

Abstract
Objective
Inflammatory signaling pathways, such as p38 MAPK play a central role in host responses to injury. In our previous studies, topical p38 MAPK inhibitors effectively attenuated inflammatory signaling in a partial-thickness scald burn model, when applied to the burn wound immediately after injury. However, clinically relevant full-thickness scald burn wounds may act as a barrier to topical immune modulators and delayed application of topical p38 MAPK inhibitors may not be effective. In this study, we evaluate the efficacy of topical p38 MAPK inhibition on full-thickness scald burns with immediate and delayed treatment.
Methods
C57/BL6 mice received `Sham' or 30% TBSA full-thickness scald burn injury. After injury, the burn wounds were treated with a topical p38 MAPK inhibitor or vehicle. The treatment group received topical p38 MAPK inhibitor either immediately after burn or 4 hours (delayed) after injury. All animals were sacrificed at 12 or 24 h. Burn wounds underwent histological analyses. Skin and plasma were analyzed by ELISA or RT-qPCR for cytokine expression.
Results
Full-thickness scald burns resulted from immersion in 62°C water for 25 s. Topical p38 MAPK inhibitor attenuated dermal IL-6, MIP-2, and IL-1β expression and plasma IL-6 and MIP-2 cytokine expression. In addition, delayed application of topical p38 MAPK inhibitors significantly reduced dermal and plasma cytokine expression compared to vehicle control.
Conclusion
Topical p38 MAPK inhibitors remain potent in reducing full-thickness burn wound inflammatory signaling, even when treatment is delayed by several hours post injury. Topical application of p38 MAPK inhibitor may be a clinically viable treatment after burn injury.
Keywords: signal transduction, mitogen activated Protein Kinase (MAPK), p38, topical treatment, burn, scald, hair follicle, apoptosis
INTRODUCTION
Approximately two million burn injuries are reported annually in the United States (1). Burn injuries account for approximately 4,000 deaths each year, making it the seventh leading cause of injury related death in the United States (1–3). Large burn injuries (extent and depth), in addition to age and concomitant inhalation injury, are associated with increasing morbidity and mortality (1, 4, 5). In a review of patients with equal or more than 20% body surface area burns, 73% had significant degrees of organ dysfunction with 28% manifesting severe multi-organ dysfunction syndrome (Marshall MOD score≥6)(6). The burn wound itself is considered to be the inflammatory source responsible for triggering the systemic inflammatory response syndrome (SIRS) and remote organ dysfunction (7–11). The skin launches a complex inflammatory response to thermal injury (9, 12), which activates SIRS via multiple mechanisms, such as the systemic liberation of pro-inflammatory mediators, attraction and activation of PMN trafficking, as well as the potential activation via sympathetic inflammatory signaling (7, 8, 10, 11, 13). Our approach is topical inflammatory source control, targeting and thereby attenuating the local dermal (burn wound) inflammatory processes through application of topical immunomodulators. In our previous studies, we demonstrated that attenuation of burn wound inflammatory signaling prevented the subsequent activation of SIRS and significantly improved end-organ function, including the heart and lungs (14–16).
Intracellular inflammatory signaling pathways, such as p38 MAPK play a central role in the host response to injury (17, 18). Local inflammatory signaling causes the production and release of pro-inflammatory mediators, which may lead to SIRS. We have previously reported that thermal injury induced p38 MAPK activation in the burn wound (19). Furthermore, post-injury topical application of p38 MAPK inhibitors directly to a partial-thickness burn wound attenuated dermal inflammatory signaling and cytokine expression (19). Topical p38 MAPK inhibitors reduced dermal inflammatory signaling, thereby shielding the body from the source of inflammation, the burn wound, and attenuating burn induced SIRS and MODS. Topical p38 MAPK inhibitors reduced post-injury systemic inflammation, pulmonary and cardiac dysfunction, and mortality in rodent burn models (14–16). Additionally, we reported that the beneficial systemic outcomes were due to topical modification of the burn wound inflammation and not due to systemic absorption of the inhibitor. Specifically, targeting the burn wound inflammatory signaling did not inhibit cardiac and pulmonary intracellular p38 MAPK, demonstrating that topical p38 MAPK inhibition works at the source of inflammation, the burn wound (14, 16). The lack of systemic inflammatory signaling inhibition may better preserve normal systemic responses to infectious challenges, such as pneumonia after burn injury.
Topical inflammatory source control represents a potential novel approach in the prevention of SIRS and SIRS related complications after thermal injury. However, our earlier experiments were carried out in a partial-thickness scald burn model with immediate application of topical p38 MAPK inhibitors post-injury. The full-thickness burn wound and eschar may act as a barrier to the topical immunomodulators. Furthermore, while the topical application can be applied easily after burn injury, even at the scene, for this treatment to be clinically practical, delayed application by several hours has to remain effective. We hypothesized that following full-thickness scald burn that both immediate topical `Treatment' and `Delayed Treatment' of p38 MAPK inhibitor will alter expression of inflammatory markers. In the current study, we first established that our mouse scald burn wounds were full-thickness injuries. Our first aim was to determine if the application of topical p38 MAPK inhibitors remain an effective treatment in the full-thickness scald burn model. Our second aim was to determine if 4 hour `Delayed Treatment' post-injury would remain effective and attenuate proinflammatory mediator production from the burn wound.
MATERIALS AND METHODS
Experimental Animals
All experiments were performed in accordance with the guidelines set forth by the National Institutes of Health for care and use of animals. Approval for the experimental protocol was obtained from the University of Washington's Institutional Animal Care and Use Committee (IACUC). Age and weight matched female C57/BL6 mice (Jackson Laboratories, Charles-River Laboratories) weighing 18–25 g were allowed to acclimate for 1 wk before being used in any experiment. Animals had unrestricted access to standard chow and water ad lib throughout the course of the study.
