

Abstract
Background
Neuro-immune interactions play a significant role in regulating the severity of inflammation. Our previous work demonstrated that neuropeptide Y (NPY) is up regulated in the enteric nervous system (ENS) during murine colitis, and that NPY knockout mice exhibit reduced inflammation. Here we investigated if NPY expression during inflammation is induced by tumor necrosis factor (TNF), the main pro-inflammatory cytokine.
Methods
Utilizing primary enteric neurons and colon explant cultures from WT and NPY knockout (NPY−/−) mice, we determined if NPY knockdown modulates TNF release and epithelial permeability. Further we assessed if NPY expression is inducible by TNF in enteric neuronal cells and mouse model of experimental colitis, utilizing the TNF inhibitors-etanercept (blocks transmembrane and soluble TNF) and XPro1595 (blocks soluble TNF only).
Results
We found that enteric neurons express TNF receptors (TNFR1 and R2). Primary enteric neurons from NPY−/− mice produced less TNF compared to WT. Further, TNF activated NPY promoter in enteric neurons via phospho-c-jun. NPY−/− mice had decreased intestinal permeability. In vitro, NPY increased epithelial permeability via phosphatidyl inositol-3-kinase (PI3-K)-induced pore-forming claudin-2. TNF inhibitors attenuated NPY expression in vitro and in vivo. TNF-inhibitor-treated colitic mice exhibited reduced NPY expression and inflammation, reduced oxidative stress, enhanced neuronal survival and improved colonic motility. XPro1595 had more protective effects on neuronal survival and motility compared to etanercept.
Conclusions
We demonstrate a novel TNF-NPY cross talk that modulates inflammation, barrier functions and colonic motility during inflammation. It is also suggested that selective blocking of soluble TNF maybe a better therapeutic option than using anti-TNF antibodies.
Keywords: NPY, TNF, enteric neurons, experimental colitis, barrier functions, colonic motility
Introduction
Neuro-immune cross talk has been identified to play a critical role in the pathogenesis of inflammatory diseases (1–3). Neuropeptide Y (NPY) is a 36 amino-acid peptide abundantly produced in the murine enteric neurons (4). NPY is implicated in inflammatory diseases (5, 6) due to its ability to regulate immune responses (7). Previously we have demonstrated that colonic NPY is upregulated during murine experimental colitis and that NPY knockout mice (NPY−/−) are protected against dextran sodium sulfate (DSS) and Salmonella typhimurium models of colitis (8). However, the mechanisms that trigger NPY expression in the enteric nervous system (ENS) during inflammation are poorly understood. The pro-inflammatory cytokine, tumor necrosis factor (TNF) plays a central role in the pathogenesis of inflammatory bowel disease (IBD). Anti-TNF antibodies (infliximab, etanercept) are the popular drugs in IBD (9) to curb the increased TNF levels during inflammation (10). Increased TNF levels has been demonstrated to increase epithelial permeability (11), induce neuropathic pain (12) and colonic motility dysfunctions (13, 14). In the present study we investigated if TNF induces NPY expression during inflammation in the ENS, and whether NPY-TNF interactions can regulate inflammation, epithelial permeability and colonic motility.
To elucidate this interaction we utilized two TNF inhibitors-etanercept (a recombinant fusion protein) that blocks both transmembrane (26 kd TNF precursor) and soluble forms of TNF (17 kd); and XPro1595 (dominant negative inhibitor of TNF) that blocks only the soluble form. Despite their popular use, the effects of TNF blockers on enteric neurons have not been investigated. We determined if NPY expression is regulated by TNF in enteric neurons in vitro and in a mouse model of colitisutilizing the TNF inhibitors.
We identified that enteric neurons express TNF receptors (TNFR1 and TNFR2), and hence are direct targets of anti-TNF drugs. TNF inhibitors attenuated NPY expression in enteric neurons, and TNF activated neuronal NPY promoter. Enteric neurons from NPY−/− mice secreted less TNF compared to WT mice, and NPY−/− mice had decreased intestinal permeability. We observed that NPY directly increased epithelial permeability via phosphatidyl inositol-3-kinase (PI3-K)-induced up regulation of claudin-2. In colitic mice, TNF inhibitors reduced NPY expression in enteric neurons, reduced oxidative stress and neuronal apoptosis and improved colonic motility. Selective blocking of soluble TNF by XPro1595 in mice proved more beneficial in attenuating oxidative stress, and improving neuronal survival and colonic motility.
Materials and Methods
Animals
All animal protocols were approved by the Emory University Institutional Animal Care and Use Committee (IACUC). Mice (Stock No.002448, 129 S3/SvImJ) were obtained from Jackson Laboratory (Bar Harbor, Maine, US).
Induction of colitis
Colitis was induced as per the documented protocols (15) in mice by administering 3% (w/v) dextran sodium sulfate (DSS, MP Biomedicals, CA) in drinking water for a week. In addition to receiving DSS, two groups of mice also received an intraperitoneal injection of one of the TNF inhibitors etanercept or XPro1595 (both from Xencor, Monrovia, CA) @ 10 mg/kg body weight) on days 1, 4 and 7. The mice were euthanized on day 8. The experimental design is given in Figure 1A. The distal colon was collected in liquid nitrogen to be used later for real-time PCR and Western blotting or cryo fixed (OCT) or paraffin embedded for hematoxylin-eosin staining or immunostaining.
Figure 1. NPY knockdown reduces TNF production from enteric neurons, and TNF activates NPY promoter.

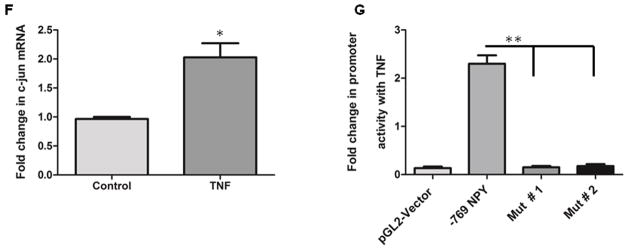
Primary enteric neuronal cultures from two-day-old WT and NPY−/− mice pups were cultured for 10 days at 37°C. Colonic explants from eight-week-old WT and NPY−/− mice were cultured in RPMI for 24 h. The supernatants from primary cultures and colon explants were assayed for TNF release by ELISA. Enteric neuronal cell lines were stimulated with TNF (25 ng) in presence or absence of TNF inhibitors (25 ng). RNA was extracted after 6 h and amplified for NPY expression by real-time PCR. Enteric neuronal cell line was transfected with various NPY deletion promoter constructs and assayed for luciferase activity in presence and absence of TNF. The chromatin from enteric neuronal cells treated with/without TNF was immunoprecipitated with phospho-c-jun antibody. Fold change in c-jun binding on NPY promoter was assessed by real-time PCR. The site directed mutagenesis of c-jun binding site was performed using a kit as detailed in methods, and TNF-induced promoter activity was monitored by luciferase assay in two mutant clones (Mut #1 & #2). (A) Primary enteric neurons in culture under light microscope (20X) (B) TNF levels in WT and NPY−/− enteric neurons (C) TNF levels in colon explants cultures from WT and NPY−/− mice (D) NPY expression in enteric neuronal cells treated with TNF and TNF inhibitors (E) luciferase assay depicting activation of NPY promoter constructs by TNF (F) Fold change in phospho-c-jun expression with TNF and (G) luciferase assay utilizing mutated constructs of c-jun binding site. Values are Mean ± SEM, n = 6, * p< 0.05, ** p < 0.01.
