

Abstract
The SLC37 family members are endoplasmic reticulum (ER)-associated sugar-phosphate/phosphate (Pi) exchangers. Three of the four members, SLC37A1, SLC37A2, and SLC37A4, function as Pi-linked glucose-6-phosphate (G6P) antiporters catalyzing G6P:Pi and Pi:Pi exchanges. The activity of SLC37A3 is unknown. SLC37A4, better known as the G6P transporter (G6PT), has been extensively characterized, functionally and structurally, and is the best characterized family member. G6PT contains 10 transmembrane helices with both N and C termini facing the cytoplasm. The primary in vivo function of the G6PT protein is to translocate G6P from the cytoplasm into the ER lumen where it couples with either the liver/kidney/intestine-restricted glucose-6-phosphatase-α (G6Pase-α or G6PC) or the ubiquitously expressed G6Pase-β (or G6PC3) to hydrolyze G6P to glucose and Pi. The G6PT/G6Pase-α complex maintains interprandial glucose homeostasis, and the G6PT/G6Pase-β complex maintains neutro-phil energy homeostasis and functionality. G6PT is highly selective for G6P and is competitively inhibited by cholorogenic acid and its derivatives. Neither SLC37A1 nor SLC37A2 can couple functionally with G6Pase-α or G6Pase-β, and the antiporter activities of SLC37A1 or SLC37A2 are not inhibited by cholorogenic acid. Deficiencies in G6PT cause glycogen storage disease type Ib (GSD-Ib), a metabolic and immune disorder. To date, 91 separate SLC37A4 mutations, including 39 missense mutations, have been identified in GSD-Ib patients. Characterization of missense mutations has yielded valuable information on functionally important residues in the G6PT protein. The biological roles of the other SLC37 proteins remain to be determined and deficiencies have not yet been correlated to diseases.
1. INTRODUCTION
The solute carrier (SLC) gene series consists of over 50 gene families that are predicted to encode membrane-bound transporters (He, Vasiliou, & Nebert, 2009). The SLC37 family consists of the following four sugar-phosphate exchange (SPX) proteins (Table 10.1): SLC37A1 (SPX1), SLC37A2 (SPX2), SLC37A3 (SPX3), and SLC37AA4 (SPX4) (Bartoloni & Antonarakis, 2004; Chou, Jun, & Mansfield, 2013). These proteins were originally grouped into the SLC37 family based on sequence homology to the bacterial organo-phosphate:phosphate (Pi) exchangers (Pao, Paulsen, & Saier, 1998), which are members of the major facilitator superfamily (MFS) (Reddy, Shlykov, Castillo, Sun, & Saier, 2012). The MFS transporters are single-polypeptide secondary carriers capable of transporting small solutes in response to chemiosmotic ion gradients, as opposed to primary carriers that create the gradients (ProSite PS50850). In the Transport Classification Database (TCDB), the SLC37 transporters are members of family 2.A.1.4 (http://www.tcdb.org/). Within the SLC37 family, the gene localization, structure, and amino acid compositions vary significantly, suggesting that the proteins have evolved independently and do not arise through gene duplication (Bartoloni & Antonarakis, 2004; Chou et al., 2013). The best characterized member is SLC37A4, which is better known as the glucose-6-phosphate (G6P) transporter (G6PT) (Chou et al., 2013; Chou, Matern, Mansfield, & Chen, 2002). In this review, we use G6PT when describing the SLC37A4 protein. Within the family, SLC37A1 and SLC37A2 are the most closely related at the protein level, while the G6PT protein is the most distant member (Fig. 10.1) (Bartoloni & Antonarakis, 2004; Chou et al., 2013; Corpet, 1988).
Table 10.1.
The SLC37 family of sugar-phosphate/phosphate exchangers
| Human gene name | SLC37A1 | SLC37A2 | SLC37A3 | SLC37A4 |
|---|---|---|---|---|
| Protein name(s) | SLC37A1, SPX1 | SLC37A2, SPX2 | SLC37A3, SPX3 | SLC37A4, G6PT, SPX4 |
| Known substrate | Glucose-6-phosphate | Glucose-6-phosphate | Unknown | Glucose-6-phosphate |
| Link to disease | None reported | None reported | None reported | GSD-Ib/non-GSD-Ia (OMIM 232220/232240) |
| Human gene locus | 21q22.3 | 11q24.2 | 7q34 | 11q23.3 |
| Sequence accession ID | NM_018964 | NM_198277 | NM_207113 | NM_001467 |
| Mouse model status (IMSR)a | Targeted mutation and gene trap—phenotype not reported | Targeted mutation—phenotype not reported | Targeted mutation and gene trap—phenotype not reported | Targeted mutation—GSD-Ib phenotype |
| Major protein isoform | 533-amino acid | 505-amino acid | 494-amino acid | 429-amino acid |
| Number of isoformsb,c | 1 | 4 | 3 | 2 |
The International Mouse Search Resource (IMSR): http://www.findmice.org/.
NCBI blast for amino acid identity and isoform prediction: http://blast.ncbi.nlm.nih.gov/Blast.cgi.
Nextprot for isoform prediction: http://www.nextprot.org/.
Figure 10.1.

Alignment of the amino acid sequences of human SLC37A1, SLC37A2, SLC37A3, and SLC37A4/G6PT by Multalin (http://multalin.toulouse.inra.fr/multalin)(Corpet, 1988). The amino acid sequences are GENBANK accession numbers NP_061837.3 (SLC37A1), NP_938018.1 (SLC37A2), AAH46567.1 (SLC37A3), and CAG33014.1 (SLC37A4/G6PT). In the consensus sequence, ! represents either I or V; $ represents either L or M; % represents either F or Y; and # represents N, D, Q, E, B, or Z.
The G6PT first came to attention as a result of studies of type I glycogen storagediseases(GSD-I),a groupofautosomalrecessivedisorderswithanoverall incidence of approximately 1 in 100,000 (Chou et al., 2002). Deficiencies in G6PT cause GSD type Ib (GSD-Ib, MIM232220), representing 15% of all GSD-I cases (Chou, Jun, & Mansfield, 2010a, 2010b; Chou et al., 2002). The primary in vivo function of G6PT is to translocate G6P from the cytoplasm into the lumen of the endoplasmic reticulum (ER) where it is hydrolyzed by a glucose-6-phosphatase (G6Pase) into glucose and Pi (Chou et al., 2002, 2010a, 2010b) (Fig. 10.2). This transport activity is dependent on the ability of G6PT toformafunctionalcomplexwithaG6Pase(Leietal.,1996).Intheabsenceofa G6Pase, the transport of G6P is minimal. There are two enzymatically active G6Pases. G6Pase-α (or G6PC) expression is restricted to the liver, kidney, and intestine (Lei, Shelly, Pan, Sidbury, & Chou, 1993), while G6Pase-β (or G6PC3) is expressed ubiquitously (Shieh, Pan, Mansfield, & Chou, 2003). Pathogenic mutations of G6Pase-α result in GSD-Ia (Chou et al., 2002, 2010b), a metabolic disorder characterized by the hallmark of fasting hypoglycemia. While GSD-Ia and GSD-Ib have similar metabolic characteristics, GSD-Ib differs by the presence of neutropenia. Notably, pathogenic mutations of G6Pase-β do not lead to the systemic metabolic disease associated with G6Pase-α mutations, but instead result in a severe congenital neutropenia syndrome type 4 (SCN4) (Boztug et al., 2009; Chou et al., 2010a, 2010b). Because of the functional coupling between G6PT and G6Pase-α/G6Pase-β, GSD-Ib provides the link between GSD-Ia and SCN4, being a G6P metabolic disease with neutropenia. The G6PT/G6Pase-α complex is critical for the maintenance of interprandial glucose homeostasis (Chou et al., 2002, 2010a, 2010b). The analogous G6PT/G6Pase-β complex plays a critical role in the maintenance of neutrophil (Cheung et al., 2007; Jun et al., 2010) and macrophage ( Jun, Cheung, Lee, Mansfield, & Chou, 2012) energy homeostasis and functionality. Three members of the SLC37 family (SLC37A1, SLC37A2, and SLC37A4/G6PT) function as Pi-linked G6P anti-porters capable of both homologous (Pi:Pi) and heterologous (G6P:Pi) exchanges, although they show differential inhibition by cholorogenic acid (Chen, Pan, Nandigama et al., 2008; Pan et al., 2011). In contrast, SLC37A3 lacks the antiporter activity (Pan et al., 2011). The molecular genetics and therapies for G6PT-deficiency/GSD-Ib have been reviewed recently (Chou et al., 2010a, 2010b, 2013) and will not be covered here. While structure–function studies have not been reported for the SLC37A1, SLC37A2, and SLC37A3 proteins, the SLC37A4/G6PT protein has been extensively characterized and is the focus of this review.
