells.
Extended Data Figure 9. A non-degradable murine NCOA4 rescues RNAi-mediated NCOA4 knockdown.
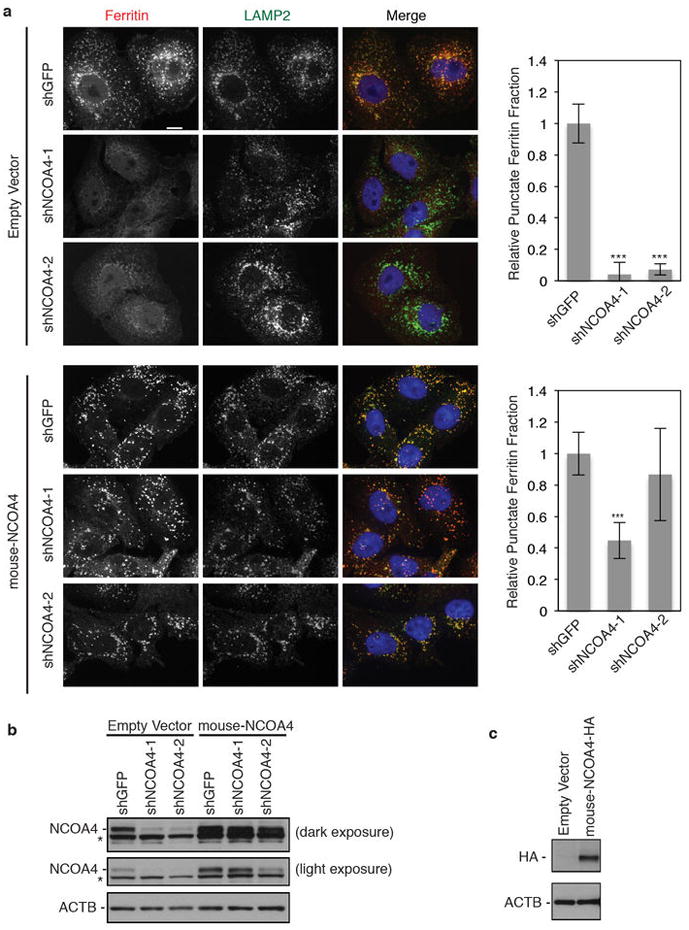
a, 8988T cells stably expressing either a control MSCV empty vector or the murine homolog of NCOA4 (selected with blasticidin) as well as stably expressing shRNAs (shGFP, shNCOA4-1, shNCOA4-2, selected with puromycin) were cultured in the presence or absence of FAC for 24 hours, washed, and subjected to DFO chelation in the presence of lysosomal protease inhibitors (E-64d and PepstatinA). Cells were fixed and immunostained using antibodies to ferritin (red) and LAMP2 (green). Scale bar, 10 μm. Punctate ferritin fraction was quantified from ≥100 cells per cell line (number of 8988T cells quantified is as follows: 8988T-control MSCV-shGFP: 100 cells, 8988T-control-MSCV-shNCOA4-1: 125 cells, 8988T-control-MSCV-shNCOA4-2: 132 cells, 8988T-mouse-NCOA4-shGFP: 151 cells, 8988T-mouse-NCOA4-shNCOA4-1: 153 cells, 8988T-mouse-NCOA4-shNCOA4-2: 172 cells). Bars and error bars represent mean values and s.d., respectively: ***denotes p<0.001 using a one-sided t-test. b, RNAi-mediated knockdown of NCOA4 in 8988T cells. 8988T cells stably expressing either a control MSCV empty vector or the murine homolog of NCOA4 (selected with blasticidin) as well as stably expressing a control shRNA (shGFP) and two independent shRNAs to NCOA4 (shNCOA4-1 and shNCOA4-2) were lysed and analyzed by immunoblotting with an antibody to NCOA4 and ACTB as a loading control. Light and dark exposures are shown. A non-specific band migrates just below the NCOA4 specific band. c, Expression of murine HA-NCOA4 protein in 8988Ts. Cell lysates were probed with HA antibody.
Extended Data Figure 10. NCOA4 mediates autophagic delivery of ferritin to lysosomes to control iron homeostasis.