Human study
The study was approved by Emory University Institutional Review Board Committee and Beth Israel Deaconess Medical Center (Boston, MA). Pre-operative medical records were reviewed for documented history of IBD. Colonic biopsies were obtained during endoscopy from healthy controls or patients with IBD (after informed consent) at Beth Israel Deaconess Medical Center, Emory University hospital, Atlanta, GA and the Atlanta VA Medical Center, Decatur, GA. These procedures were performed for polyps or malignancy. Real time PCR was used for analysis of NPY and catalase (antioxidant enzyme) expression.
Primary enteric neuronal culture
Primary enteric neuronal cultures were established from two-day-old WT and NPY−/− pups as described previously (16).
Enzyme Linked Immunosorbent Assay (ELISA)
Primary enteric neurons from WT and NPY−/− mice (129S-Npytm1Rpa/J, Stock no. 004545, Jackson Laboratory, Maine, US) were cultured as described previously (16). The colon from eight-week old WT and NPY−/− mice was harvested and cultured in RPMI medium for 24 h. TNF levels in the culture supernatants were determined by ELISA (Ray BioTech Inc, Norcross, GA).
Immunostaining
Colonic sections were immunostained with anti-NPY, anti-nNOS and anti-Peripherin antibodies (1:500, BD Laboratories) as described previously (16). The cleaved caspase-3 staining (1: 300) was expressed as a percentage of peripherin staining. The images were captured using a Zeiss fluorescence microscope, and analyzed by Metamorph (Molecular Devices, Silicon Valley, CA, USA) as described previously (17).
Laser capture microdissection (LCM)
Enteric ganglia were captured from cryo sections by LCM as described previously (17). The RNA was isolated from the ganglia using Arcturus Pico-Pure RNA Extraction Kit (Grand Island, NY) and amplified for NPY expression by real time PCR. GAPDH was used as loading control.
Luciferase assay
Enteric neurons were transfected with full length (−1078) and various deletion sequences of the NPY promoter (−278, −728, −867 and −1078 bp) in a pGL2-luciferase reporter system kindly provided by Dr. Denise Belsham (University of Toronto, Ontario, Canada)(18, 19). The cells were then stimulated with TNF (25 ng) for 6 h, and NPY promoter activity was determined by Dual Luciferase Assay Kit (Promega, Madison, WI), and expressed as Relative Luciferase Units (RLU) as compared to the empty pGL2 vector.
Site-directed mutagenesis
Site-directed mutagenesis was performed using the QuikChange Site Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The c-jun binding site (consensus sequence-5′-TGAGTC-3′) within the −769 NPY-Luc construct was mutated to generate mutant constructs Mut # 1 (CGATTC) and Mut # 2 (GAAGCC). Primers were designed to amplify the mutated region (S. Table 1). The plasmid DNA of the −769 NPY-Luc construct was amplified with mutant primers and mutant clone plasmid DNA was isolated (Sigma Gen-Elute Midi prep Kit), and sequenced to confirm the mutation (Eurofins MWG Operon sequencing, Des Moines, IA).
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed using manufacturer’s protocol (Upstate Biotechnology, Lake Placid, NY) using anti-c-Jun, anti-c-fos, anti-phospho-c-jun or anti-ATF (Cell Signaling) antibodies. For PCR, the primers: 5′-GGG AAG TGT TCT CAT CTA TG-3′ and antisense, 5′-GGC CAC TTCCAA CCT TTA AA-3′) encompassing a 220 bp fragment (from −398 to −624 bp) of the mouse NPY promoter were used.
Transcription Element Search System (TESS)
The NPY promoter sequence (−2000 base pairs from the transcription start site of mouse NPY mRNA, NM_023456.2) was analyzed for putative transcription factor binding sites by TESS (http://www.cbil.upenn.edu/tess/).
Transepithelial electrical Resistance (TER)
Caco2-BBE cells were seeded on transwell permeable filters (pore size 0.4 μm) at 40,000 cells/0.33 cm2, (Corning, Lowell, MA). TER was measured using Millicel-ERS (Electrical Resistance System from Millipore, Billerica, MA) before and after basolateral addition of NPY (0.1 μM) at 1, 2, 24 and 48 h.
Paracellular permeability
FITC-dextran (4 kD) was added to the apical compartment of Caco2-BBE monolayer. At 0 and 24 h, 100 μl of the basolateral medium was sampled for FD4 translocation (mg of FITC-dextran release/ml) by measuring the absorbance (Excitation λ450 nm, Emission λ510 nm).
Colonic permeability
WT and NPY −/− mice were orally gavaged with FD-4 and colonic permeability was measured as described previously (20). The colonic muscle strips from WT and NPY −/− mice were tested ex vivo for FD-4 (22 mg/ml) permeability in an Ussing chamber (20). TER across the muscle strips was simultaneously monitored at 10, 20, 30, 45 and 60 min (T-10 to T-60).
Western blotting
Caco2-BBE/T84 monolayers on transwell permeable supports were exposed to NPY (0.1μM), and probed for claudins-1, 2 and 4 after 24 h (Life Technologies, 1: 2500), and pAkt after 1h (Cell Signaling, 1: 1000).
Colonic motility assessment
Colonic motility was evaluated by isometric muscle recording with electrical field stimulation as described previously (17). The longitudinal muscle strips with intact innervation were used for the experiment.
Whole mount staining
The myenteric plexus was peeled off from the formalin-fixed proximal colon, and was used for NADPH diaphorasestaining. The neuronal numbers were counted using a 1mm2 grid as described previously (16).
Statistical analysis
Data were analyzed using one-way ANOVA (Graph Pad Software, San Diego, CA), followed by Tukey’s multiple comparison tests, or Student’s t-test (two tailed unpaired) as applicable. A p value < 0.05 was considered statistically significant.
Results
NPY knockdown is associated with attenuated TNF production
Based on our previous observations that NPY knockdown mitigates experimental colitis (8), we investigated if NPY knockdown would modulate TNF production. We observed that primary enteric neuronal cultures from two-day-old WT mouse pups produced more TNF compared to NPY−/− pups (Figure 1A–B, p < 0.01). In addition, explant colon cultures from WT mice also produced more TNF compared to NPY−/− (Figure 1C, p< 0.05). We have previously demonstrated that NPY increases TNF production from enteric neuronal cells in vitro (8). Collectively our data suggests a cross talk between TNF-NPY in enteric neurons. To understand the relevance of this cross talk during inflammation, we next investigated whether TNF can directly activate NPY expression in enteric neurons.