Figure 10.2.

The G6PT/G6Pase complex. The diagram shows a cross-section of the ER. The cell cytoplasm lies outside the ER membrane that encircles the ER lumen. In the gluconeogenic tissues of the liver, kidney, and intestine, G6PT couples with G6Pase-α, while in nongluconeogenic tissues (including neutrophils and macrophages), G6PT couples with G6Pase-β. The proteins are shown as spatially separated for clarity, but evidence suggests that they reside in physical contact with each other as a G6PT/ G6Pase complex. The G6PT/G6Pase-α complex expressed in gluconeogenic tissues maintains interprandial blood glucose homeostasis. The analogous G6PT/G6Pase-β complex, which is ubiquitously expressed, maintains neutrophil/macrophage energy homeostasis and functionality.
2. G6PT (SLC37A4 OR SPX4)
G6PT was the first SLC37 member identified and is the best characterized protein in this family (Chou et al., 2002, 2010a, 2010b, 2013). Human SLC37A4 is a single copy gene consisting of nine exons (Gerin, Veiga-da-Cunha, Noel, & Van Schaftingen, 1999; Hiraiwa, Pan, Lin, Moses, & Chou, 1999) on chromosome 11q23 (Annabi et al., 1998). The G6PT proteins from human (Gerin, Veiga-da-Cunha, Achouri, Collet, & Van Schaftingen, 1997), mouse (Lin, Annabi, Hiraiwa, Pan, & Chou, 1998), and rat (Lin et al., 1998) are predicted to be very similar, with mouse and rat proteins sharing 95% and 93% amino acid sequence identity to the human protein, respectively. Transcription of the human SLC37A4 gene gives rise to two alternately spliced transcripts, G6PT and variant G6PT (vG6PT) that encode nonglycosylated polypeptides of 429 and 451 amino acids, respectively, and reflect the absence or presence of exon-7 encoded sequences (Gerin et al., 1997; Hiraiwa et al., 1999; Lin, Pan, & Chou, 2000). The SLC37A4 gene is ubiquitously expressed (Lin et al., 1998), while the variant SLC37A4 is expressed primarily in the brain, heart, and skeletal muscle (Lin et al., 2000). The significance of these expression patterns is not understood. Both G6PT and vG6PT are equally active in G6P uptake (Lin et al., 2000) and so further discussion will refer to both species as G6PT, although most experiments use G6PT, not the variant.
2.1. The function of the G6PT protein
The primaryin vivo functionof theG6PTprotein is totranslocate G6P fromthe cytoplasm into the lumen of the ER (see Fig. 10.2). Inside the lumen, G6P is hydrolyzed to glucose and Pi either by G6Pase-α (Chou et al., 2010a, 2010b; Lei et al., 1993) or G6Pase-β (Chou et al., 2010a, 2010b; Shieh et al., 2003). G6Pase-α (Ghosh, Shieh, Pan, Sun, & Chou, 2002) and G6Pase-β (Ghosh, Shieh, Pan, & Chou, 2004) are ER transmembrane proteins with their active sites situated inside the ER lumen. Both G6Pases depend upon the G6PT for theiractivityintwodistinctways.First,G6PTprovidestheenzymesubstrateby transporting cytoplasmic G6P into the ER lumen. Second, G6PT provides a physical interaction that increases G6P transport activity, presumably by some allosteric mechanism. This functional coupling of G6PT and G6Pase-α activities has been demonstrated in several ways. The coupling was first revealed by characterizing hepatic microsomal G6P uptake activity in GSD-Ia mice (G6Pase-α −/− , G6PT+/+) (Lei et al., 1996). While hepatic microsomes from wild-typemicewereobservedtotransportG6Pefficiently,hepaticmicrosomes from GSD-Ia mice transported G6P inefficiently despite the presence of the transporter (Lei et al., 1996) (Fig. 10.3A). However, the G6P transport activity could be increased markedly if hepatic G6Pase-α activity was restored in GSD Ia mice via gene transfer (Zingone et al., 2000) (Fig. 10.3A).
Figure 10.3.

The function of G6PT. (A) Uptake of G6P into hepatic microsomes of wild-type mice (○), GSD-Ia mice (•), or GSD-Ia mice at day 14 post Ad-G6Pase-α infusion (△). (B) The cell-based microsomal G6P uptake activity. Uptake of G6P into microsomes prepared from COS-1 cells transfected with cDNA encoding G6PT alone (△), G6Pase-α alone (□), or G6PT and G6Pase-α (○). (C) The antiporter activity of G6PT. G6P uptake (left) and Pi uptake (right) were determined in proteoliposomes expressing G6PT preloaded with 50 mM Pi (○) or with MOPS buffer (•). Data are presented as the mean SEM.
Cell-based activity assays for recombinant G6PT proteins have also been developed (Chen, Lin, Pan, Hiraiwa, & Chou, 2000; Chen, Pan, Shieh, & Chou, 2002; Hiraiwa et al., 1999). Similarly, these assays showed that G6Pase-α-expressing G6PT-deficient microsomes (G6Pase-α+/+/G6PT −/− ) exhibited little or no G6P uptake activity (Fig. 10.3B), G6PT-expressing G6Pase-α-deficient microsomes (G6Pase-α −/− /G6PT+/+) exhibited low levels of G6P uptake activity (Fig. 10.3B), and G6Pase-α/ G6PT-expressing microsomes (G6Pase-α+/+/G6PT+/+) had markedly increased G6P transport activity (Fig. 10.3B). This suggests that the role of a coexpressed G6Pase-α is to increase the Pi concentration in the ER lumen to generate a driving Pi gradient required for antiporter activity of the G6PT protein. Further evidence came from studying reconstituted proteoliposomes preloaded with Pi. Using this system, G6PT was shown to be a Pi-linked G6P antiporter that catalyzes G6P:Pi and Pi:Pi exchanges (Chen, Pan, Nandigama, et al., 2008) (Fig. 10.3C). Preloading of proteoliposomes with Pi eliminates the need for a coexpressing G6Pase, and the Pi-loaded G6PT-proteoliposomes transport both G6P and Pi efficiently without a coexpressing G6Pase (Fig. 10.3C) (Chen, Pan, Nandigama, et al., 2008).
When the 23 SLC37A4 mutations identified in GSD-Ib patients were characterized using the cell-based assay and proteoliposome assay (see Fig. 10.8) (Chen, Pan, Lee, Peng, & Chou, 2008) similar results have been seen for 22 of the 23 SLC37A4 mutations, confirming the validity of each assay system. Interestingly, the outstanding mutation, p.Q133P, identified in a patient originally classified as a nonGSD-Ia (Veiga-da-Cunha et al., 1999), is devoid of G6P transport activity regardless of the assay methods, but retains 5% wild-type Pi transport activity (Chen, Pan, Lee, et al., 2008). The results suggest that further characterization of the G6P and Pi transport activities of G6PT mutations may yield insights into this genetic disorder. It is possible that while the active sites for the G6P and Pi transport activity overlap, they may also depend on residues that are not shared.
Figure 10.8.