Burn Injury
C57/BL6 female mice were anesthetized via inhalation of isofluorane in a chamber with supplemental oxygen. The dorsal hairs were clipped, and animals were placed in an insulating mold device with an opening calculated to expose 30% total body surface area (20). After immersion in 62°C water for 25 s, the burn area was scrub debrided with dry sterile gauze and rinsed with 0.9% sterile saline. Animals were resuscitated with 4 ml/percentage of total body surface area burn/kg Ringer's lactate by i.p. injection (21). Each animal received 0.3 mg/kg buprenorphine (Buprenex; Reckitt & Coleman Pharmaceuticals) s.c. injection. Animals were housed by experimental group with no more than five per cage. Sham animals were subjected to identical procedure, resuscitation, and buprenorphine, but immersed in room temperature water.
For topical inhibition of p38 MAPK three inhibitors were used, one for each of the three separate mouse experiments. The inhibitors were 1 × 10−4 M SB202190 (Calbiochem) or 3% ARRY-371797 (Array Biopharma). Topical inhibitor was applied post-injury either: immediately `Treatment', or after 4 h `Delayed Treatment'. Dose-response studies were performed for each inhibitor prior to this current study (data not shown). Treatments were applied on a q8 schedule until sacrifice at 12 or 24 h. `Sham' and `Burn' controls received the drug vehicle. The lipid soluble SB202190 vehicle consisted of a 4:1 mixture of acetone/olive oil, a combination widely used in topical applications (19), and ARRY-371797 vehicle consisted of an inert drug delivery gel liposomal pluronic lecithin organogel transdermal gel (PLO) (Transdermal Pharmaceuticals Inc). Wounds were covered with gauze and held in place with `band net' (Surgilast; Derma Sciences). For retro-orbital phlebotomy animals were anesthetized with i.p. 40 mg/kg pentobarbital (Abbot Laboratories). Whole blood was collected and placed in sodium citrate solution on ice for further processing. Animals were exsanguinated by incision of the right atrium and tissues were collected. Dermal tissue for ELISA were immediately frozen on dry ice and stored at −80°C, while tissue for RT-qPCR were immediately submerged in RNAlater (Ambion) and stored at 4°C until further processing.
To determine that the 25 s immersion time in 62°C water (used above), results in a full-thickness scald burn wound a parallel study was conducted. The aforementioned mouse protocol was followed, with two exceptions: 1) animals received no topical applications, 2) 62°C water immersion groups: Sham (room temperature water 25 s), or 62°C water (17 s, 20 s, 25 s, or 30 s). All animals were sacrificed at 24 h post scald injury; the skin was harvested and placed in 10% formalin for histological examination.
Determination of Burn Wound Depth
Paraffin-embedded sections were fixed to slides, dewaxed, rehydrated and underwent traditional H&E staining. These slides were viewed in 40× HPF and evaluated by three blinded, independent investigators for characteristics consistent with partial or full-thickness scald burns. A total of 2 slides per animal were analyzed.
Fluorescent-labeled TUNEL assay apoptosis was detected in situ with fluorescein-based labeling of DNA strand breaks using the in situ terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay (Roche Applied Sciences). Skin samples were fixed in 10% buffered formalin (Sigma Aldridge) and embedded in paraffin, cut in 5-μm-thick sections, and fixed to slides for indirect immunofluorescence (according to manufactures instructions). Briefly, paraffin-embedded sections were dewaxed, rehydrated, and incubated with proteinase K (35 μg/mL in 10 mmol/L Tris/HCl, pH 7.4–8) for 25 min at 37°C in a humidified chamber. Slides underwent 4 washes with 1× PBS. Hereafter, sections were incubated with the TUNEL reaction mixture in a humidified chamber for 1 h at 37°C. The reaction was terminated by rinsing the sections in a stop/wash buffer. Sections were incubated in a humidified chamber at room temperature with antidigoxigenin fluorescein (fluorescein isothiocyanate; FITC) for 30 min and rinsed 3 times in PBS. After blot drying slides, each received a 4, 6-diamidino-2-phenylindole dihydrochloride (DAPI) counter stain and a cover slip was placed. DNase I (Roche Applied Sciences) was used to make a set of positive controls with mouse skin samples. Negative controls were made with labeling solution minus terminal transferase on sham and burn subjects. TUNEL-labeled slides were analyzed by fluorescent microscopy and images captured at 40× magnification.
Image analysis
Fluorescent-labeled TUNEL slides were captured digitally at identical time post labeling to control fading of fluorescence using a Nikon Eclipse TE2000-S fluorescence microscope (Nikon Inc) at fixed image capture settings and 40× magnification. The analysis was randomized to order and treatment groups to control for systemic error. Five hair follicles per slide were randomly selected and digitally captured visualizing counterstained nuclei in the DAPI excitation/emission channel. Next, a region of interest (ROI) was digitally defined, set to include only high power fields (HPF) at the dermal/subcutis junction or subcutis/subcutaneous muscle/fascia junction and exclude bright fluorescing hair shafts and surrounding cells (MetaMorph v.5.0r4; Universal Imaging Corporation). Hereafter, the excitation/filter channel was changed to visualize fluorescein-labeled TUNEL-positive cells, and images were digitally captured. Fluorescence of TUNEL (+) cells was quantified, normalized to ROI size, and expressed as TUNEL (+) cells/HPF/Pixel area, controlling differences in ROI size. TUNEL-positive cells per randomly chosen high power field were counted by three independent investigators. A total of two slides per animal and at least 5 high power fields per slide per experimental group were analyzed.