TNF inhibitors attenuate NPY expression in enteric neurons, and TNF activates NPY promoter
Studies on primary cells and the enteric neuronal cell line developed in our laboratory (21) demonstrated that enteric neurons express both TNF receptors TNFR1 and TNFR2 (Supplemental Figure 1A), and TNF induced NPY expression in enteric neuronal cells (Figure 1D, p < 0.01). Further studies utilizing the neuronal cell line indicated that both TNF inhibitors etanercept (p < 0.05) and XPro1595 (Figure 1D, p < 0.01) inhibited NPY expression. These results indicate that NPY expression is inducible by TNF, suggestive of dynamic NPY-TNF cross-talk during inflammation in the ENS. We next assessed if TNF can regulate NPY promoter. We transiently transfected enteric neurons with pGL2-reporter plasmids containing various human NPY promoter deletion constructs as described in Methods. Luciferase assays demonstrated an increase in the reporter gene activity (2.5 ± 0.7, p < 0.05) in the region between −728 and −867 region upon TNF stimulation (Figure 1E). To investigate the downstream effectors of TNF signaling in enteric neurons, we analyzed the TNF-responsive region (−728 and −867 bp) for putative transcription factor binding sites utilizing the TESS database. Interestingly we found numerous putative binding sites for Activator Protein-1 (AP-1, includes c-jun, c-fos and Activating Transcription Factor, ATF), a well-known downstream effector of TNF, on NPY promoter. ChIP assays showed increased phospho-c-Jun binding to the −769bp region of the NPY promoter after TNF stimulation. Further, real-time PCR demonstrated a significant increase in the phospho-c-jun promoter occupancy (2.2 ± 0.5, p < 0.05, Figure 1F) in TNF-stimulated cells compared to control. To further ensure the requirement of phospho-c-jun binding in mediating TNF-induced promoter activity, mutations were introduced in the c-jun binding site (consensus sequence TGAGTCA) on NPY promoter. Our results indicate that mutations in the consensus sequence completely abolished the TNF-responsiveness, implying that phospho-c-jun binding is essential for TNF-induced NPY expression in enteric neurons (p< 0.01, Figure 1G). Collectively, these results support that TNF activates NPY promoter in enteric neurons.
NPY−/− mice display decreased intestinal permeability compared to WT
Increased epithelial permeability is a risk factor as well as a hallmark of IBD pathophysiology (22) and TNF has been demonstrated to increase epithelial permeability via claudin-2 (11). It has also been demonstrated that anti-TNF drugs can prevent epithelial barrier dysfunctions (23). Having established that NPY knockdown inhibits TNF production, we next investigated the relevance of this interaction in regulating epithelial permeability. We first compared the colonic paracellular permeability in WT and NPY−/− mice. We gavaged mice with FITC-Dextran (4 kD, FD-4) and serum levels of FD-4 (ng/gm serum protein) were measured after 4 h. We observed that serum FD-4 levels were lower in NPY−/− mice compared to WT, suggestive of decreased epithelial paracellular permeability in these mice (Figure 2A, p<0.05). We next performed ex vivo permeability studies on isolated distal colonic strips from WT and NPY−/− mice using an Ussing chamber. A significantly higher short circuit current (ISc) was observed in WT mice (12.39 ± 0.5 μA) compared to NPY−/− (3 ± 0.2μA, Figure 2B, p< 0.05), suggestive of increased permeability in WT mice. Further, reduced FD-4 flux was observed across the colonic muscle strips at 1 h in NPY−/− mice (90 ± 15 μg/cm2, Figure 2C, p< 0.01) compared to WT mice (550 ± 165 μg/cm2). These data suggest that NPY may compromise the epithelial integrity.
Figure 2. NPY increases permeability of colonic epithelial monolayer via pore-forming claudin-2.

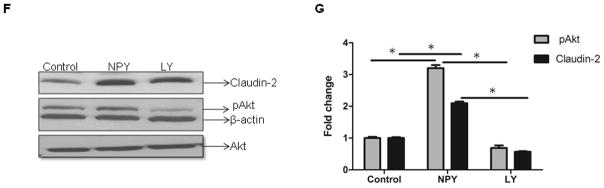
(A) WT and NPY −/− mice were gavaged with FITC Dextran (FD4, 4 kD) and amount of FD4 released into the serum (ng/gm serum protein) was measured after 4 h in a spectrofluorimeter (excitation λ 450 nm, emission λ 510 nm). The colonic muscle strips from WT and NPY −/− mice were tested ex vivo in Ussing chamber for (B) short circuit current, ISc (μA) and (C) FD-4 (22mg/ml) translocation over a time period of 0–60 min as described in Methods. (D) Caco2-BBE cells on transwell supports were treated with NPY (0.1 μM) and TER measured at 1, 2, 24 and 48h. (E) Caco2-BBE cells were treated with NPY and the translocation of apically added FD-4 into the basolateral medium was measured after 24 h relative to control. (F) Caco2-BBE cells were cultured on transwell filter supports and treated with NPY (0.1 μM) in presence or absence of PI3-K inhibitor (LY-294002) and cell lysates were analyzed for claudin-2, pAKt and Akt by Western blotting. Results are graphically represented in (G). Values are Mean ± SEM, n = 6, * p < 0.05, **p< 0.01, *** p< 0.001.
NPY increases permeability of the epithelial monolayer via pore-forming Claudin-2
Having observed a difference in colonic permeability in mouse models and to define the mechanisms involved, we next assessed the effect of treatment with NPY (24) on colonic epithelial permeability in vitro (Caco2-BBE) using TER and macromolecular flux (FD-4). We observed a significant decrease in TER in NPY-treated cells compared to control in 24 h (54 ± 5.0 %, P < 0.01) and 48 h (45 ± 8%, p<0.001, Figure 2D), indicative of increased permeability. We observed increased flux of FD-4 in NPY-treated Caco2-BBE cells after 24 h (144.17 ± 12.58 μg/ml, p< 0.01) compared to untreated controls (34.92 ± 1.57 μg/ml, Figure 2E). Collectively, our in vitro and in vivo data indicate a central role of NPY in increasing epithelial permeability and subsequent susceptibility to inflammation. Given that NPY can increase permeability, we next investigated whether NPY could alter the expression of tight junction proteins in the epithelium. Among the tight junction proteins, claudin-2 has been associated with a “leaky” barrier and is up regulated in human IBD (25). Also it is reported that TNF induces claudin-2 in epithelia via PI3-K (11). Hence we assessed whether treatment with NPY affects claudin-2. We observed a modest increase in claudin-2 protein (2 ± 0.3, p< 0.05) after NPY treatment in Caco2-BBE cells (Figure 2F, p < 0.05). Further experiments revealed that effects of NPY were mediated in part by Akt activation (3.2 ± 0.1, p< 0.05, Figure 2F–G) as it was sensitive to inhibition by the phosphatidyl-inositol-3-kinase (PI3-K) inhibitor (LY 294002, LY). We also noticed that NPY induced similar Akt activation in T84 cells (Supplemental Figure 2A–B). We did not notice changes in other claudins like claudins- 1 and -4 on NPY treatment (Supplemental Figure C–D). Taken together our results indicate that in addition to mediating TNF effects, NPY can also have direct effects on permeability via PI3-K.