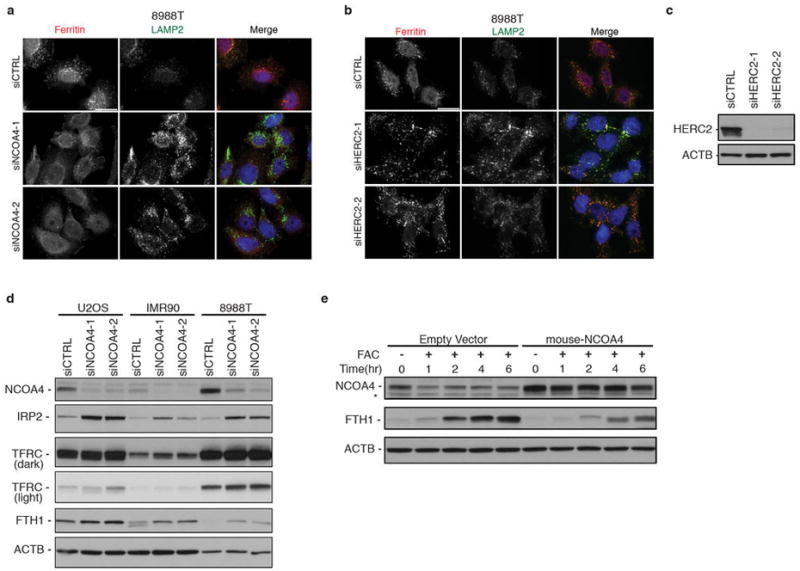
a, Immunostaining of 8988T cells transfected with luciferase control siRNA or two independent siRNAs to NCOA4 and subjected to DFO chelation in the presence of lysosomal protease inhibitors for 9 hours. Scale bar, 20 μm. b, Immunostaining of 8988T cells transfected with luciferase control siRNA or two independent siRNAs to HERC2 and subjected to DFO chelation in the presence of lysosomal protease inhibitors for 9 hours. Scale bar, 20 μm. c, 8988T cells were transfected with luciferase control siRNA or two independent siRNAs to HERC2. Lysates were immunoblotted using antibodies to HERC2 and ACTB. d, siRNA-mediated knockdown of NCOA4 in U2OS, IMR90 and, 8988T cells leads to increase in IRP2, FTH1, and TFRC levels. Cells were transfected with luciferase control siRNA or two independent siRNAs to NCOA4. Lysates were immunoblotted using antibodies to NCOA4, IRP2, TFRC, FTH1, and ACTB. Light and dark exposures are shown for TFRC. e, NCOA4 expression levels are affected by iron levels and NCOA4 levels affect ferritin levels in response to iron load. 8988T cells stably expressing either a control MSCV empty vector or mouse NCOA4 were cultured in the presence of FAC for the indicated times. Cells were lysed and analyzed by immunoblotting with antibodies to NCOA4, FTH1 and ACTB as a loading control. A non-specific band migrates just below the NCOA4 specific band.
Supplementary Material
Acknowledgments
We thank R. Everley and E. Huttlin for assistance with LC-MS/MS, A. Cuervo for assistance with autophagosome purification protocol development, Y. Liu for technical support with cell culture, A. White, J. Lydeard, and S. Hayes for interaction proteomics support, M. Haigis for access to nitrogen cavitation equipment, and the Nikon Imaging and Electron Microscopy Centers (Harvard Medical School) for imaging support. We thank Millennium Pharmaceuticals for the GABARAPL2 antibody. Support by NIH grants GM070565 and GM095567 to J.W.H. and National Cancer Institute Grant R01CA157490, ACS Research Scholar Grant RSG-13-298-01-TBG, and the Lustgarten Foundation, to A.C.K. J.D.M. was supported by an American Board of Radiology Holman Research Pathway Post-doctoral Fellowship and an American Society of Radiation Oncology Junior Faculty Career Research Training Award. J.W.H. is a Consultant for Millennium: The Takeda Oncology Company and Biogen Idec. A.C.K. is a Consultant for Forma Therapeutics.
Footnotes
Supplementary Information is available in a separate attachment.
Author Contributions: J.D.M., J.W.H., and A.C.K. conceived the experiments. J.D.M. performed all experiments. X.W. assisted with cell line generation. S.P.G. provided proteomics software and analysis support. J.D.M., J.W.H., and A.C.K. analyzed data and wrote the manuscript. All authors edited the manuscript.