Isolation and detection of dermal proinflammatory cytokines (ELISA)
For measurement of mouse IL-6, MIP-2, and IL-1β by sandwich ELISA, we used ELISA kits (Quantikine Immunoassay; R&D Systems) and 96-well micro plates (Costar EIA/RIA; Corning Inc) according to manufacturer's instructions. Exact tissue weight was recorded (~100 mg), tissue was bead homogenized (Bullet Blender; Next Advance, Inc) in 1000 μl of ice-cold lysis buffer consisting of 50 ml 0.9% sterile saline, protease inhibitor (Complete X; Roche Applied Sciences), and 50 μl Triton X-100 (Sigma-Aldrich). A total of 400 ul of whole blood was collected by retro-orbital phlebotomy and placed on ice in 200 ul 3.6% sodium citrate solution. Blood samples were centrifuged at 14,000 × g for 5 min at 4°C, the serum supernatant was stored at −80°C until use. Tissue homogenates were centrifuged at 13,900 × g for 5 min at 4°C, tissue supernatants were stored at −80°C until use. Microplates were read using a microplate reader (SPECTRAmax Plus; Molecular Devices) at 450 nm. Cytokine concentrations were calculated using four parameter logistic (4-PL) curve fit, concentration expressed as pg/ml. The individual tissues weights were used to normalize final concentrations to 100 mgs.
Isolation and detection of dermal IL-6 mRNA expression by real-time quantitative PCR (RT-qPCR)
Samples were processed in small batches to minimize handling time. Approximately 50–100 mg of fresh tissue was homogenized in 1 ml of TRIzol (Invitrogen by Life Technologies) using the guanidine isothiocyanate/chloroform mRNA extraction method according to manufactures instructions. Additionally, to ensure mRNA of highest purity and highest integrity mRNA was further purified with either: RNeasy Mini Kit (Qiagen) or Purelink RNA Mini Kit (Ambion) following manufacturer's guidelines. Purity of the sample with respect to protein contamination was assessed spectrophotometrically by measuring the OD 260/280 ratio; acceptable OD 260/280 ratio was 1.8–2.0. A few small groups were further tested for RNA integrity through miscrofluidics-based electrophoreses system (Agilent 2100 Bioanalyzer; Agilent). A virtual gel, electropherogram, ratio 28S/18S, and RNA quality indicator (RQI) was determined, an acceptable RQI values was >5.
For reverse transcription (RT), 250 ng/12 μL total mRNA was heated for 3 min at 70°C then cooled on ice to denature secondary structures. Omniscript RT kit's (Qiagen) reverse transcription master mix was added as follows: 2 μL of 10× RT Buffer, 2 μL of dNTP mix, 1 μL of Omniscript reverse transcriptase, 2 μL of Oligo (dt) primers, and 1 μL (10 U/ul) of Rnase inhibitor (Promega). Final concentrations were adjusted by adding Rnase-free H20. Samples were incubated at 37°C for 1 h. The cDNA was stored at −80°C until use.
For RT-qPCR, 2.0 μL of cDNA was added to the following master mix: 2.5 μL of Rnase-free H2O, 5.0 μL of SYBR Green (Applied Biosystems), 0.25 μL of forward primer (10 μmol/L) and 0.25 μL of reverse primer (10 μmol/L) for a total 10μL RT-qPCR reaction/sample. For mRNA target gene amplification, a thermal cycler (ABI Prism 7900HT; Applied Biosystems) was programmed as follows: “hot start” 95°C (15 min), followed by 45 amplification cycles with 95°C melting temperature (15 s), 60°C annealing temperature (30 s), and 72°C extension temperature (30 s), proceeded by the thermal cyclers default melting curve protocol. Amplification plot was used to determine optimal annealing temperature. Appropriate primer design was evaluated using Primer-Blast (www.ncbi.nlm.nih.gov/tools/primers-blast/). The following `target gene' primers were used: IL-6 (forward primer) 5'-ACTTCACAGAGGATACCACTC -3', (reverse primer) 5'-TCTGCAAGTGCATCATCGTTG - 3' (Invitrogen by Life Technologies), resulting in 154 bp amplicon. β-actin was chosen as a `reference gene' because it does not exhibit changes in expression between samples from various experimental conditions or time points in the mouse skin. The following β-actin primer set was used: (forward primer) 5'-AAGAGCTATGAGCTGCCTGA -3', (reverse primer) 5'- TACGGATGTCAACGTCACAC -3' (www.Realtimeprimers.com), resulting in 160 bp amplicon. All sample from an experiment were tested at the same time on the same 383-well plate (MicroAmp Optical 383-Well Reaction Plate; Applied Biosystems). Each sample was plated in 4 replicate wells, with both `target' and `reference' genes on this same plate. Product purity was verified by the thermocycler generate melting curves. Random samples were selected for assessing specificity of RT-qPCR products via 1.2% agarose gel electrophoresis, thus confirming amplicon size (data not shown). CT values were generated using the software provided by the manufacture (ABI Prism 7900HT). Results are presented as the ratio of the `target gene' IL-6 to the `reference gene' β-actin.
Statistical analysis
All experiments receiving topical applications were replicated and representative findings are shown. Data were analyzed using one way ANOVA followed by Kruskal-Wallis post-test or by paired Student t test. Values are expressed as mean ± SEM or SD (Prism 5 Software). Statistical significance is assigned at a P value of <0.05.