TNF inhibitors attenuated NPY expression in enteric ganglia during experimental colitis
Several studies have shown that TNF levels are upregulated in human IBD (10, 26). Having demonstrated TNF-regulation of NPY promoter in vitro, we next assessed if administration of TNF inhibitors can attenuate NPY expression during experimental colitis in mice. The in vivo study design utilizing a dextran sodium sulfate (DSS) model of colitis is represented in Figure 3A. We have previously demonstrated that NPY is upregulated in the colon during two experimental colitis models, and that NPY−/− mice are resistant to colitis (8). Now we observed that in the DSS colitis model, mice receiving intraperitoneal administration of TNF inhibitors exhibited attenuated NPY expression in the enteric ganglia as demonstrated by immunostaining and quantified by Metamorph (Figure 3B–C, p< 0.05). To further validate this finding, we isolated enteric ganglia by laser capture micro dissection and assessed NPY expression by real-time PCR. There was a 2 fold up regulation (± 0.1, p < 0.05) of NPY in the enteric ganglia of DSS-treated mice, which was significantly reduced by TNF inhibitors (Figure 3D, p <0.01). Further we observed that mice receiving the TNF inhibitors also exhibited less inflammation as assessed from loss of body weight (Figure 3E), colon length (Figure 3F) and histological damage (Figure 3G–H). In addition, we observed a difference in the anti-inflammatory effects of the two TNF inhibitors tested. Etanercept is a recombinant fusion protein that acts as a decoy receptor that binds TNF, thereby reducing its availability. On the other hand, XPro1595 is a dominant negative inhibitor of TNF which depletes the TNF pool (composed of active TNF homotrimers) by replacing it with heterotrimers that do not bind to the receptor. A comparison of the inhibitors revealed that soluble TNF inhibition was more beneficial considering changes in colon length and (Figure 3E, p < 0.05) and histological damage (Figure 3G–H, p < 0.05).
Figure 3. TNF inhibitors attenuate enteric neuronal NPY expression and inflammation in murine experimental colitis.


Mice were given DSS in drinking water for seven days, with three intraperitoneal injections of either etanercept or XPro1595at 10 mg/kg body weight starting from day 1 as outlined in (A). On day 8, mice were euthanized and colon was collected. The paraffin-embedded colon sections were assessed for (B) NPY expression (red) by immunostaining using peripherin (green) as general neuronal marker, and the staining intensity arbitrarily quantified by Metamorph is graphically represented in (C). The colonic ganglia were isolated by LCM from cryo sections, and NPY expression was assessed by real-time PCR in (D). Markers of inflammation like (E) Body weight loss (F) Colon length (cm) and Histological damage (G–H) after hematoxylin-eosin staining were also determined in mice receiving DSS alone or both DSS and TNF inhibitors. Values are Mean ± SEM, n = 6, * p < 0.05, ** p< 0.01. Magnification 20 X, Scale bar 50 μm.
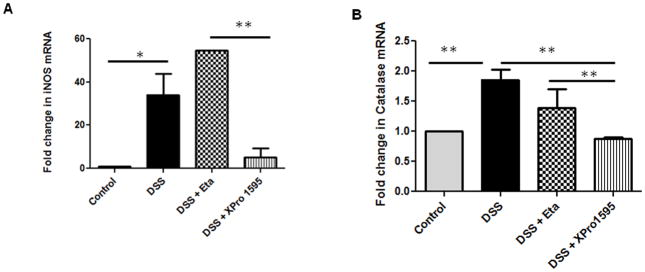
TNF inhibitors reduced oxidative stress and neuronal apoptosis in colitic mice, thereby improving neuronal survival
Oxidative stress is a characteristic feature of IBD (27, 28) and inducible nitric oxide synthase (iNOS) expression is an established marker of inflammation and oxidative stress (29). We have previously demonstrated that NPY−/− mice exhibit less nitrosative stress and hence attenuated inflammation compared to WT. In this study we observed that NPY levels are up regulated in biopsies from IBD patients (Supplemental Figure 3A) and also there was increased oxidative stress in biopsies as evident from increased catalase expression (Supplemental Figure 3B). Analysis of oxidative stress markers revealed that XPro1595-treated mice had significant reduction in the expression of colonic iNOS as compared to DSS or DSS/etanercept-treated mice (Figure 4A, p < 0.05). We also observed an increase in the expression of the antioxidant enzyme catalase in DSS and DSS-etanercept treated mice, suggestive of increased oxidative stress (Figure 4B, p < 0.01). Since oxidative stress has been linked to increased neuronal apoptosis (30), we next investigated whether TNF inhibitors could attenuate apoptosis in the enteric ganglia (using cleaved caspase-3 immunostaining with peripherin as a general neuronal marker). We observed a significant reduction in apoptosis in colitic mice treated with TNF inhibitors as compared to DSS alone (p < 0.05, Figure 4C). A quantitative analysis of the immunostaining data is presented in Figure 4D. We next investigated whether a reduction in neuronal apoptosis would result in improved neuronal survival. We observed that the number of diaphorase (blue) and nNOS-positive (red) neurons in the myenteric plexi of mice treated with etanercept (p < 0.05) and XPro1595 (p< 0.001) were significantly higher when compared to DSS-treated mice (Figure 4E–G). Moreover we also observed that XPro1595 had more beneficial neuro-protective effects (neuronal survival, motility) compared to etanercept.
Figure 4. TNF inhibitors reduce enteric neuronal apoptosis and enhance neuronal survival via attenuation of oxidative stress.


Mice were administered DSS in drinking water for seven days; they also received three intraperitoneal injections (every third day) of either etanercept or XPro1595 at 10 mg/kg body weight. Oxidative stress was evaluated from inducible nitric oxide synthase (iNOS) and antioxidant catalase expression in mouse colonic mucosa by real time PCR. Paraffin-embedded colonic sections were immuno stained for cleaved caspase-3, and peripherin was used as a general neuronal marker. Intensity of staining was assessed by Metamorph. Myenteric plexi peeled from the formalin-fixed colonic tissues of mice were stained for diaphorase and neuronal nitric oxide synthase (nNOS) positive neurons as described in Methods (A) iNOS, and (B) catalase expression. (C) Representative images of cleaved caspase-3 (red) staining in colonic sections, ganglia are represented by arrows, and graphical representation of staining intensity as quantified by Metamorph in (D). Representative images of diaphorase (blue) and nNOS staining (red) is represented in (E) and neuronal counts are represented in (F) and (G). Values are Mean ± SEM, n = 6, * p < 0.05, *** p< 0.001. Magnification 20X, Scale bar 50 μm.