Author Information: The processed proteomics data reported in the paper are available in the Supplementary Information; raw data files are available upon request.
The authors declare no competing financial interests.
References
- 1.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nature Cell Biology. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Molecular Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Molecular Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 4.Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry. 2012;51:5705–5724. doi: 10.1021/bi300752r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asano T, et al. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Molecular and Cellular Biology. 2011;31:2040–2052. doi: 10.1128/MCB.01437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kidane TZ, Sauble E, Linder MC. Release of iron from ferritin requires lysosomal activity. American Journal of Physiology Cell Physiology. 2006;291:C445–455. doi: 10.1152/ajpcell.00505.2005. [DOI] [PubMed] [Google Scholar]
- 7.Yang S, et al. Pancreatic cancers require autophagy for tumor growth. Genes & Development. 2011;25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sandilands E, et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nature Cell Biology. 2012;14:51–60. doi: 10.1038/ncb2386. [DOI] [PubMed] [Google Scholar]
- 9.Kimmelman AC. The dynamic nature of autophagy in cancer. Genes & Development. 2011;25:1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nature reviews Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Overbye A, Fengsrud M, Seglen PO. Proteomic analysis of membrane-associated proteins from rat liver autophagosomes. Autophagy. 2007;3:300–322. doi: 10.4161/auto.3910. [DOI] [PubMed] [Google Scholar]
- 12.Gao W, et al. Biochemical isolation and characterization of the tubulovesicular LC3-positive autophagosomal compartment. The Journal of Biological Chemistry. 2010;285:1371–1383. doi: 10.1074/jbc.M109.054197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dengjel J, et al. Identification of autophagosome-associated proteins and regulators by quantitative proteomic analysis and genetic screens. Molecular & Cellular Proteomics : MCP. 2012;11:M111 014035. doi: 10.1074/mcp.M111.014035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marzella L, Ahlberg J, Glaumann H. Isolation of autophagic vacuoles from rat liver: morphological and biochemical characterization. The Journal of Cell Biology. 1982;93:144–154. doi: 10.1083/jcb.93.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24:3052–3065. doi: 10.1096/fj.09-144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huttlin EL, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature Protocols. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 18.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeh S, Chang C. Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:5517–5521. doi: 10.1073/pnas.93.11.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao T, Brantley K, Bolu E, McPhaul MJ. RFG (ARA70, ELE1) interacts with the human androgen receptor in a ligand-dependent fashion, but functions only weakly as a coactivator in cotransfection assays. Molecular Endocrinology. 1999;13:1645–1656. doi: 10.1210/mend.13.10.0352. [DOI] [PubMed] [Google Scholar]
- 21.von Muhlinen N, et al. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Molecular Cell. 2012;48:329–342. doi: 10.1016/j.molcel.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Noel G, et al. Identification and proteomic analysis of distinct UBE3A/E6AP protein complexes. Molecular and Cellular Biology. 2012;32:3095–3106. doi: 10.1128/MCB.00201-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trump BF, Valigorsky JM, Arstila AU, Mergner WJ, Kinney TD. The relationship of intracellular pathways of iron metabolism to cellular iron overload and the iron storage diseases. Cell sap and cytocavitary network pathways in relation to lysosomal storage and turnover of iron macromolecules. The American Journal of Pathology. 1973;72:295–336. [PMC free article] [PubMed] [Google Scholar]
- 25.De Domenico I, et al. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. The EMBO Journal. 2006;25:5396–5404. doi: 10.1038/sj.emboj.7601409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurz T, Gustafsson B, Brunk UT. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. The FEBS Journal. 2006;273:3106–3117. doi: 10.1111/j.1742-4658.2006.05321.x. [DOI] [PubMed] [Google Scholar]
- 27.Nilsson R, et al. Discovery of genes essential for heme biosynthesis through large-scale gene expression analysis. Cell Metabolism. 2009;10:119–130. doi: 10.1016/j.cmet.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffiths RE, et al. The ins and outs of human reticulocyte maturation: autophagy and the endosome/exosome pathway. Autophagy. 2012;8:1150–1151. doi: 10.4161/auto.20648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods Reference
- 29.Lopes CT, et al. Cytoscape Web: an interactive web-based network browser. Bioinformatics. 2010;26:2347–2348. doi: 10.1093/bioinformatics/btq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.