RESULTS
The full-thickness scald burn model causes deep burn injury
To demonstrate that our chosen burn model did in fact create a deep full-thickness dermal injury, we conducted a parallel study to evaluate burn depth at various time-exposures to hot water. Animals were separated into sham or burn groups and subjected to a 62°C scald burn for 17, 20, 25 or 30 seconds (n=5 in each group). Sham animals were placed in room temperature water. Burn wound skin was harvested at 24 h and analyzed using fluorescent-labeled TUNEL assays. The FITC stain was used as a surrogate for cell injury (Fig. 1). Intense staining that included skin appendages and the dermal/subcutis junction was noted for all burn groups receiving >20 s burns. The 17 s, partial-thickness model, showed moderate staining throughout the deep dermis. The 25 s, full-thickness model, showed significantly increased deep dermal staining. Burn animals subjected to 25 s burns also demonstrated injury involving the deep subcutaneous tissue, vessels, glands and subcutaneous muscle and fascia (Fig. 1). When counting the number of TUNEL-positive cells/high-power field, the 25 s (full-thickness model) resulted in a significant increase in cell death compared to the 17 s burn injury in both superficial and deep levels (17 s Burn vs. 25 s Burn, p<0.001) (Fig. 2); additionally, while there was extensive deep damage in the 25 s burn, there was essentially no quantifiable deep damage in the 17 s burn group (Fig. 2). Furthermore, skin was evaluated by traditional H&E staining for evidence of cell necrosis and destruction. The 25 s and 30 s burns demonstrated classic full-thickness injury (Fig. 3).
Figure 1. The 25 second mouse scald burn model causes an extensive full-thickness burn injury.

Examples of TUNEL assay of high-power fields in sham, 17sec burn, and 25 second burn skin. The 25 second burn images show more extensive damage at the dermal/subcutis junction compared with 17s. There is extensive deep injury demonstrated in the subcutaneous tissue. The DAPI (41 6-diamidino – 2 – phenylindole dihydrochloride) nuclear stain shows cells in blue; FITC-TUNEL positive labeled cells appear green, demonstrating cells in apoptotic or non-apoptotic cell death. (Images captures at 40× magnification)
Figure 2. The full-thickness scald burn model markedly increases dermal tissue cell death.

Skin from sham, 17s and 25s burn animals was sectioned for fluorescein-labeled TUNEL as described. Sections were randomized; per section, 3 hair follicles were randomly selected, the ROI was defined and analyzed according to the described protocol using digital image software. A significant increase in cell death was seen in the 25 s burn group vs. the 17s burn group at both superficial (dermal/subcutis junction) and deep (subcutis/subQ muscle fascia) levels. Values are expressed as mean ± 95% confidence interval (* comparing the 25 s to 17 s superficial and ** comparing the 25 s to 17 s deep levels, p<0.001 for both comparison, ANOVA, n=15 HPF per group).
Fig. 3. Scald burn injury for greater than 25 seconds results in a full-thickness burn.

Examples of H&E stained burn tissue. (A) Sham demonstrates the normal skin structure with intact hair follicles (HF). (B)17 second burn tissue slide demonstrates destruction of epidermal layer with extensive dermal cell damage. (C) 25 second full-thickness burn with only deep dermis(arrow) remaining and congealed eosinophilic appearance consistent with extensive cell death. There are few definable cellular elements and no intact hair follicles. (D) 30 second full-thickness burn shows more extensive injury with deep injury in subcutis (SC).
Topical p38 MAPK inhibitors attenuate full-thickness scald burn wound's dermal proinflammatory cytokine production
Burn injury induces dermal expression of inflammatory mediators (7, 22, 23). Our previous work demonstrated that topical p38 MAPK inhibition effectively attenuated dermal IL-6, MIP-2 and IL-1β cytokine expression (important p38 MAPK dependent cytokines) in a partial-thickness scald burn model (14, 15, 19). Here, in a full-thickness scald burn model, immediate `Treatment' and 4 h `Delayed Treatment' of topical p38 MAPK inhibition resulted in a significant reduction in cytokine production of IL-6 (Sham 22.06 ± 2.56; Burn 859.97 ± 100.09 vs. Treatment 302.17 ± 58.33 or Delayed Treatment 616.54 ± 136.53) (Fig. 4A), MIP-2 (Sham 14.55 ± 11.91; Burn 478.53 ± 74.8 vs. Treatment 47.95 ± 16.45 or Delayed Treatment 192.51 ± 72.09) (Fig. 4B), and IL-1β (Sham 43.19 ± 4.16; Burn 235.91 ± 28.8 vs. Treatment 76.40 ± 14.97 or Delayed Treatment 109.30 ± 29.09) (Fig. 4C) (n=5 animals per group with 3 separate experiments for total of 15 animals per group). Immediate `Treatment' of the topical inhibitor had a greater attenuating effect on cytokine production than 4 h `Delayed Treatment'. However, 4 h `Delayed Treatment' significantly reduces inflammatory cytokine expression.
Fig. 4. Topical p38MAPK inhibition attenuates acute dermal proinflammatory cytokine expression.
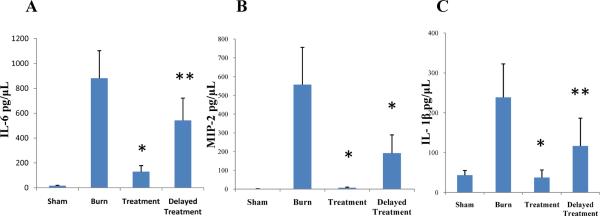
Skin samples, harvested at 12 or 24 hours post-injury, were homogenized and supernatants were processed by ELISA for IL-6 (A), MIP-2 (B) and IL-1β (C) detection. Topical treatment with a p38MAPK inhibitor resulted in significant inhibition of dermal proinflammatory cytokine expression versus untreated burn control with both immediate and delayed (4h post-injury) application. Data presented as mean ± 95% confidence interval. *p < 0.001, **p< 0.03, ANOVA, n= 15 per group (5 animals/group × 3 experiments).
Topical p38 MAPK inhibitors attenuate full-thickness scald burn wound's proinflammatory cytokine gene expression
Dermal IL-6 (ELISA) results were further supported by IL-6 gene expression data. The expression of IL-6 mRNA is markedly increased after burn injury (Fig. 5). In contrast, modulating the inflammatory signaling by applying a topical p38 MAPK inhibitor immediate `Treatment' and 4 h `Delayed Treatment' reduced dermal IL-6 gene expression (Sham normalized to 1; Burn vs. Treatment or Delayed treatment;).