TNF inhibitors improved colonic motility during experimental colitis
Colonic motility dysfunctions are commonly associated with IBD (31–33). Also inducible nitric oxide (iNOS)- induced over production of nitric oxide has been implicated in the suppression of colonic contractility during IBD (29). Having demonstrated the protective role of TNF inhibitors against neuronal apoptosis, we next assessed if enhanced neuronal survival would improve colonic motility. We have previously demonstrated that WT mice exhibit severe colonic motility impairments during acute DSS colitis (8). We found that TNF inhibitors significantly improved neuronal relaxation responses (tetradotoxin-sensitive) from longitudinal muscle strips in mice. Non-adrenergic non-cholinergic (NANC) relaxation improved with etanercept treatment (22.57 ± 7.25 %), and was even better in XPro1595-treated mice (31.57 ± 6.34 %, p < 0.05, Figure 5A). The mice exposed to DSS alone (4.98 ± 2.0%) exhibited significantly reduced relaxation due to extensive inflammation-induced neuronal loss. The representative tracings for relaxation are given in Figure 5B. Similarly we found that colonic contraction (induced after treatment of muscle strips with L-NAME, nitric oxide inhibitor) improved in both etanercept and XPro1595-treated mice (p < 0.05,Figure 5C) as compared to mice on DSS alone. Representative tracings for contraction are given in Figure 5D. Colonic motility is coordinated by complex neuron-smooth muscle interactions; hence we also investigated the relaxation and contraction responses of the colonic smooth muscle. We found that smooth muscle relaxation (induced by sodium nitroprusside, SNP) significantly improved in TNF-inhibitor treated mice as compared to the DSS-treated mice (p < 0.05, Figure 5E). However, smooth muscle contraction responses significantly improved only in XPro1595-treated mice (p < 0.05, Figure 5F). The representative tracings are given in Figure 5G. These data suggest that inhibition of solTNF with XPro1595 improved both neuronal and smooth muscle responses that ultimately coordinate colonic motility. In aggregate, our findings indicate that TNF inhibitors decreased oxidative stress and improved neuronal survival, which in turn, improved the colonic motility, during experimental colitis in mice.
Figure 5. Administration of TNF inhibitors improves colonic motility in mice with colitis.


Mice were given DSS in drinking water for seven days, with three intraperitoneal injections of either etanercept or XPro1595at 10 mg/kg body weight starting from day 1. On day 8, mice were euthanized and the longitudinal muscle strips peeled from the colon were subjected to isometric muscle recording (as detailed in Methods) with electrical field stimulation for assessment of neuronally-mediated colonic relaxation and contraction. In addition, these muscle strips were stimulated with carbachol to assess smooth muscle contraction, and with sodium nitroprusside (SNP) to evaluate smooth muscle relaxation responses. (A) Neuronally mediated relaxation, the representative tracings are given in (B). Colonic contraction responses in mice are plotted in (C), the representative tracings are shown in (D). Smooth muscle relaxation and contraction responses are given in (E) and (F) respectively, with the tracings represented in (G). Values are Mean ± SEM, n = 6, * p < 0.05, **p< 0.01, *** p< 0.001.
Discussion
The role of neuro-immune interactions has gained significant attention in the pathogenesis of inflammatory diseases such as IBD (34, 35). Our current study reveals a whole new perspective on the role of NPY during inflammation as represented in Figure 6. We have demonstrated previously that NPY is up regulated during experimental colitis, and NPY knockout mice exhibit less inflammation using two models of experimental colitis (8). In the present study we demonstrate that primary enteric neuronal cells from NPY−/− mice produce less TNF than WT mice. This finding suggests a strong cross talk between NPY-TNF in the enteric neurons. We have also demonstrated previously that NPY co-localizes with neuronal nitric oxide synthase containing neurons (nNOS)(16). We identified that up regulation of NPY during inflammation also involves nNOS upregulation, leading to increased levels of nitric oxide and oxidative stress via creation of a double knockout mice for NPY−/−/nNOS−/−(8). Since nNOS–derived nitric oxide also regulates colonic motility under normal conditions, decreased nNOS in NPY−/− mice accounts for reduced colonic relaxation in these mice. Collectively, these observations indicate that NPY can regulate TNF production, oxidative stress and colonic motility. However the trigger for NPY up regulation during inflammation was unclear.
Figure 6. Modeling TNF-NPY interactions.

Proposed scheme depicting how TNF-NPY cross talk regulates inflammation, epithelial permeability and colonic motility. The possible mechanism of regulation of these effects by TNF-inhibitors is also indicated. The enteric neurons express both the TNF receptors (TNFR1 and TNFR2). XPro1595 inhibits trimerization of soluble TNF and its interaction with TNFR1, whereas etanercept inhibits transmembrane TNF actions through TNFR1 and TNFR2. TNF mediated effects are represented by red arrows and NPY induced effects are shown by purple arrows. TNF induces NPY expression in the ENS, and vice versa. NPY up regulation can have downstream effects of increasing epithelial permeability through phosphatidyl-inositol-3-kinase (PI3-K)-induced claudin-2 up regulation. Inflammation-induced oxidative stress in the enteric neurons leads to enteric neuronal apoptosis and impairments in colonic motility. Both TNF inhibitors attenuate NPY expression, oxidative stress and enteric neuronal apoptosis, thereby enhancing neuronal survival and improving colonic motility.
TNF is a key pro-inflammatory cytokine in IBD and increased expression of TNF has been reported in the colonic biopsies from IBD patients (10). In addition, TNF is the popular drug target in IBD. Hence we investigated if increased TNF levels during inflammation could up regulate NPY expression. In vitro studies demonstrated that TNF induces NPY expression in enteric neuronal cell lines and primary enteric neuronal cultures. Further we validated that TNF-induced NPY expression is highly sensitive to inhibition by TNF inhibitors. Further studies revealed that TNF regulates NPY promoter in enteric neurons via c-jun phosphorylation. This is important as c-jun expression is highly induced in response to neuronal injury and c-jun activation is reported to up regulate the expression of neuropeptides such as vasoactive intestinal peptide (VIP) and NPY in the dorsal root ganglia (36, 37). This finding suggests that NPY in enteric neurons is inducible by TNF during inflammation, and NPY neurons are an immediate sensor of upregulated TNF signaling.