A summary of mutations of the SLC37A4 gene that affect G6P uptake activity. The G6PT protein is shown embedded in the ER membrane. The 10-helical trans-membrane domains of G6PT are marked as boxes H1 to H10, the cytoplasmic loops are marked as C1 to C4, and the luminal loops are marked as L1 to L4. Mutations that have been shown to obliterate G6PT activity are listed without parentheses after. Mutants that retain residual activity are listed with the percent of wild-type transport activity retained noted in parentheses. The three naturally occurring signature motif mutations (p.Q133P, p.A148V, and p.G149E) identified to date in GSD-Ib patients are shown in boldface. Mutations that have not been functionally characterized are boxed. Mutations that have been characterized by both cell-based assays and proteoliposomal assays are shown in italic.
In addition to GSD-Ia (G6Pase-α deficiency), and GSD-Ib (G6PT deficiency), early biochemical classifications of GSD-I subtypes, based on limited kinetic analysis of the activity of the G6PT/G6Pase-α complex in patient's hepatic microsomes, implied there was a third form of GSD-I—GSD-Ic (Nordlie, Sukalski, Munoz, & Baldwin, 1983) characterized by a lack of Pi transport activity (Chou et al., 2002). Initially proposed to represent a third gene implicated in GSD-I, more recent genotyping of GSD-Ic or nonGSD-Ia patients has revealed deleterious SLC37A4 mutations (Galli et al., 1999; Janecke et al., 2000; Veiga-da-Cunha et al., 1998, 1999), consistent with the dual role of G6PT as both a G6P and Pi transporter.
2.2. Competitive inhibitors for G6PT
G6Pase-α is a nonspecific phosphatase and the substrate specificity of the G6PT/G6Pase-α complex is conferred by the high specificity of G6PT for G6P (Arion, Wallin, Carlson, & Lange, 1972). Chlorogenic acid, the ester of caffeic acid and (–)quinic acid (Fig. 10.4), is a reversible, competitive inhibitor for G6PT (Arion et al., 1998; Hemmerle et al., 1997). S3483 (Arion et al., 1998) and S4048 (Herling et al., 1999) (Fig. 10.4) are cholorogenic acid derivatives that are 2–3 orders of magnitude more potent as competitive inhibitors for G6PT. These inhibitors have been used in mechanistic studies on the metabolic defects in GSD-1 (Bandsma et al., 2001; Grefhorst et al., 2010). As expected, G6P transport activity in mouse hepatic microsomes (Lei et al., 1996) and microsomes and proteoliposomes containing recombinant G6PT protein is markedly inhibited by chlorogenic acid (Chen, Pan, Nandigama, et al., 2008; Hiraiwa et al., 1999).
Figure 10.4.
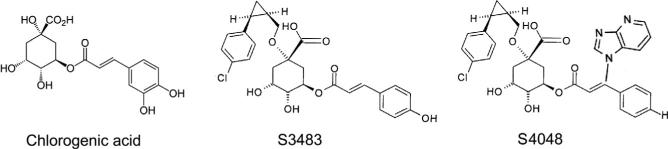
The structure of chlorogenic acid, S3483, and S4048.
2.3. Transmembrane topology of the G6PT protein
Hydropathy profile analysis predicts that G6PT contains either 10 (Fig. 10.5) (Hoffman & Stoffel, 1993) or 12 (Gerin et al., 1997) putative transmembrane helices. Along with G6PT, both the glycerol-3-phosphate transporter (GlpT) and the bacterial hexose-6-phosphate transporter (UhpT) are members of the organo-phosphate:Pi antiporter family (He et al., 2009; Pao et al., 1998). The X-ray crystallography structure of Escherichia coli GlpT has been solved and shown to contain 12 transmembrane helices (Huang, Lemieux, Song, Auer, & Wang, 2003). Homology modeling (Almqvist, Huang, Hovmöller, & Wang, 2004) lends support to G6PT having a similar 12-helix structure. To address the transmembrane structure of G6PT experimentally, investigators have performed mutational glycosylation and protease protection analyses. Protease protection confirmed that G6PT contains an even number (but not the exact number) of transmembrane helices, with both N and C termini facing the cytoplasm (Pan, Lin, & Chou, 1999). Because G6PT is a nonglycosylated protein, mutational glycosylation was used to differentiate between the 10 and 12 transmembrane domain models. The region of G6PT corresponding to amino acid residues 50–71 is predicted to form a transmembrane segment in the 12-domain model that would be resistant to glycosylation, while it is predicted to lie within a 51-residue lumenal loop available for glycosylation (Landolt-Marticorena & Reithmeier, 1994; Nilsson & von Heijne, 1993) in the 10-domain model (Fig. 10.5). It has been shown that glycosylation of membrane proteins is critical for their membrane targeting and folding (Goder & Spiess, 2001; Parodi, 2000), and incorrect glycosylation can cause misfolding and preferential degradation of the misfolded proteins (Caramelo & Parodi, 2007; Molinari, 2007). It was predicted that G6PT-p.T53N and G6PT-p.S55N glycoprotein mutants would be misfolded and degraded in the ER (Almqvist et al., 2004) if there were 12 helices, but expressed without disruption if the 10 helix model was correct. Contrary to the predictions based on protein homology, transient expression of G6PT, G6PT-p.T53N, and G6PT-p.S55N mutants showed that all were expressed at similar levels (Fig. 10.6A) (Pan et al., 1999, 2009), suggesting that the 10-helical model of G6PT is correct.
Figure 10.5.

The topology of human G6PT. The 429 amino acid residues are denoted by circles. The 34 amino acid residues altered by the 39 missense SLC37A4 mutations identified in GSD-Ib patients are shown in black. The signature motif (composed of amino acids 133–149) and the ER retention signal KKAE (composed of amino acids 426–429) are denoted by shaded circles. Three residues (p.Q133, p.A148, and p.G149) in the signature motif altered by missense SLC37A4 mutations identified in GSD-Ib patients are shown in dark shaded circles. The residues mutagenized for generating the glycosylation mutants p.T53N and p.S55N are denoted by arrows. Amino acids corresponding to essential residues in UhpT are indicated by arrowheads.
Figure 10.6.
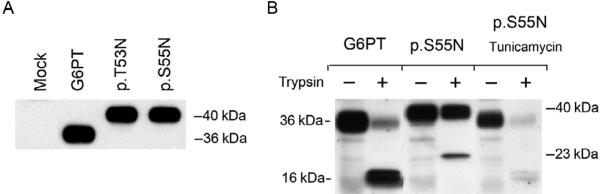
Protease sensitivity of G6PT wild-type and glycosylation mutants. Intact microsomes were isolated from COS-1 cells infected with Ad-G6PT, Ad-G6PT-p.T53N, or Ad-G6PT-p.S55N (Pan, Chen, Lee, & Chou, 2009). (A) Western blot analysis of the synthesis of G6PT, G6PT-p.T53N, and G6PT-p.S55N. (B) Western blot analysis of G6PT and G6PT-p.S55N after limited trypsin digestion in the absence and presence of tunicamycin.
Protease sensitivity assays were then used to confirm that the expressed proteins were embedded in the ER membrane. Intact microsomes expressing the wild-type or mutant G6PTs were subjected to limited trypsin digestion and analyzed by Western blot (Pan et al., 2009). G6PT wild-type and p.S55N (or p.T53N) mutant migrated as 36-kDa and 40-kDa polypep-tides, respectively, prior to trypsin digestion and 36-kDa/16-kDa and 40-kDa/23-kDa polypeptides, respectively, following limited trypsin digestion (Fig. 10.6B). In the presence of the glycosylation inhibitor tunicamycin, the G6PT-p.S55N (or p.T53N) mutant directed the synthesis of a 36-kDa polypeptide (Fig. 10.6B), similar to, the size of wild-type G6PT (Pan et al., 1999). As expected, the G6PT-p.S55N mutant synthesized in the presence of tunicamycin migrated as 36-kDa/16-kDa polypeptides after limited trypsin digestion similar to, wild-type G6PT (Fig. 10.6B). These data further supported a 10-domain model for the G6PT protein (Pan et al., 1999, 2009).