Fig. 5. Topical p38 MAPK inhibition attenuates dermal IL-6 mRNA expression.
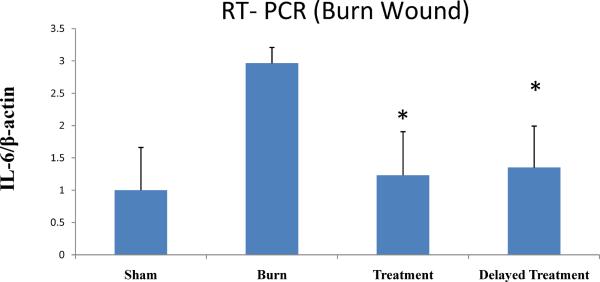
RT-qPCR results demonstrated significant reduction of dermal IL-6 mRNA expression for treatment vs. untreated burn control. mRNA was isolated from skin at 24 h post-injury. Data are represented as mean ± 95% confidence interval. *P< 0.001, ANOVA, n= 10 per group (5 animals/group × 2 experiments).
Circulating plasma proinflammatory cytokines are attenuated after topical modulation of burn wound inflammation
At 12 and 24 h post burn injury, plasma showed a significant increase in proinflammatory cytokine levels for both IL-6 and MIP-2 in the `Burn' vs. `Sham' animals. IL-6 levels were significantly reduced in both the immediate `Treatment' and 4 h `Delayed Treatment' groups, (Sham 4.58 ± 0.74; Burn 163.1 ± 35.89 vs. Treatment 71.44 ± 15.88 or Delayed Treatment 64.95 ± 19.95) (Fig. 6A). MIP-2 levels were significantly decreased in both treatment groups (Sham 33.75 ± 16.15; Burn 97.2 ± 24.65 vs. Treatment 31.15 ± 9.77 or Delayed treatment 38.84 ± 7.35) (Fig. 6B).
Fig. 6. Topical p38 MAPK inhibition reduces serum IL-6 and MIP-2 levels.

Serum samples were obtained at 24 h post-injury from Sham, untreated burn (Burn), treatment (immediate post-injury) or delayed treatment (4 h post-injury). Serum IL-6 and MIP-2 levels were significantly increased in Burn as compared to sham. Topical p38 MAPK inhibition in burn wounds significantly reduced serum IL-6 and MIP-2 levels in all the treatment groups. Data are represented as mean ± 95% confidence interval. For figure 6(A) * denotes p< 0.001 and ** p<0.05 vs. Burn; for figure 6(B) * denotes p< 0.002 and ** p<0.02 vs. Burn, ANOVA, n= 15 per group (5 animals/group × 3 experiments).
DISCUSSION
We define “inflammatory source control” to be all the maneuvers that can be used to control a focus of inflammation, which is thought to be the initial source of systemic immune activation. The hyper-activated local inflammatory process may be the trigger for SIRS and subsequent multisystem organ dysfunction; therefore, controlling the local inflammatory signaling may attenuate subsequent complications such as acute lung injury. Thermal injury represents an excellent model by which to investigate the interaction between local and systemic inflammatory responses (6, 19, 24). In a burn injury model, the burn wound inflammatory response is considered to be the inflammatory source responsible for remote organ dysfunction (7–11, 25). We and others have established that burn induced local dermal inflammatory signaling is associated with local and systemic inflammatory mediator release that may lead to SIRS and SIRS related complications (7, 15). Burns can lead to an exuberant over stimulated inflammatory response that causes acute lung injury and organ dysfunction even in the absence of inhalation injury (23).
A therapeutic approach, often contemplated to address burn induced SIRS has been to administer systemic immunomodulators (e.g. anti-TNFα and anti-IL-1β) in order to disrupt the over stimulated inflammatory response. So far, this approach has been unpredictable in improving morbidity or mortality (26, 27). In several cases, administration of systemic immunomodulators has actually increased morbidity and mortality (28). The likely explanation for these disappointing results is multifactorial. Administration of a systemic immunomodulator is very nonspecific. These substances are active in multiple organ systems and tissues, sometimes with competing and contradictory effects (29, 30). As a result, this approach often interferes with normal physiologic and inflammatory processes in addition to modulating the desired target. To further complicate the issue, the inflammatory signaling pathways remain a highly complex and poorly understood system, inhibiting one particular pathway may be effectively bypassed through another pathway (12, 18).
Our group has taken a novel approach. We target the burn wound, the actual source of inflammatory signaling in order to block the initiation of SIRS. In determining a key inflammatory pathway target, p38 MAPK was identified as central in the host response to burn injury (31–34). By applying a topical p38 MAPK inhibitor directly to the burn wound, we hypothesized that local inflammatory source control would lead to improved survival and organ function by blocking the initiation of SIRS. This approach avoids the systemic immunosuppression and potential deleterious effects of inhibiting normal systemic inflammatory and physiologic responses. In a mouse scald burn model, our previously published work has demonstrated that topical p38 MAPK inhibition attenuates partial-thickness burn wound inflammatory signaling, leading to a significant reduction in acute lung injury and improved pulmonary and cardiac function (14–16). Markedly, we demonstrated a significant survival advantage in a rodent partial-thickness burn and pneumonia model for animals treated with a topical p38 MAPK inhibitor (15). Though our results with this approach thus far have been promising, two major questions remained prior to this current study. First, all the above studies were carried out on an established partial-thickness burn model. Due to the importance of full-thickness burn injury in the clinical sphere, it is crucial to elucidate the efficacy of topical inflammatory source control in a full-thickness burn model. Second, in order for this treatment to be of practical use, we need to establish a post-injury time window in which application may remain effective. Previous work centered on immediate (< 1 min) post-injury application of topical inflammatory inhibitor. To these ends, we established a mouse full-thickness scald burn model to test topical p38 MAPK inhibitors applications post burn, both immediate `Treatment' and 4 h `Delayed Treatment.'