The breach of epithelial barrier is a critical event that precedes inflammation and is implicated in the pathogenesis of IBD (38). A leaky barrier leads to increased exposure of the mucosal immune systems to pro-inflammatory molecules like TNF, and bacterial antigens adding to the severity of inflammation (39). In addition, pro-inflammatory cytokines like TNF can induce barrier dysfunctions via disrupting tight junctions (11). Having established that NPY knockdown inhibits TNF production, we next investigated the relevance of NPY-TNF cross talk in regulating epithelial permeability. This is important because NPY-producing sub mucosal neurons are in close proximity with the epithelium and under normal conditions NPY is an anti-secretory peptide (24, 40, 41). However studies have shown that anti-secretory effects of NPY are subjugated during inflammation as demonstrated (6, 42). We demonstrate that NPY−/− mice had improved barrier functions compared to WT mice, which might have reduced their susceptibility to inflammation as reported by us previously (8). We also observed that NPY increased the permeability of Caco2-BBE cells in vitro as demonstrated by a decrease in TER, which may be the result of alterations in tight junction size or ion selectivity. A further study revealed that like TNF, NPY also up regulates‘claudin-2, a pore-forming tight junction protein, in Caco2-BBE cells in vitro. Thus our data suggest that NPY-induced claudin-2 may be one of the contributing factors to enhanced epithelial permeability, and may involve other tight junction proteins.
TNF can regulate neuronal apoptosis via oxidative stress (43) and is a major cause of autonomic dysfunction during diseases (13). We have previously demonstrated that inflammation damages the enteric neurons resulting in chronic colonic motility impairments in mouse models of colitis (8). In the current study, we demonstrate that TNF-induced oxidative stress results in enteric neuronal apoptosis that contributes to motility defects post inflammation. Accordingly we identified that enteric neurons from mice receiving TNF inhibitors had significantly reduced oxidative stress and neuronal apoptosis leading to enhanced neuronal survival and significant improvements in colonic motility. Further, a comparison between the two TNF inhibitors revealed that selective inhibition of soluble TNF by XPro1595 resulted in a significant reduction of inflammation as demonstrated by reduction in colon length, oxidative stress and histological damage. Studies utilizing mouse models of inflammatory diseases have demonstrated that selective inhibition of soluble TNF is beneficial since sparing of transmembrane TNF preserves the innate immune functions (44). Similar beneficial results in neuronal survival with soluble TNF inhibition have also been demonstrated in the CNS in diseases like Alzheimer’s (45–47).
Despite being a pro-inflammatory cytokine, TNF has been identified to regulate enteric neuronal development in vitro (48). Also, TNF can regulate neuronal apoptosis via oxidative stress (43). Similarly, we and others have shown that NPY has neurotrophic effects (16, 49, 50). However despite their protective roles under normal conditions, we demonstrate that during inflammation TNF up regulation induces NPY, and TNF-NPY cross talk aggravates inflammatory signaling, induces barrier dysfunctions and colonic motility impairments. Our data highlights the significant role of NPY-producing neurons in the ENS as a mediator of the TNF effects. Further, NPY-producing neurons in the ENS are immediate targets and sensors of pro-inflammatory TNF signals. Our results suggest that NPY antagonists might also offer some therapeutic benefits in the treatment of IBD by preventing the propagation of TNF signals. We also suggest that selective inhibition of soluble TNF may be a more efficacious approach compared to non-selective blockade with etanercept or other anti-TNF biological therapies.
Supplementary Material
Figure 1. Enteric neuronal cells express TNF receptors TNFR1 and TNFR2. The enteric neuronal cell line was tested for the presence of TNF receptors R1 and R2 by PCR using respective primers.
Figure 2. NPY increases epithelial permeability via claudin-2. (A) Caco2-BBE or T-84 cells on transwell supports were treated with NPY (0.1 μM) in presence or absence of PI3-K inhibitor (LY-294002), and cell lysates were probed for claudins 1 and 4, or pAKt by western blotting. Scion Image analysis was used to measure band intensities and β-actin was used as loading control. (A) T84 cells treated with NPY showed increased Akt phosphorylation. and (B) claudin- 1 and claudin- 4 were unchanged upon NPY treatment in Caco2-BBE cells. Values are Mean ± SEM, n = 3, * p< 0.05.
Figure 3. NPY and oxidative stress is upregulated in human IBD. Colonic biopsies were obtained from normal and IBD patients. The study was approved by Emory University Institutional Review Board Committee and Beth Israel Deaconess Medical Center (Boston, MA). Pre-operative medical records were reviewed for documented history of IBD. Colonic biopsies were obtained during endoscopy from patients with IBD or disease controls at Beth Israel Deaconess Medical Center, Emory University hospital, Atlanta, GA and the Atlanta VA Medical Center, Decatur, GA. These procedures were performed for polyps or malignancy. RNA was extracted from the biopsies using Qiagen Kit. (A) NPY expression and (B) antioxidant catalase expression was evaluated by real time PCR. Values are Mean ± SEM, n = 10–12, **p < 0.01.
Supplemental Table 1. Design of primers flanking the c-jun binding site for assessing the effects of site-directed mutagenesis. The sequences are represented from 5′-3′ end starting from −769 to −709 bp of the mouse NPY promoter (NM_023456.2).