According to homology modeling (Almqvist et al., 2004), amino acid residues in G6PT that correspond to the amino acid residues essential for the activity of the 12-domain UhpT (Lloyd & Kadner, 1990) would also play a vital role in G6PT if it contained 12 domains. The amino acid residues absolutely required for transport activities in UhpT include p.R46 and p. R275 that are proposed to form the substrate-binding site (Fann et al., 1998), and p.D388 and p.K391 that are proposed to form an intra-helical salt bridge (Hall, Fann, & Maloney, 1999). The corresponding residues in G6PT are p.R28, p.K240, p.H366, and p.V369 (Fig. 10.5). Site-directed mutagenesis and transient expression analyses showed that while amino acid residue p.R28 is an essential residue in G6PT, p.K240, p.H366, and p.V369 are not (Pan et al., 2009), indicating that the structural requirements of G6PT and UhpT differ. In UhpT, the p.R46K and p.R275K mutations destabilize the protein (Fann et al., 1998). In contrast, the p.R28C, p.R28H, p.K240C, and p.K240R mutants support the synthesis of wild-type levels of G6PT proteins (Chen et al., 2000; Pan et al., 2009). Taken together, the experimental data solidly support the conclusion that G6PT is anchored in the ER by 10 transmembrane helices (Fig. 10.5).
2.4. Structure–function analyses of G6PT
G6PT deficiency gives rise to GSD-Ib, an autosomal recessive metabolic and immune disorder with an incidence of 1 in 700,000 (Chou et al., 2002 2010a, 2010b)., The GSD-Ib metabolic phenotype is impaired glucose homeostasis, characterized by fasting hypoglycemia, hepatomegaly, nephromegaly, growth retardation, hyperlipidemia, and lactic acidemia. The GSDIb immune phenotype is neutropenia and neutrophil dysfunction. The latter is characterized by impaired respiratory bust, chemotaxis, calcium mobilization, and phagocytic activities. A large number of mutations have been identified in the SLC37A4 gene of GSD-Ib patients. Characterizing these mutations may yield insights to this genetic disorder including the genotype–phenotype relationship for SLC37A4 mutations.
2.4.1 Mutations that cause GSD-Ib
To date, 91 separate mutations have been identified in the SLC37A4 gene of GSD-Ib/nonGSD-Ia patients (Dissanayake, Jayasinghe, Thilakaratne, & Jayasekara, 2011; Qiu, Lu, Wang, Qiu, & Wei, 2011; reviewed in Chou et al., 2013). These include 39 missense, 11 nonsense, 21 insertion/deletion (including 2 codon deletion mutations), and 19 splicing mutations that are distributed across the entire coding and exon–intron junction regions with the exception of the small exon VII (Fig. 10.7). There is also one gross deletion mutation of sequences upstream of exon 7. Characterization of missense and codon deletion mutations that result in single amino acid alterations provides valuable information on functionally important residues in a protein. In G6PT, 31 missense and 2 codon deletion mutations have been characterized functionally via site-directed mutagenesis and transient expression assays and were shown to abolish or reduce greatly microsomal G6P uptake activity (Chen et al., 2000, 2002; Chen, Pan, Lee, et al., 2008; Hiraiwa et al., 1999). These mutations are grouped into the following three categories: helical (H), nonhelical that includes cytoplasmic (C) and luminal (L) loops, and terminal that includes N- (p.MIV) and C- (p.R415X) terminal domains (Fig. 10.8).
Figure 10.7.

Mutations identified in the SLC37A4 gene of GSD-Ib patients. The SLC37A4 gene is shown as a line diagram with the nine exons marked as boxes I to IX. Black boxes represent coding regions, and white boxes represent the 5′- and 3′-untranslated regions of the SLC37A4 transcript. The positions of all known mutations are listed from left to right as insertion/deletion, splicing, missense, and nonsense mutations.
Among the 18 helical mutations, 9 missense and 2 codon deletion mutations are devoid of microsomal G6P uptake activity, while 7 missense mutations retain residual activities (Fig. 10.8). Among the 14 nonhelical missense mutations, 9 are devoid of microsomal G6P uptake activity (Fig. 10.8). It is interesting to note that of the 7 missense mutations characterized in luminal loop-1, 6 are null mutations—a finding consistent with this loop being critical for G6P uptake activity. Similarly, the N-terminus appears critical for function. The p.MIV N-terminal mutation that lacks the entire N-terminal domain (amino acids 1–7) and part of helix 1 (amino acids 8–16) is devoid of microsomal G6P transport activity (Chen et al., 2002). However, surprisingly, the p.R415X C-terminal mutation that lacks the entire C-terminal tail containing the ER retention signature motif (KKAE) ( Jackson, Nilsson, & Peterson, 1990, 1993) at amino acids 426–429 (Fig. 10.5) retains 47% of wild-type G6PT activity (Fig. 10.8) (Chen et al., 2000).
The following seven helical mutations lying within transmembrane domains destabilize the G6PT protein: p.G20D (H1), p.F93del (H2), p. C176R (H4), p.V235del (H5), p.I278N (H6), p.G339C (H8), and p.G339D (H8). p.G50R is the only nonhelical mutation that destabilizes the G6PT protein (Chen et al., 2002). The results suggest the structural integrity of the transmembrane helices is critical for the stability of the G6PT protein. The p.W393X nonsense mutation was identified in the SLC37A4 gene of a GSD-Ib patient (Hiraiwa et al., 1999). Transient expression analysis showed that the p.W393X mutant supported the synthesis of little or no G6PT. Two additional nonsense mutations (p.E401X and p.T408X) that delete or disrupt the structure of helix 10 were generated. Again, transient expression assays showed that the p.E401X and p.T408X mutants supported the synthesis of little or no G6PT protein (Chen et al., 2000). While the data show that G6PT mutants lacking an intact helix-10 are misfolded and undergo degradation within cells, additional nonsense mutations need to be characterized to delineate the role of transmembrane helices in the stability of G6PT.
2.4.2 The N- and C-terminal domains of G6PT
The N-terminal cytoplasmic domain of human G6PT consists of seven amino acids (Fig. 10.5). To examine the role of the N domain in G6P transport function, mutants were generated with sequential deletions (Chen et al., 2002). The p.A3M, p.Q4M, p.G5M, p.Y6M, p.G7M, and p.Y8M mutants retained 44.5%, 20.2%, 15.1%, 17.4%, 16.5%, and 8.7% of wild-type G6PT activity, respectively. Because all these N-terminal deletion mutants expressed similar amounts of G6PT proteins as the wild-type transporter, the N domain appears essential for optimal microsomal G6P uptake, but not G6PT stability.
The C-terminal cytoplasmic domain (amino acids 415–429) of human G6PT consists of 15 amino acids (Fig. 10.5) and appears important for protein stability. The naturally occurring R415X mutant lacking the entire cytoplasmic tail is degraded more rapidly than wild-type G6PT (Chen et al., 2000). To determine the minimal length of the cytoplasmic tail required for G6PT folding and/or stability, G6PT mutants were generated that lacked the last 14 (p.N416X), 13 (p.I417X), 12 (R418X), 11 (p.T419X), and 10 (p.K420X) amino acids (Chen et al., 2000). The length of the first three residues at amino acids 415–417 of the cytoplasmic tail appeared to correlate with stability, with p.R415X<p.N416<p.I417X to wild-type G6PT stability, while p.R418X, p.T419X, and p.K420X mutants supported the synthesis of similar amounts of proteins as wild-type G6PT (Chen et al., 2000).