To establish a full-thickness burn, we lengthened the time of burn to 25 s with scald temperature at 62°C, as described in materials and methods. Burn depth is poorly understood phenomenon. After burn injury, there is the initial cell necrosis and death that represents the zone of coagulation (35). The zone of stasis is just adjacent to the zone of coagulation, which may include the hair follicles (36). The cells in the zone of stasis are not necrotic, but may be apoptotic and could progress to cell death. The apoptotic cells in the zone of stasis that surround burn injuries usually die over a period of 2–3 days post injury with progressive increase in the wound depth (37). If the hair-cell follicles are well preserved without apoptosis, the burn wound is considered partial –thickness (38). To diagnose the depth of the wound, several methods have been advocated, and investigating the apoptotic status of the hair-cell follicles is one such method that is accepted in the experimental studies (39). In addition to H&E slide analyses, we used TUNEL assay to investigate the apoptotic status of the hair follicles as a surrogate to detect the depth of burn injuries. We found that our current burn model created a full-thickness injury appropriate for our experimental aims (Figs. 1, 2, 3).
Our first aim was to establish that a topical p38 MAPK inhibitor remains effective on full-thickness burn wounds. Utilizing ELISA to measure IL-6, MIP-2 and IL-1β cytokine levels, we were able to demonstrate a significant reduction of cytokine expression in the targeted dermis with topical p38 MAPK inhibitor treatment for both immediate `Treatment' and 4 h `Delayed Treatment' (Fig. 4). Cytokine IL-6 (ELISA) results were further confirmed via RT-qPCR, which demonstrated attenuation of IL-6 mRNA expression (Fig. 5). ELISA results for IL-6 and MIP-2 in serum samples showed significant attenuation of cytokine levels for both immediate `Treatment' and 4 h `Delayed Treatment' of topical inhibitors (Fig. 6). Attenuating the burn wound local inflammatory signaling with topical p38 MAPK inhibitors reduces the systemic release of pro-inflammatory mediators in a full-thickness burn model. The goal is not systemic absorption but penetration of the wound only, and the zone of coagulation and the eschar did not act as a barrier to the inhibitor, which was able to act on the viable cells in the wound with attenuation of expression of inflammatory mediators. Even in the delayed application group, topical p38 MAPK inhibitors are applied early in the course of wound healing, potentially before full eschar formation.
A second aim was to define a reasonable window of time post-injury that the topical inhibitor would remain effective if application were delayed. This is of practical importance, as an emergency responder or ER admission might be delayed for several minutes to several hours. In this study, we arbitrarily selected a 4 h delay in application. Evaluating burn wound tissue by ELISA for IL-6, MIP-2 and IL-1β cytokine production, we showed significant attenuation of inflammatory signaling with delayed inhibitor application (Fig. 4). Though the degree of attenuation was noted to be less dramatic with 4 hours delayed treatment versus immediate post-injury treatment, the resulted inhibition in cytokine expression remained significant. These 4 h `Delayed Treatment' findings were similarly confirmed by RT-qPCR IL-6 mRNA expression (Fig. 5).
There are several limitations to the current study. First, we used a short course experimental model with analyses of inflammatory mediator expression 24 hours after initial injury. Long-term outcome data with organ function analyses, similar to our previous experiments (14, 15), may be required. Moreover, a murine model does not fully represent human response, especially as it relates to wound healing. For future long-term wound healing experiments a female red Duroc pig model may better represent human wound healing (40, 41).
The concept of an effective topical treatment for burn induced SIRS is an exciting proposition. This form of drug delivery has the advantage of directly targeting the source of inflammation. We previously established that there is no significant functional absorption of a topical p38 MAPK inhibitor, and the effect of topical treatment is due to inhibition of burn wound inflammatory signaling (14). Topical p38 MAPK inhibition is more cell-specific and avoids the deleterious effects of systemic administration. This approach is also easily integrated into standard burn care practices with the use of topical bacteriostatic medications. In this study, we presented data that demonstrates the efficacy of this treatment in full-thickness scald burns and with a several hours delay in treatment application. These data further elucidate the viability and practicality of topical inflammatory source control in the eventual clinical treatment of severely burned patients.