Acknowledgments
Funding This work is funded by the Crohn’s and Colitis Foundation of America Career Development Award (BC), NIH-RO1 (DK080684, SS), VA-MERIT award (SS) and DDRDC (DK064399, SS), DK06411 (SVS), DK59888 (AN) and DK080058 (EK)
Abbreviations used in this paper
- DSS
dextran sodium sulfate
- ENS
enteric nervous system
- ELISA
enzyme-linked immunosorbent assay
- FD-4
FITC-dextran-4
- LY
LY-294002
- NPY
neuropeptide Y
- nNOS
neuronal nitric oxide synthase
- PD
PD-98059
- TER
transepithelial resistance
- TNF
tumor necrosis factor alpha
Footnotes
Competing Interests None
References
- 1.Gallowitsch-Puerta M, Pavlov VA. Neuro-immune interactions via the cholinergic anti-inflammatory pathway. Life Sci. 2007;80(24–25):2325–9. doi: 10.1016/j.lfs.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gad M, Pedersen AE, Kristensen NN, de Fernandez CF, Claesson MH. Blockage of the neurokinin 1 receptor and capsaicin-induced ablation of the enteric afferent nerves protect SCID mice against T-cell-induced chronic colitis. Inflamm Bowel Dis. 2009;15(8):1174–82. doi: 10.1002/ibd.20902. [DOI] [PubMed] [Google Scholar]
- 3.Margolis KG, Stevanovic K, Karamooz N, et al. Enteric neuronal density contributes to the severity of intestinal inflammation. Gastroenterology. 2011;141(2):588–98. 98 e1–2. doi: 10.1053/j.gastro.2011.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sang Q, Young HM. Chemical coding of neurons in the myenteric plexus and external muscle of the small and large intestine of the mouse. Cell Tissue Res. 1996;284(1):39–53. doi: 10.1007/s004410050565. [DOI] [PubMed] [Google Scholar]
- 5.Wheway J, Herzog H, Mackay F. The Y1 receptor for NPY: a key modulator of the adaptive immune system. Peptides. 2007;28(2):453–8. doi: 10.1016/j.peptides.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 6.Klompus M, Ho W, Sharkey KA, McKay DM. Antisecretory effects of neuropeptide Y in the mouse colon are region-specific and are lost in DSS-induced colitis. Regul Pept. 2010;165(2–3):138–45. doi: 10.1016/j.regpep.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 7.Wheway J, Mackay CR, Newton RA, et al. A fundamental bimodal role for neuropeptide Y1 receptor in the immune system. J Exp Med. 2005;202(11):1527–38. doi: 10.1084/jem.20051971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandrasekharan B, Bala V, Kolachala VL, et al. Targeted deletion of neuropeptide Y (NPY) modulates experimental colitis. PLoS One. 2008;3(10):e3304. doi: 10.1371/journal.pone.0003304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colombel JF, Sandborn WJ, Reinisch W, et al. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N Engl J Med. 2010;362(15):1383–95. doi: 10.1056/NEJMoa0904492. [DOI] [PubMed] [Google Scholar]
- 10.Matsuda R, Koide T, Tokoro C, et al. Quantitive cytokine mRNA expression profiles in the colonic mucosa of patients with steroid naive ulcerative colitis during active and quiescent disease. Inflamm Bowel Dis. 2009;15(3):328–34. doi: 10.1002/ibd.20759. [DOI] [PubMed] [Google Scholar]
- 11.Mankertz J, Amasheh M, Krug SM, et al. TNFalpha up-regulates claudin-2 expression in epithelial HT-29/B6 cells via phosphatidylinositol-3-kinase signaling. Cell Tissue Res. 2009;336(1):67–77. doi: 10.1007/s00441-009-0751-8. [DOI] [PubMed] [Google Scholar]
- 12.Czeschik JC, Hagenacker T, Schafers M, Busselberg D. TNF-alpha differentially modulates ion channels of nociceptive neurons. Neurosci Lett. 2008;434(3):293–8. doi: 10.1016/j.neulet.2008.01.070. [DOI] [PubMed] [Google Scholar]
- 13.Hermann GE, Rogers RC. TNFalpha: a trigger of autonomic dysfunction. Neuroscientist. 2008;14(1):53–67. doi: 10.1177/1073858407305725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rehn M, Hubschle T, Diener M. TNF-alpha hyperpolarizes membrane potential and potentiates the response to nicotinic receptor stimulation in cultured rat myenteric neurones. Acta Physiol Scand. 2004;181(1):13–22. doi: 10.1111/j.1365-201X.2004.01269.x. [DOI] [PubMed] [Google Scholar]
- 15.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2(3):541–6. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 16.Anitha M, Chandrasekharan B, Salgado JR, et al. Glial-derived neurotrophic factor modulates enteric neuronal survival and proliferation through neuropeptide Y. Gastroenterology. 2006;131(4):1164–78. doi: 10.1053/j.gastro.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandrasekharan BP, Kolachala VL, Dalmasso G, et al. Adenosine 2B receptors (A(2B)AR) on enteric neurons regulate murine distal colonic motility. FASEB J. 2009;23(8):2727–34. doi: 10.1096/fj.09-129544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayer CM, Cai F, Cui H, Gillespie JM, MacMillan M, Belsham DD. Analysis of a repressor region in the human neuropeptide Y gene that binds Oct-1 and Pbx-1 in GT1-7 neurons. Biochemical and biophysical research communications. 2003;307(4):847–54. doi: 10.1016/s0006-291x(03)01289-0. [DOI] [PubMed] [Google Scholar]
- 19.Titolo D, Cai F, Belsham DD. Coordinate regulation of neuropeptide Y and agouti-related peptide gene expression by estrogen depends on the ratio of estrogen receptor (ER) alpha to ERbeta in clonal hypothalamic neurons. Mol Endocrinol. 2006;20(9):2080–92. doi: 10.1210/me.2006-0027. [DOI] [PubMed] [Google Scholar]
- 20.Laukoetter MG, Nava P, Lee WY, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204(13):3067–76. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anitha M, Joseph I, Ding X, et al. Characterization of fetal and postnatal enteric neuronal cell lines with improvement in intestinal neural function. Gastroenterology. 2008;134(5):1424–35. doi: 10.1053/j.gastro.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hollander D. Crohn’s disease, TNF-alpha, and the leaky gut. The chicken or the egg? Am J Gastroenterol. 2002;97(8):1867–8. doi: 10.1111/j.1572-0241.2002.05895.x. [DOI] [PubMed] [Google Scholar]
- 23.Fischer A, Gluth M, Pape UF, Wiedenmann B, Theuring F, Baumgart DC. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-alpha on tight junction proteins and signaling pathways in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2013;304(11):G970–9. doi: 10.1152/ajpgi.00183.2012. [DOI] [PubMed] [Google Scholar]
- 24.Saksena S, Tyagi S, Goyal S, et al. Stimulation of apical Cl/HCO(OH) exchanger, SLC26A3 by neuropeptide Y is lipid raft dependent. Am J Physiol Gastrointest Liver Physiol. 2010;299(6):G1334–43. doi: 10.1152/ajpgi.00039.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prasad S, Mingrino R, Kaukinen K, et al. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest. 2005;85(9):1139–62. doi: 10.1038/labinvest.3700316. [DOI] [PubMed] [Google Scholar]
- 26.Franze E, Caruso R, Stolfi C, et al. High expression of the “A Disintegrin And Metalloprotease” 19 (ADAM19), a sheddase for TNF-alpha in the mucosa of patients with inflammatory bowel diseases. Inflamm Bowel Dis. 2013;19(3):501–11. doi: 10.1097/MIB.0b013e31828028e8. [DOI] [PubMed] [Google Scholar]
- 27.Knutson CG, Mangerich A, Zeng Y, et al. Chemical and cytokine features of innate immunity characterize serum and tissue profiles in inflammatory bowel disease. Proc Natl Acad Sci U S A. 2013;110(26):E2332–41. doi: 10.1073/pnas.1222669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krzystek-Korpacka M, Neubauer K, Berdowska I, et al. Enhanced formation of advanced oxidation protein products in IBD. Inflamm Bowel Dis. 2008;14(6):794–802. doi: 10.1002/ibd.20383. [DOI] [PubMed] [Google Scholar]