2.4.3 The signature motif in G6PT
The organo-phosphate:Pi antiporter family members (G6PT, GlpT, and uhpT), the G6P receptor (uhpC), and the phosphoglycerate transporter (PgtP) share a signature motif (ProSite PDOC00726), which lies between amino acids 133–149 in human G6PT (Fig. 10.9A). One SLC37A4 mutation identified in GSD-Ib/nonGSD-Ia patients, p.Q133P (Veiga-da-Cunha et al., 1999), which alters the first amino acid in this motif, was shown to be devoid of G6P transport activity but retains 5% wild-type Pi transport activity (Chen, Pan, Lee, et al., 2008; Chen et al., 2002). The other SLC37A4 mutation identified in GSD-Ib patients, p.G149E (Hiraiwa et al., 1999), which alters the last amino acid in this motif, was shown to be a null mutation that completely abolished microsomal G6P and Pi uptake (Chen et al., 2002). Although the p.A148V SLC37A4 mutation identified in a GSD-Ib patient has not been functionally characterized, the p.A148D mutation was shown to be null (Fig. 10.9B). To evaluate the role of the signature motif in G6PT activity, G6PT mutants involving the invariant residues (p.G135D and p.N146D), the near invariant residues (p.L147A and p.A148D), and the uhpC invariant residues (p.W137A, p.W138A, and p.A139D) were generated and functionally characterized (Pan et al., 2003). None of the signature mutants destabilize the G6PT protein (Fig. 10.9B). However, the p.G135D, p.W138A, p.A139D, p.N146D, and p.A148D mutants, along with p.Q133P and p.G149E mutants are devoid of microsomal G6P uptake activity (Fig. 10.9B), suggesting that in G6PT the signature motif is a functional element required for optimal microsomal G6P transport.
Figure 10.9.

The signature motif in G6PT. (A) Alignment of the signature motifs of mammalian G6PT, uhpT, uhpC, GlpT, and PgtP. Amino acid residues mutated in human G6PT are shown in bold. (B) Western blot analysis of synthesis and microsomal G6P uptake activity of the G6PT signature motif mutants. Mutations that have been shown to obliterate G6PT activity are listed without parentheses after. Mutants that retain residual activity are listed with the percent of wild-type transport activity retained noted in parentheses.
3. SLC37A1, SLC37A2, AND SLC37A3
SLC37A1 or SPX1 (Bartoloni & Antonarakis, 2004; Bartoloni et al., 2000; Chou et al., 2013; Iacopetta et al., 2010; Pan et al., 2011), SLC37A2 or SPX2 (Bartoloni & Antonarakis, 2004; Chou et al., 2013; Kim, Tillison, Zhou, Wu, & Smas, 2007; Pan et al., 2011; Takahashi et al., 2000), and SLC37A3 or SPX3 (Bartoloni & Antonarakis, 2004; Chou et al., 2013; Pan et al., 2011) are the less characterized members of the SLC37 family. Human SLC37A1, SLC37A2, and SLC37A3 are similar to G6PT in being ER-associated transmembrane proteins (Pan et al., 2011).
3.1. SLC37A1 (SPX1)
The human SLC37A1 gene consists of 19 coding exons on chromosome 21q22.3 that encodes a 533 amino acid polypeptide with a calculated molecular weight of 58 kDa (Table 10.1) (Bartoloni et al., 2000). The SLC37A1 protein shares 59% homology to SLC37A2, 35% homology to SLC37A3, and 22% homology to SLC37A4 (Fig. 10.1) (Bartoloni & Antonarakis, 2004; Chou et al., 2013). The SLC37A1 protein also shares 30% sequence identity to GlpT, suggesting that SLC37A1 could be a mammalian glycerol-3-phosphate transporter (Bartoloni et al., 2000). The SLC37A1 gene is widely expressed, with the highest transcript levels in adult kidney, bone marrow, intestine, spleen, and liver (Bartoloni et al., 2000). In the liver and kidney, the SLC37A1 transcripts constitute less than 2% of the levels seen for SLC37A4 (Pan et al., 2011). In the intestine, pancreas, and macrophage, the SLC37A1 transcripts constitute 60%, 69%, and 43%, respectively, of the levels seen for SLC37A4 (Chou et al., 2013). In neutrophils, the SLC37A1 transcripts are significantly higher (2.8-fold) than that of SLC37A4. The expression of the SLC37A1 transcript is upregulated by epidermal growth factor in breast cancer and endometrial cancer cells (Iacopetta et al., 2010). One hypothesis is that SLC37A1 is involved in phospholipid biosynthesis (Iacopetta et al., 2010), which has a role in the proliferation of tumor cells. However, to date, there is no direct evidence for a role of SLC37A1 in breast cancer, or any other disease. Because the levels of the SLC37A1 transcript in neutrophils is higher than that of SLC37A4, which is known to play a vital role in neutrophil homeostasis (Chou et al., 2010a, 2010b, 2013), neutrophils may be an attractive cell type to initiate a search for the biological role of SLC37A1.
Figure 10.10.

The activities of SLC37A1, SLC37A2, and G6PT, and the organo-phosphate:Pi antiporter family consensus signature motif. (A) The antiporter activity. The activity was determined in 50 mM Pi-preloaded proteoliposomes expressing SLC37A1, SLC37A2, or G6PT in the absence or presence of 2 mM chlorogenic acid. Data are presented as the mean SEM. (B) Effects of G6Pase-α on microsomal G6P transport activity of SLC37A1, SLC37A2, or G6PT. (C) The organo-phosphate:Pi antiporter family signature motif, ProSite PDOC00726, present in the SLC37 members. The motif lies at residues 188–204 in SLC37A1, residues 176–192 in SLC37A2, residues 174–190 in SLC37A3, and residues 133–149 in SLC37A4/G6PT protein sequences. Only SLC37A4 shows an exact match with the ProSite motif. While only one residue (p.G135) is invariant across the family, 12 of the 17 residues in the ProSite motif are conserved in all members. The five nonconserved residues that cluster at the N and C termini of the motif are boxed. The two residues in SLC37A3 (p.G174 and p.C185) with significantly different characteristics are boxed and shaded. The redundancy at each position of the ProSite motif is shown. X denotes any amino acid.
3.2. SLC37A2 (SPX2)
The human SLC37A2 gene consists of 18 coding exons on chromosome 11q24.2 that encodes four alternative spliced transcripts (Table 10.1). The 505-residue SLC37A2 shares 59% homology to SLC37A1, 36% homology to SLC37A3, and 23% homology to SLC37A4 (Fig. 10.1) (Bartoloni & Antonarakis, 2004; Chou et al., 2013). The murine Slc37a2 was first identified as a cAMP-inducible gene in RAW264 macrophages (Takahashi et al., 2000). The 510 amino acid murine Slc37a2 protein contains three potential N-linked glycosylation sites and migrates as a heterogeneous species of 55–75 kDa (Kim et al., 2007). In this respect, it differs from the nonglycosylated SLC37A1, SLC37A3, and SLC37A4. The Slc37a2 transcript in mice is primarily expressed in the spleen, thymus, and macrophages (Kim et al., 2007). Compared to Slc37a4, the Slc37a2 transcript is highly expressed in neutrophils and macrophages. Human SLC37A2 is post-translationally modified by N-linked glycosylation, and the human SLC37A2 transcript increases markedly during differentiation of THP-1 human monocytes to macrophages (Kim et al., 2007). The tissue-specific expression of SLC37A2 in macrophages and neutrophils may point to a biological transport role in these cells.
3.3. SLC37A3 (SPX3)
The human SLC37A3 gene consists of 17 coding exons on chromosome 7q34 that encodes three alternatively spliced transcripts (Table 10.1) (Bartoloni & Antonarakis, 2004). The SLC37A3 transcripts are expressed in liver, kidney, intestine, pancreas, neutrophils, and macrophages (Pan et al., 2011). Interestingly, the levels of the SLC37A3 transcript in the pancreas and neutrophils are significantly higher than the levels of other SLC37 members (Chou et al., 2013), suggesting a yet undiscovered role for this protein in the pancreas and neutrophils. Indeed, all SLC37 members seem to be expressed well in neutrophils, suggesting a possible common role in the immune system.