Acknowledgements
This work was supported by NIH GM084132-01 (PI: Arbabi)
Footnotes
Conflict of interest: Dr Arbabi has received a grant from Array BioPharma Inc to study Post Burn Application of Arry-797 in a pig model of wound healing.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Runyan CW, Casteel C, Perkis D, Black C, Marshall SW, Johnson RM, Coyne-Beasley T, Waller AE, Viswanathan S. Unintentional injuries in the home in the United States - Part I: Mortality. American Journal of Preventive Medicine. 2005;28(1):73–9. doi: 10.1016/j.amepre.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 2.Ryan CM, Schoenfeld DA, Thorpe WP, Sheridan RL, Cassem EH, Tompkins RG. Objective estimates of the probability of death from burn injuries (vol 338, pg 362, 1998) New England Journal of Medicine. 1998;338(25):1849–50. doi: 10.1056/NEJM199802053380604. [DOI] [PubMed] [Google Scholar]
- 3.McGwin G, George RL, Cross JM, Reiff DA, Chaudry IH, Rue LW. Gender differences in mortality following burn injury. Shock. 2002;18(4):311–5. doi: 10.1097/00024382-200210000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Horan JM, Mallonee S. Injury surveillance. Epidemiologic Reviews. 2003;2(5):5–24. doi: 10.1093/epirev/mxg010. [DOI] [PubMed] [Google Scholar]
- 5.Mosier MJ, Pham TN, Klein MB, Gibran NS, Arnoldo BD, Gamelli RL, Tompkins RG, Herndon DN. Early acute kidney injury predicts progressive renal dysfunction and higher mortality in severely burned adults. J Burn Care Res. 2010;31(1):83–92. doi: 10.1097/BCR.0b013e3181cb8c87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cumming J, Purdue GF, Hunt JL, O'Keefe GE. Objective estimates of the incidence and consequences of multiple organ dysfunction and sepsis after burn trauma. J Trauma. 2001;50(3):510–5. doi: 10.1097/00005373-200103000-00016. [DOI] [PubMed] [Google Scholar]
- 7.Hansbrough JF, Wikstrom T, Braide M, Tenenhaus M, Rennekampff OH, Kiessig V, Bjursten LM. Neutrophil activation and tissue neutrophil sequestration in a rat model of thermal injury. Journal of Surgical Research. 1996;61(1):17–22. doi: 10.1006/jsre.1996.0074. [DOI] [PubMed] [Google Scholar]
- 8.Till GO, Johnson KJ, Kunkel R, Ward PA. Intravascular activation of complement and acute lung injury. Dependency on neutrophils and toxic oxygen metabolites. J Clin Invest. 1982;69(5):1126–35. doi: 10.1172/JCI110548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piccolo MT, Wang Y, Sannomiya P, Piccolo NS, Piccolo MS, Hugli TE, Ward PA, Till GO. Chemotactic mediator requirements in lung injury following skin burns in rats. Exp Mol Pathol. 1999;66(3):220–6. doi: 10.1006/exmp.1999.2263. [DOI] [PubMed] [Google Scholar]
- 10.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31(4 Suppl):S195–9. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 11.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405(6785):458–62. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 12.Bos JD. The skin as an organ of immunity. Clinical and Experimental Immunology. 1997;107:3–5. [PubMed] [Google Scholar]
- 13.Piccolo MT, Wang Y, Verbrugge S, Warner RL, Sannomiya P, Piccolo NS, Piccolo MS, Hugli TE, Ward PA, Till GO. Role of chemotactic factors in neutrophil activation after thermal injury in rats. Inflammation. 1999;23(4):371–85. doi: 10.1023/a:1020213717336. [DOI] [PubMed] [Google Scholar]
- 14.Ipaktchi K, Mattar A, Niederbichler AD, Hoesel LM, Vollmannshauser S, Hemmila MR, Su GL, Remick DG, Wang SC, Arbabi S. Attenuating burn wound inflammatory signaling reduces systemic inflammation and acute lung injury. Journal of Immunology. 2006;177(11):8065–71. doi: 10.4049/jimmunol.177.11.8065. [DOI] [PubMed] [Google Scholar]
- 15.Ipaktchi K, Mattar A, Niederbichler AD, Kim J, Hoesel LM, Hemmila MR, Su GL, Remick DG, Wang SC, Arbabi S. Attenuating burn wound inflammation improves pulmonary function and survival in a burn-pneumonia model. Crit Care Med. 2007;35(9):2139–44. doi: 10.1097/01.ccm.0000280568.61217.26. [DOI] [PubMed] [Google Scholar]
- 16.Hoesel LM, Mattar AF, Arbabi S, Niederbichler AD, Ipaktchi K, Su GL, Westfall MV, Wang SC, Hemmila MR. Local wound p38 MAPK inhibition attenuates burn-induced cardiac dysfunction. Surgery. 2009;146(4):775–86. doi: 10.1016/j.surg.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cowan KJ, Storey KB. Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. Journal of Experimental Biology. 2003;206(7):1107–15. doi: 10.1242/jeb.00220. [DOI] [PubMed] [Google Scholar]
- 18.Arbabi S, Maier RV. Mitogen-activated protein kinases. Critical Care Medicine. 2002;30(1):S74–S9. [PubMed] [Google Scholar]
- 19.Ipaktchi K, Mattar A, Niederbichler AD, Hoesel LM, Hemmila MR, Su GL, Remick DG, Wang SC, Arbabi S. Topical p38MAPK inhibition reduces dermal inflammation and epithelial apoptosis in burn wounds. Shock. 2006;26(2):201–9. doi: 10.1097/01.shk.0000225739.13796.f2. [DOI] [PubMed] [Google Scholar]
- 20.Gilpin DA. Calculation of a new Meeh constant and experimental determination of burn size. Burns. 1996;22(8):607–11. doi: 10.1016/s0305-4179(96)00064-2. [DOI] [PubMed] [Google Scholar]
- 21.Ioannovich I, Alexakis D, Kotzampassakis S, Harkiolakis G. The Use of a Myocutaneous Flap of the Platysma Muscle and the Cervical Skin for Reconstruction of Cheek Defects - Technical Approach and Results - 5 Cases. Annales De Chirurgie Plastique Et Esthetique. 1989;34(1):43–7. [PubMed] [Google Scholar]