- 29.Lundberg S, Holst M, Hellstrom PM. Expression of iNOS mRNA associated with suppression of colonic contraction in rat colitis. Acta Physiol (Oxf) 2006;187(4):489–94. doi: 10.1111/j.1748-1716.2006.01576.x. [DOI] [PubMed] [Google Scholar]
- 30.Chandrasekharan B, Anitha M, Blatt R, et al. Colonic motor dysfunction in human diabetes is associated with enteric neuronal loss and increased oxidative stress. Neurogastroenterol Motil. 2011;23(2):131–8. e26. doi: 10.1111/j.1365-2982.2010.01611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gulbransen BD, Bashashati M, Hirota SA, et al. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat Med. 2012;18(4):600–4. doi: 10.1038/nm.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camilleri M, Carlson P, McKinzie S, et al. Genetic susceptibility to inflammation and colonic transit in lower functional gastrointestinal disorders: preliminary analysis. Neurogastroenterol Motil. 2011;23(10):935–e398. doi: 10.1111/j.1365-2982.2011.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al-Jarallah A, Oriowo MA, Khan I. Mechanism of reduced colonic contractility in experimental colitis: role of sarcoplasmic reticulum pump isoform-2. Mol Cell Biochem. 2007;298(1–2):169–78. doi: 10.1007/s11010-006-9363-8. [DOI] [PubMed] [Google Scholar]
- 34.Goyal RK, Hirano I. The enteric nervous system. N Engl J Med. 1996;334(17):1106–15. doi: 10.1056/NEJM199604253341707. [DOI] [PubMed] [Google Scholar]
- 35.Elson CO, Sartor RB, Tennyson GS, Riddell RH. Experimental models of inflammatory bowel disease. Gastroenterology. 1995;109(4):1344–67. doi: 10.1016/0016-5085(95)90599-5. [DOI] [PubMed] [Google Scholar]
- 36.Herdegen T, Leah JD. Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res Brain Res Rev. 1998;28(3):370–490. doi: 10.1016/s0165-0173(98)00018-6. [DOI] [PubMed] [Google Scholar]
- 37.Son SJ, Lee KM, Jeon SM, Park ES, Park KM, Cho HJ. Activation of transcription factor c-jun in dorsal root ganglia induces VIP and NPY upregulation and contributes to the pathogenesis of neuropathic pain. Exp Neurol. 2007;204(1):467–72. doi: 10.1016/j.expneurol.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 38.Gassler N, Rohr C, Schneider A, et al. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol Gastrointest Liver Physiol. 2001;281(1):G216–28. doi: 10.1152/ajpgi.2001.281.1.G216. [DOI] [PubMed] [Google Scholar]
- 39.Weber CR, Raleigh DR, Su L, et al. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. J Biol Chem. 2010;285(16):12037–46. doi: 10.1074/jbc.M109.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cox HM, Cuthbert AW. Neuropeptide Y antagonises secretagogue evoked chloride transport in rat jejunal epithelium. Pflugers Arch. 1988;413(1):38–42. doi: 10.1007/BF00581226. [DOI] [PubMed] [Google Scholar]
- 41.Cox HM, Cuthbert AW, Hakanson R, Wahlestedt C. The effect of neuropeptide Y and peptide YY on electrogenic ion transport in rat intestinal epithelia. J Physiol. 1988;398:65–80. doi: 10.1113/jphysiol.1988.sp017029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klompus M, Ho W, Sharkey KA, McKay DM. Antisecretory effects of neuropeptide Y in the mouse colon are region-specific and are lost in DSS-induced colitis. Regul Pept. 2010 doi: 10.1016/j.regpep.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 43.Talley AK, Dewhurst S, Perry SW, et al. Tumor necrosis factor alpha-induced apoptosis in human neuronal cells: protection by the antioxidant N-acetylcysteine and the genes bcl-2 and crmA. Mol Cell Biol. 1995;15(5):2359–66. doi: 10.1128/mcb.15.5.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zalevsky J, Secher T, Ezhevsky SA, et al. Dominant-negative inhibitors of soluble TNF attenuate experimental arthritis without suppressing innate immunity to infection. J Immunol. 2007;179(3):1872–83. doi: 10.4049/jimmunol.179.3.1872. [DOI] [PubMed] [Google Scholar]
- 45.McAlpine FE, Lee JK, Harms AS, et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009;34(1):163–77. doi: 10.1016/j.nbd.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez TN, Chen X, Bandyopadhyay S, Merrill AH, Tansey MG. Ceramide sphingolipid signaling mediates Tumor Necrosis Factor (TNF)-dependent toxicity via caspase signaling in dopaminergic neurons. Mol Neurodegener. 2012;7:45. doi: 10.1186/1750-1326-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel UL. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. The Journal of biological chemistry. 2004;279(31):32869–81. doi: 10.1074/jbc.M311766200. [DOI] [PubMed] [Google Scholar]
- 48.Gougeon PY, Lourenssen S, Han TY, Nair DG, Ropeleski MJ, Blennerhassett MG. The pro-inflammatory cytokines IL-1beta and TNFalpha are neurotrophic for enteric neurons. J Neurosci. 2013;33(8):3339–51. doi: 10.1523/JNEUROSCI.3564-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agasse F, Bernardino L, Kristiansen H, et al. Neuropeptide Y promotes neurogenesis in murine subventricular zone. Stem Cells. 2008;26(6):1636–45. doi: 10.1634/stemcells.2008-0056. [DOI] [PubMed] [Google Scholar]
- 50.Alvaro AR, Martins J, Araujo IM, Rosmaninho-Salgado J, Ambrosio AF, Cavadas C. Neuropeptide Y stimulates retinal neural cell proliferation--involvement of nitric oxide. J Neurochem. 2008;105(6):2501–10. doi: 10.1111/j.1471-4159.2008.05334.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1. Enteric neuronal cells express TNF receptors TNFR1 and TNFR2. The enteric neuronal cell line was tested for the presence of TNF receptors R1 and R2 by PCR using respective primers.
Figure 2. NPY increases epithelial permeability via claudin-2. (A) Caco2-BBE or T-84 cells on transwell supports were treated with NPY (0.1 μM) in presence or absence of PI3-K inhibitor (LY-294002), and cell lysates were probed for claudins 1 and 4, or pAKt by western blotting. Scion Image analysis was used to measure band intensities and β-actin was used as loading control. (A) T84 cells treated with NPY showed increased Akt phosphorylation. and (B) claudin- 1 and claudin- 4 were unchanged upon NPY treatment in Caco2-BBE cells. Values are Mean ± SEM, n = 3, * p< 0.05.
Figure 3. NPY and oxidative stress is upregulated in human IBD. Colonic biopsies were obtained from normal and IBD patients. The study was approved by Emory University Institutional Review Board Committee and Beth Israel Deaconess Medical Center (Boston, MA). Pre-operative medical records were reviewed for documented history of IBD. Colonic biopsies were obtained during endoscopy from patients with IBD or disease controls at Beth Israel Deaconess Medical Center, Emory University hospital, Atlanta, GA and the Atlanta VA Medical Center, Decatur, GA. These procedures were performed for polyps or malignancy. RNA was extracted from the biopsies using Qiagen Kit. (A) NPY expression and (B) antioxidant catalase expression was evaluated by real time PCR. Values are Mean ± SEM, n = 10–12, **p < 0.01.
Supplemental Table 1. Design of primers flanking the c-jun binding site for assessing the effects of site-directed mutagenesis. The sequences are represented from 5′-3′ end starting from −769 to −709 bp of the mouse NPY promoter (NM_023456.2).