3.4. Functional analysis of the SLC37A1, SLC37A2, and SLC37A3 proteins
Using reconstituted proteoliposomes, investigators demonstrated that SLC37A1 and SLC37A2 are Pi-linked G6P antiporters capable of G6P:Pi and Pi:Pi exchanges, as seen with G6PT (Fig. 10.10A) (Pan et al., 2011). The SLC37A3 isoform of 494 amino acids lacks G6P antiporter activity (Pan et al., 2011), and there are no other reported activities for the protein. The G6PT-mediated microsomal G6P uptake activity, which is co-dependent upon G6Pase-α activity (Lei et al., 1996) and sensitive to cholorgenic acid inhibition (Arion et al., 1998), is the rate-limiting step in endogenous glucose production (Arion, Lange, & Ballas, 1976). Despite SLC37A1, SLC37A2, and G6PT/SLC47A4 being Pi-linked G6P antiporters, the transport activity of SLC37A1 and SLC37A2 differs from that of G6PT. First, the transport activities of SLC37A1 and SLC37A2 are not sensitive to chlorogenic acid inhibition (Pan et al., 2011) (Fig. 10.10A). Second, neither SLC37A1 nor SLC37A2 can couple functionally with either G6Pase-α (Fig. 10.10B) or G6Pase-β to mediate micro-somal G6P uptake (Pan et al., 2011). Studies have shown that in human cell lines, there is an additional microsomal G6P transporter activity that is insensitive to chlorogenic acid inhibition (Leuzzi et al., 2001). In contrast, to G6PT that is highly selective for G6P, the G6PT-unrelated microsomal transport activity is equally responsive to G6P and glucose 1-phosphate (Leuzzi et al., 2001). Presently, it is not known whether these activities are in fact due to the SLC37 family proteins or not. Taken together, multiple sugar-phosphate transporters exist in the ER, although the physiological substrate for SLC37A1 and SLC37A2 remains to be determined.
Interestingly, the 17-residue signature motif (ProSite PDOC00726) shared by G6PT, GlpT, uhpT, uhpC, and PgtP (Fig. 10.9A) is also shared by SLC37A1, SLC37A2, and SLC37A3 (Fig. 10.10C). While SLC37A4/ G6PT shows an exact match to the consensus sequence, SLC37A1 and SLC37A2 differ from the consensus in five positions but contain residues with similar characteristics (Fig. 10.10C). Between SLC37A1 and SLC37A2, this motif is almost entirely conserved. The signature motif in SLC37A3 also differs from SLC37A4 at the same five positions but has two residues with significantly different characteristics, including a small noncharged Gly in place of the charged residue at position 1, and a Cys at position 12 (Fig. 10.10C). It is possible that this motif in SLC37A3 forms a different tertiary structure that may contribute to the lack of antiporter activity of this protein. It would be of interest to determine the impact of mutations in these residues on the transport activity of SLC37A3.
4. SYNOPSIS
A deficiency in G6PT causes the genetic disorder GSD-Ib, characterized by impaired glucose homeostasis, neutropenia, and myeloid dysfunction. A large number of SLC37A4 mutations identified in GSD-Ib patients have been functionally characterized and shown to reduce or abolish G6PT activity. G6PT is anchored in the ER by 10 transmembrane helices with both N and C termini facing the cytoplasm. Despite the identification of cholorogenic acid and its derivatives as potent competitive inhibitors for G6PT, the G6P-binding motif in the G6PT protein remains to be identified. SLC37A1, SLC37A2, and SLC37A3 are all predicted to contain 10–12 transmembrane domains. However, very little is known about the other SLC37 proteins beyond the P:P and G6P:P exchange activities of SLC37A1 and SLC37A2. The inability of SLC37A1 and SLC37A2 to couple functionally with a G6Pase, compounded with their lack of sensitivity to inhibition by cholorogenic acid, suggests that they are not the physiological ER G6P transporter. Despite expressing similar or higher levels than G6PT in key tissues, including neutrophils and macrophages, SLC37A1, SLC37A2, and SLC37A3 do not appear to have roles in regulating neutrophil/macrophage glucose metabolism or blood glucose homeostasis. Future structure analysis of these proteins may yield clues to their function. The mouse model of GSD-Ib that mimics the phenotype of human GSD-Ib has been generated (Chen et al., 2003) and is now being used to develop new therapies for this disorder. Mouse models of the other SLC37 family members do exist but their phenotypes remain to be characterized. Studying these models may give valuable insights into the biological and functional roles of the other SLC37 family members. In summary, there is still much to learn about the other ER transmembrane proteins of the SLC37 family.
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
REFERENCES
- Almqvist J, Huang Y, Hovmöller S, Wang DN. Homology modeling of the human microsomal glucose 6-phosphate transporter explains the mutations that cause the glycogen storage disease type Ib. Biochemistry. 2004;43(29):9289–9297. doi: 10.1021/bi049334h. [DOI] [PubMed] [Google Scholar]
- Annabi B, Hiraiwa H, Mansfield BC, Lei K-J, Ubagai T, Polymeropoulos MH, et al. The gene for glycogen storage disease type 1b maps to chromosome 11q23. American Journal of Human Genetics. 1998;62(2):400–405. doi: 10.1086/301727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arion WJ, Canfield WK, Ramos FC, Su ML, Burger HJ, Hemmerle H, et al. Chlorogenic acid analogue S 3483: A potent competitive inhibitor of the hepatic and renal glucose-6-phosphatase systems. Achieves of Biochemistry and Biophysics. 1998;351(2):279–285. doi: 10.1006/abbi.1997.0563. [DOI] [PubMed] [Google Scholar]
- Arion WJ, Lange AJ, Ballas LM. Quantitative aspects of relationship between glucose 6-phosphate transport and hydrolysis for liver microsomal glucose-6-phosphatase system. Selective thermal inactivation of catalytic component in situ at acid pH. Journal of Biological Chemistry. 1976;251(16):6784–6790. [PubMed] [Google Scholar]
- Arion WJ, Wallin BK, Carlson PW, Lange AJ. The specificity of glucose 6-phosphatase of intact liver microsomes. Journal of Biological Chemistry. 1972;247(8):2558–2565. [PubMed] [Google Scholar]
- Bandsma RH, Wiegman CH, Herling AW, Burger HJ, ter Harmsel A, Meijer AJ, et al. Acute inhibition of glucose-6-phosphate translocator activity leads to increased de novo lipogenesis and development of hepatic steatosis without affecting VLDL production in rats. Diabetes. 2001;50(11):2591–2597. doi: 10.2337/diabetes.50.11.2591. [DOI] [PubMed] [Google Scholar]
- Bartoloni L, Antonarakis SE. The human sugar-phosphate/phosphate exchanger family SLC37. Pflügers Archiv. 2004;447(5):780–783. doi: 10.1007/s00424-003-1105-0. [DOI] [PubMed] [Google Scholar]
- Bartoloni L, Wattenhofer M, Kudoh J, Berry A, Shibuya K, Kawasaki K, et al. Cloning and characterization of a putative human glycerol 3-phosphate permease gene (SLC37A1 or G3PP) on 21q22.3: Mutation analysis in two candidate phenotypes, DFNB10 and a glycerol kinase deficiency. Genomics. 2000;70(2):190–200. doi: 10.1006/geno.2000.6395. [DOI] [PubMed] [Google Scholar]