- 22.Rodriguez JL, Miller CG, Garner WL, Till GO, Guerrero P, Moore NP, Corridore M, Normolle DP, Smith DJ, Remick DG, Baker CC, Wiles CE, Pruitt BA, Deitch EA, Rodriguez JL. Correlation of the Local and Systemic Cytokine Response with Clinical Outcome Following Thermal-Injury. Journal of Trauma-Injury Infection and Critical Care. 1993;34(5):684–95. doi: 10.1097/00005373-199305000-00011. [DOI] [PubMed] [Google Scholar]
- 23.Piccolo MTS, Wang Y, Verbrugge S, Warner RL, Sannomiya P, Piccolo NS, Piccolo MS, Hugli TE, Ward PA, Till GO. Role of chemotactic factors in neutrophil activation after thermal injury in rats. Inflammation. 1999;23(4):371–85. doi: 10.1023/a:1020213717336. [DOI] [PubMed] [Google Scholar]
- 24.Fitzwater J, Purdue GF, Hunt JL, O'Keefe GE. The risk factors and time course of sepsis and organ dysfunction after burn trauma. J Trauma. 2003;54(5):959–66. doi: 10.1097/01.TA.0000029382.26295.AB. [DOI] [PubMed] [Google Scholar]
- 25.Cohen MJ, Carroll C, He LK, Muthu K, Gamelli RL, Jones SB, Shankar R. Severity of burn injury and sepsis determines the cytokine responses of bone marrow progenitor-derived macrophages. J Trauma. 2007;62(4):858–67. doi: 10.1097/01.ta.0000222975.03874.58. [DOI] [PubMed] [Google Scholar]
- 26.Alejandria MM, Lansang MA, Dans LF, Mantaring JB. Intravenous immunoglobulin for treating sepsis and septic shock. Cochrane Database Syst Rev. 2002;(1):CD001090. doi: 10.1002/14651858.CD001090. [DOI] [PubMed] [Google Scholar]
- 27.Annane D, Bellissant E, Bollaert PE, Briegel J, Confalonieri M, De Gaudio R, Keh D, Kupfer Y, Oppert M, Meduri GU. Corticosteroids in the treatment of severe sepsis and septic shock in adults: a systematic review. JAMA. 2009;301(22):2362–75. doi: 10.1001/jama.2009.815. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura A, Niimi R, Yanagawa Y. Renal beta2-adrenoceptor blockade worsens the outcome of an induced Escherichia coli renal infection. J Nephrol. 2010;23(3):341–9. [PubMed] [Google Scholar]
- 29.Bolla M, Matrougui K, Loufrani L, Maclouf J, Levy BI, Levy-Toledano S, Habib A, Henrion D. p38 mitogen-activated protein kinase activation is required for thromboxane-induced contraction in perfused and pressurized rat mesenteric resistance arteries. Journal of Vascular Research. 2002;39(4):353–60. doi: 10.1159/000065547. [DOI] [PubMed] [Google Scholar]
- 30.Houliston RA, Pearson JD, Wheeler-Jones CPD. Agonist-specific cross talk between ERKs and p38(mapk) regulates PGI(2) synthesis in endothelium. American Journal of Physiology-Cell Physiology. 2001;281(4):C1266–C76. doi: 10.1152/ajpcell.2001.281.4.C1266. [DOI] [PubMed] [Google Scholar]
- 31.Adediran SG, Dauplaise DJ, Kasten KR, Tschop J, Dattilo J, Goetzman HS, England LG, Cave CM, Robinson CT, Caldwell CC. Early infection during burn-induced inflammatory response results in increased mortality and p38-mediated neutrophil dysfunction. Am J Physiol Regul Integr Comp Physiol. 2010;299(3):R918–25. doi: 10.1152/ajpregu.00132.2010. [DOI] [PubMed] [Google Scholar]
- 32.Bulger EM, Edwards T, Klotz P, Jurkovich GJ. Epidural analgesia improves outcome after multiple rib fractures. Surgery. 2004;136(2):426–30. doi: 10.1016/j.surg.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 33.Costantini TW, Peterson CY, Kroll L, Loomis WH, Eliceiri BP, Baird A, Bansal V, Coimbra R. Role of p38 MAPK in burn-induced intestinal barrier breakdown. J Surg Res. 2009;156(1):64–9. doi: 10.1016/j.jss.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q, Carter EA, Ma B, Fischman AJ, Tompkins RG. Burn-related metabolic and signaling changes in rat brain. J Burn Care Res. 2008;29(2):346–52. doi: 10.1097/BCR.0b013e3181667387. [DOI] [PubMed] [Google Scholar]
- 35.Shupp JW, Nasabzadeh TJ, Rosenthal DS, Jordan MH, Fidler P, Jeng JC. A review of the local pathophysiologic bases of burn wound progression. J Burn Care Res. 2010;31(6):849–73. doi: 10.1097/BCR.0b013e3181f93571. [DOI] [PubMed] [Google Scholar]
- 36.Penington AJ, Craft RO, Morrison WA. A defined period of sensitivity of an experimental burn wound to a second injury. J Burn Care Res. 2006;27(6):882–8. doi: 10.1097/01.BCR.0000245655.16595.E0. [DOI] [PubMed] [Google Scholar]
- 37.Morykwas MJ, David LR, Schneider AM, Whang C, Jennings DA, Canty C, Parker D, White WL, Argenta LC. Use of subatmospheric pressure to prevent progression of partial-thickness burns in a swine model. J Burn Care Rehabil. 1999;20(1 Pt 1):15–21. doi: 10.1097/00004630-199901001-00003. [DOI] [PubMed] [Google Scholar]
- 38.Uygur F, Evinc R, Urhan M, Celikoz B, Haholu A. Salvaging the zone of stasis by simvastatin: an experimental study in rats. J Burn Care Res. 2009;30(5):872–9. doi: 10.1097/BCR.0b013e3181b47eb8. [DOI] [PubMed] [Google Scholar]
- 39.Harada T, Izaki S, Tsutsumi H, Kobayashi M, Kitamura K. Apoptosis of hair follicle cells in the second-degree burn wound unders hypernatremic conditions. Burns. 1998;24(5):464–9. doi: 10.1016/s0305-4179(98)00034-5. [DOI] [PubMed] [Google Scholar]
- 40.Zhu KQ, Carrougher GJ, Couture OP, Tuggle CK, Gibran NS, Engrav LH. Expression of collagen genes in the cones of skin in the Duroc/Yorkshire porcine model of fibroproliferative scarring. J Burn Care Res. 2008;29(5):815–27. doi: 10.1097/BCR.0b013e3181848141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu KQ, Engrav LH, Gibran NS, Cole JK, Matsumura H, Piepkorn M, Isik FF, Carrougher GJ, Muangman PM, Yunusov MY, Yang TM. The female, red Duroc pig as an animal model of hypertrophic scarring and the potential role of the cones of skin. Burns. 2003;29(7):649–64. doi: 10.1016/s0305-4179(03)00205-5. [DOI] [PubMed] [Google Scholar]