- Boztug K, Appaswamy G, Ashikov A, Schäffer AA, Salzer U, Diestelhorst J, et al. A syndrome with congenital neutropenia and mutations in G6PC3. New England Journal of Medicine. 2009;360(1):32–43. doi: 10.1056/NEJMoa0805051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramelo JJ, Parodi AJ. How sugars convey information on protein conformation in the endoplasmic reticulum. Seminars in Cell and Developmental Biology. 2007;18(6):732–742. doi: 10.1016/j.semcdb.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L-Y, Lin B, Pan C-J, Hiraiwa H, Chou JY. Structural requirements for the stability and microsomal transport activity of the human glucose-6-phosphate transporter. Journal of Biological Chemistry. 2000;275(44):34280–34286. doi: 10.1074/jbc.M006439200. [DOI] [PubMed] [Google Scholar]
- Chen SY, Pan CJ, Lee S, Peng W, Chou JY. Functional analysis of mutations in the glucose-6-phosphate transporter that cause glycogen storage disease type Ib. Molecular Genetics and Metabolism. 2008;95(4):220–223. doi: 10.1016/j.ymgme.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SY, Pan CJ, Nandigama K, Mansfield BC, Ambudkar SV, Chou JY. The glucose-6-phosphate transporter is a phosphate-linked antiporter deficient in glycogen storage disease type Ib and Ic. FASEB Journal. 2008;22(7):2206–2213. doi: 10.1096/fj.07-104851. [DOI] [PubMed] [Google Scholar]
- Chen L-Y, Pan C-J, Shieh J-J, Chou JY. Structure-function analysis of the glucose-6-phosphate transporter deficient in glycogen storage disease type Ib. Human Molecular Genetics. 2002;11(25):3199–3207. doi: 10.1093/hmg/11.25.3199. [DOI] [PubMed] [Google Scholar]
- Chen L-Y, Shieh J-J, Lin B, Pan C-J, Gao J-L, Murphy PM, et al. Impaired glucose homeostasis, neutrophil trafficking and function in mice lacking the glucose-6-phosphate transporter. Human Molecular Genetics. 2003;12(19):2547–2558. doi: 10.1093/hmg/ddg263. [DOI] [PubMed] [Google Scholar]
- Cheung YY, Kim SY, Yiu WH, Pan CJ, Jun HS, Ruef RA, et al. Impaired neutrophil activity and increased susceptibility to bacterial infection in mice lacking glucose-6-phosphatase-beta. Journal of Clinical Investigation. 2007;117(3):784–793. doi: 10.1172/JCI30443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JY, Jun HS, Mansfield BC. Neutropenia in type Ib glycogen storage disease. Current Opinion in Hematology. 2010a;17(1):36–42. doi: 10.1097/MOH.0b013e328331df85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JY, Jun HS, Mansfield BC. Glycogen storage disease type I and G6Pase-β deficiency: Etiology and therapy. Nature Reviews. Endocrinology. 2010b;6(12):676–688. doi: 10.1038/nrendo.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JY, Jun HS, Mansfield BC. The SLC37 family of phosphate-linked sugar phosphate antiporters. Molecular Aspects of Medicine. 2013;34(2–3):601–611. doi: 10.1016/j.mam.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JY, Matern D, Mansfield BC, Chen Y-T. Type I glycogen storage diseases: Disorders of the glucose-6-phosphatase complex. Current Molecular Medicine. 2002;2(12):121–143. doi: 10.2174/1566524024605798. [DOI] [PubMed] [Google Scholar]
- Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Research. 1988;16(22):10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissanayake VH, Jayasinghe JD, Thilakaratne V, Jayasekara RW. A novel mutation in SLC37A4 gene in a Sri Lankan boy with glycogen storage disease type Ib associated with very early onset neutropenia. Journal of Molecular and Genetic Medicine: An International Journal of Biomedical Research. 2011;5:262–263. doi: 10.4172/1747-0862.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fann MC, Davies AH, Varadhachary A, Kuroda T, Sevier C, Tsuchiya T, et al. Identification of two essential arginine residues in UhpT, the sugar phosphate antiporter of Escherichia coli. Journal of Membrane Biology. 1998;164(2):187–195. doi: 10.1007/s002329900404. [DOI] [PubMed] [Google Scholar]
- Galli L, Orrico A, Marcolongo P, Fulceri R, Burchell A, Melis D, et al. Mutations in the glucose-6-phosphate transporter (G6PT) gene in patients with glycogen storage diseases type 1b and 1c. FEBS Letters. 1999;459(2):255–258. doi: 10.1016/s0014-5793(99)01248-x. [DOI] [PubMed] [Google Scholar]
- Gerin I, Veiga-da-Cunha M, Achouri Y, Collet JF, Van Schaftingen E. Sequence of a putative glucose-6-phosphate translocase, mutated in glycogen storage disease type 1b. FEBS Letters. 1997;419(2–3):235–238. doi: 10.1016/s0014-5793(97)01463-4. [DOI] [PubMed] [Google Scholar]
- Gerin I, Veiga-da-Cunha M, Noel G, Van Schaftingen E. Structure of the gene mutated in glycogen storage disease type Ib. Gene. 1999;227(2):189–195. doi: 10.1016/s0378-1119(98)00614-3. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Shieh J-J, Pan C-J, Chou JY. Histidine-167 is the phosphate acceptor in glucose-6-phosphatase-β forming a phosphohistidine-enzyme intermediate during catalysis. Journal of Biological Chemistry. 2004;279(13):12479–12483. doi: 10.1074/jbc.M313271200. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Shieh J-J, Pan C-J, Sun M-S, Chou JY. The catalytic center of glucose-6-phosphatase: His176 is the nucleophile forming the phosphohistidine-enzyme intermediate during catalysis. Journal of Biological Chemistry. 2002;277(36):32837–32842. doi: 10.1074/jbc.M201853200. [DOI] [PubMed] [Google Scholar]
- Goder V, Spiess M. Topogenesis of membrane proteins: Determinants and dynamics. FEBS Letters. 2001;504(3):87–93. doi: 10.1016/s0014-5793(01)02712-0. [DOI] [PubMed] [Google Scholar]
- Grefhorst A, Schreurs M, Oosterveer MH, Cortés VA, Havinga R, Herling AW, et al. Carbohydrate-response-element-binding protein (ChREBP) and not the liver X receptor α (LXRα) mediates elevated hepatic lipogenic gene expression in a mouse model of glycogen storage disease type 1. Biochemical Journal. 2010;432(2):249–254. doi: 10.1042/BJ20101225. [DOI] [PubMed] [Google Scholar]
- Hall JA, Fann MC, Maloney PC. Altered substrate selectivity in a mutant of an intrahelical salt bridge in UhpT, the sugar phosphate carrier of Escherichia coli. Journal of Biological Chemistry. 1999;274(10):6148–6153. doi: 10.1074/jbc.274.10.6148. [DOI] [PubMed] [Google Scholar]
- He L, Vasiliou K, Nebert DW. Analysis and update of the human solute carrier (SLC) gene superfamily. Human Genomics. 2009;3(2):195–206. doi: 10.1186/1479-7364-3-2-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmerle H, Burger HJ, Below P, Schubert G, Rippel R, Schindler PW, et al. Chlorogenic acid and synthetic chlorogenic acid derivatives: Novel inhibitors of hepatic glucose-6-phosphate translocase. Journal of Medicinal Chemistry. 1997;40(2):137–145. doi: 10.1021/jm9607360. [DOI] [PubMed] [Google Scholar]
- Herling AW, Burger H, Schubert G, Hemmerle H, Schaefer H, Kramer W. Alterations of carbohydrate and lipid intermediary metabolism during inhibition of glucose-6-phosphatase in rats. European Journal of Pharmacology. 1999;386(1):75–82. doi: 10.1016/s0014-2999(99)00748-7. [DOI] [PubMed] [Google Scholar]
- Hiraiwa H, Pan C-J, Lin B, Moses SW, Chou JY. Inactivation of the glucose-6-phosphate transporter causes glycogen storage disease type 1b. Journal of Biological Chemistry. 1999;274(9):5532–5536. doi: 10.1074/jbc.274.9.5532. [DOI] [PubMed] [Google Scholar]
- Hoffman K, Stoffel W. TMbase—A database of membrane spanning protein segments. Biological Chemistry Hoppe-Seyler. 1993;347:166–170. [Google Scholar]
- Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301(5633):616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- Iacopetta D, Lappano R, Cappello AR, Madeo M, De Francesco EM, Santoro A, et al. SLC37A1 gene expression is up-regulated by epidermal growth factor in breast cancer cells. Breast Cancer Research and Treatment. 2010;122(3):755–764. doi: 10.1007/s10549-009-0620-x. [DOI] [PubMed] [Google Scholar]
- Jackson MR, Nilsson T, Peterson PA. Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO Journal. 1990;9(10):3153–3162. doi: 10.1002/j.1460-2075.1990.tb07513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MR, Nilsson T, Peterson PA. Retrieval of transmembrane proteins to the endoplasmic reticulum. Journal of Cell Biology. 1993;121(2):317–333. doi: 10.1083/jcb.121.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janecke AR, Linder M, Erdel M, Mayatepek E, Moslinger D, Podskarbi T, et al. Mutation analysis in glycogen storage disease type 1 non-a. Human Genetics. 2000;107(3):285–289. doi: 10.1007/s004390000371. [DOI] [PubMed] [Google Scholar]
- Jun HS, Cheung YY, Lee YM, Mansfield BC, Chou JY. Glucose-6-phosphatase-β, implicated in a congenital neutropenia syndrome, is essential for macrophage energy homeostasis and functionality. Blood. 2012;119(17):4047–4055. doi: 10.1182/blood-2011-09-377820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun HS, Lee YM, McDermott DH, DeRavin SS, Murphy PM, Mansfield BC, et al. Lack of glucose recycling between endoplasmic reticulum and cytoplasm underlies cellular dysfunction in glucose-6-phosphatase-beta-deficient neutrophils in a congenital neutropenia syndrome. Blood. 2010;116(15):2783–2792. doi: 10.1182/blood-2009-12-258491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Tillison K, Zhou S, Wu Y, Smas CM. The major facilitator super-family member Slc37a2 is a novel macrophage- specific gene selectively expressed in obese white adipose tissue. American Journal of Physiology. Endocrinology and Metabolism. 2007;293(1):E110–E120. doi: 10.1152/ajpendo.00404.2006. [DOI] [PubMed] [Google Scholar]
- Landolt-Marticorena C, Reithmeier RAF. Asparagine-linked oligosaccha-rides are localized to single extracytosolic segments in multi-span membrane glycoproteins. Biochemical Journal. 1994;302(Pt 1):253–260. doi: 10.1042/bj3020253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K-J, Chen H, Pan C-J, Ward JM, Mosinger B, Lee EJ, et al. Glucose-6-phosphatase dependent substrate transport in the glycogen storage disease type 1a mouse. Nature Genetics. 1996;13(2):203–209. doi: 10.1038/ng0696-203. [DOI] [PubMed] [Google Scholar]
- Lei K-J, Shelly LL, Pan C-J, Sidbury JB, Chou JY. Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a. Science. 1993;262(5133):580–583. doi: 10.1126/science.8211187. [DOI] [PubMed] [Google Scholar]
- Leuzzi R, Fulceri R, Marcolongo P, Bánhegyi G, Zammarchi E, Stafford K, et al. Glucose 6-phosphate transport in fibroblast microsomes from glycogen storage disease type 1b patients: Evidence for multiple glucose 6-phosphate transport systems. Biochemical Journal. 2001;357(Pt 2):557–562. doi: 10.1042/0264-6021:3570557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Annabi B, Hiraiwa H, Pan C-J, Chou JY. Cloning and characterization of cDNAs encoding a candidate glycogen storage disease type 1b protein in rodents. Journal of Biological Chemistry. 1998;273(48):31656–31670. doi: 10.1074/jbc.273.48.31656. [DOI] [PubMed] [Google Scholar]
- Lin B, Pan C-J, Chou JY. Human variant glucose-6-phosphate transporter is active in microsomal transport. Human Genetics. 2000;107(5):526–529. doi: 10.1007/s004390000404. [DOI] [PubMed] [Google Scholar]
- Lloyd AD, Kadner RJ. Topology of the Escherichia coli UhpT sugar-phosphate transporter analyzed by using TnphoA fusions. Journal of Bacteriology. 1990;172(4):1688–1693. doi: 10.1128/jb.172.4.1688-1693.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari M. N-glycan structure dictates extension of protein folding or onset of disposal. Nature Chemical Biology. 2007;3(6):313–320. doi: 10.1038/nchembio880. [DOI] [PubMed] [Google Scholar]
- Nilsson IM, von Heijne G. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. Journal of Biological Chemistry. 1993;268(8):5798–5801. [PubMed] [Google Scholar]
- Nordlie RC, Sukalski KA, Munoz JM, Baldwin JJ. Type 1c, a novel glycogenosis. Journal of Biological Chemistry. 1983;258:9739–9744. [PubMed] [Google Scholar]
- Pan CJ, Chen SY, Jun HS, Lin SR, Mansfield BC, Chou JY. SLC37A1 and SLC37A2 are phosphate-linked, glucose-6-phosphate antiporters. PLoS One. 2011;6(9):e23157. doi: 10.1371/journal.pone.0023157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan CJ, Chen SY, Lee S, Chou JY. Structure-function study of glucose-6-phosphate transporter, an eukaryotic antiporter deficient in glycogen storage disease type Ib. Molecular Genetics and Metabolism. 2009;96(1):32–37. doi: 10.1016/j.ymgme.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan CJ, Chen LY, Mansfield BC, Salani B, Varesio L, Chou JY. The signature motif in human glucose-6-phosphate transporter is essential for microsomal transport of glucose-6-phosphate. Human Genetics. 2003;112(4):430–433. doi: 10.1007/s00439-002-0903-3. [DOI] [PubMed] [Google Scholar]
- Pan C-J, Lin B, Chou JY. Transmembrane topology of human glucose-6-phosphate transporter. Journal of Biological Chemistry. 1999;274(20):13865–13869. doi: 10.1074/jbc.274.20.13865. [DOI] [PubMed] [Google Scholar]
- Pao SS, Paulsen IT, Saier MH., Jr. Major facilitator superfamily. Microbiology and Molecular Biology Reviews. 1998;62(1):1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi AJ. Protein glucosylation and its role in protein folding. Annual Review of Biochemistry. 2000;69:69–93. doi: 10.1146/annurev.biochem.69.1.69. [DOI] [PubMed] [Google Scholar]
- Qiu ZQ, Lu CX, Wang W, Qiu JJ, Wei M. Mutation in the SLC37A4 gene of glycogen storage disease type Ib in 15 families of the mainland of China. Zhonghua Er Ke Za Zhi. Chinese Journal of Pediatrics. 2011;49(3):203–208. [PubMed] [Google Scholar]
- Reddy VS, Shlykov MA, Castillo R, Sun EI, Saier MH., Jr. The major facilitator superfamily (MFS) revisited. FEBS Journal. 2012;279(11):2022–2035. doi: 10.1111/j.1742-4658.2012.08588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh J-J, Pan C-J, Mansfield BC, Chou JY. Glucose-6-phosphate hydro-lase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycemia in glycogen storage disease type Ia. Journal of Biological Chemistry. 2003;278(47):47098–47103. doi: 10.1074/jbc.M309472200. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Miyata M, Zheng P, Imazato T, Horwitz A, Smith JD. Identification of cAMP analogue inducible genes in RAW264 macrophages. Biochimica et Biophysica Acta. 2000;1492(2–3):385–394. doi: 10.1016/s0167-4781(00)00133-0. [DOI] [PubMed] [Google Scholar]
- Veiga-da-Cunha M, Gerin I, Chen Y-T, de Barsy T, de Lonlay P, Dionisi-Vici C, et al. A gene on chromosome 11q23 coding for a putative glucose-6-phosphate translocase is mutated in glycogen-storage disease types Ib and Ic. American Journal of Human Genetics. 1998;63(4):976–983. doi: 10.1086/302068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-da-Cunha M, Gerin I, Chen Y-T, Lee PJ, Leonard JV, Maire I, et al. The putative glucose 6-phosphate translocase gene is mutated in essentially all cases of glycogen storage disease type I non-a. European Journal of Human Genetics. 1999;7(16):717–723. doi: 10.1038/sj.ejhg.5200366. [DOI] [PubMed] [Google Scholar]
- Zingone A, Hiraiwa H, Pan C-J, Lin B, Chen H, Ward JM, et al. Correction of glycogen storage disease type 1a in a mouse model by gene therapy. Journal of Biological Chemistry. 2000;275(2):828–832. doi: 10.1074/jbc.275.2.828. [DOI] [PubMed] [Google Scholar]