

Abstract
Hypertrophic cardiomyopathy (HCM), characterized by myocardial hypertrophy, is the most common cause of sudden cardiac arrest in young individuals. More than 270 mutations have been found to be responsible for familial HCM to date; mutations in MYH7, which encodes the β-myosin heavy chain (β-MHC) and MYBPC3, which encodes the myosin binding protein C, are seen most often. This study aimed to screen a pathogenic mutation causing HCM in a large family and assess its possible impact on the function of the specific protein. Exome sequencing was applied in the proband for searching a novel mutation; segments bearing the specific mutation were analyzed by polymerase chain reaction and direct sequencing. A novel p.G407C mutation in the β-MHC gene (MYH7) was identified to be responsible for familial HCM in this family. The mutation may cause damage to the second structure of the protein despite the fact that patients bearing the mutation may have a relatively benign prognosis in this family. The clinical details of the p.G407C mutation are described for the first time in this study. Our report shows a good genotype–phenotype consistency and makes it possible for genetic counseling in this family.
Introduction
Hypertrophic cardiomyopathy (HCM), characterized by myocardial hypertrophy, usually of the left ventricle, is the most common cause of sudden cardiac arrest (SCA) in young individuals (Maron et al., 1986). The prevalence of HCM is 1 in 500, and it affects individuals of all ages (Maron et al., 1995, 2003; Maron, 2002).
Familial hypertrophic cardiomyopathy (FHC) is now considered to be an autosomal dominant disease. Although more than 270 mutations have been found to be responsible for HCM to date (Bashyam et al., 2003), mutations in MYH7, which encodes the β-myosin heavy chain and MYBPC, which encodes myosin binding protein C, are seen most often (accounting for 60–70% of FHC cases) (Spirito et al., 1997; Seidman and Seidman, 2001; Xu et al., 2010). Among these pathogenic mutations, some are defined as benign, while others result in severe clinical presentation. A number of mutations in MYH7, located at 14q12, are associated with early onset and high incidence of SCA, while other mutations are benign (Arad, et al., 2002; Richard et al., 2003). These differences are the result of the varying effects of the properties (type, location, and dosage) of the mutations reflected by their different effects on the structure and function of the encoded protein (Rayment et al., 1995; Ho et al., 2000). Even people harboring the same mutation can show different clinical features affected by a variety of environmental and genetic factors.
Whole-exome sequencing developed rapidly in recent years with the development of the next-generation sequencing technology. This high-throughput sequencing technology, which is highly effective to detect common and rare variations, is applied for sequencing the exome (1% of genome) to discover most of the disease-related variations in exons (Metzker 2009; Mamanova et al., 2010). We studied the proband of a Chinese family with FHC by exome sequencing (Ng et al., 2009). After sequencing the whole exome of the proband, we identified a new variation mapped to MYH7 to be responsible for myocardial hypertrophy.
Materials and Methods
Ethics statement
The study was approved by local ethics committees as per the revised Declaration of Helsinki (2004). Patients and members of the control group were recruited in the Xin Hua Hospital and Shanghai Children's Medical Center. Consent was obtained from all the participants being informed of the purposes and risks of this study.
Patients and samples
Twelve individuals (five affected and seven unaffected) were enrolled for clinical and genetic studies from a HCM family (Fig. 1). Echocardiography, electrocardiography, and physical examinations were carried out for all family members by us and local hospitals. FHC diagnosis was mainly based on echocardiography, an interventricular septal diameter of greater than 1.2 cm, electrocardiography, and family history.
FIG. 1.

Pedigree of the familial hypertrophic cardiomyopathy family (I, I, III refer to the first, second, and third generations of the family, respectively). 22 is the proband. 4, 6, 7, 8, 9, 13, 17, 18, 21, 23, 24 are members screened for mutations.
Genomic DNA was isolated from the blood samples of the FHC family members and the control group as previously described (Bashyam et al., 2004).
Exome sequencing and analysis
Genomic DNA of the proband was applied into exome sequencing using the HiSeq 2000 Genome Analyzer (Illumina, Inc.). Standard shot-gun sequencing library is prepared from genomic DNA using SeqCap EZ Oligo pool or an array made against target regions in the genome. The Illumina CASAVA v1.7 pipeline was used to produce 101 bp sequence reads. Paired-end sequences were first mapped against the human reference genome UCSC hg19 (http://genome.ucsc.edu/), and the reads are mapped by BWA (http://bio-bwa.sourceforge.net/). The SNPs and Indels are detected by SAMTOOLS (http://samtools.sourceforge.net/) and compared with common variant databases, including dbSNP135 (http://ncbi.nlm.nih.gov/), and 1000 genomes project database (www.1000genomes.org/).
Mutation detection
Sanger sequencing was used to confirm candidate variants through whole-exome sequencing. We designed the primers of exon 13 of MYH7 (NM_000257) and exon 3 of JPH2 (NM_020433) for polymerase chain reaction amplification. The sequence of the primers and corresponding annealing temperatures can be seen in Table 1. An ABI 3730 genetic analyzer (Applied Biosystems) was used for DNA sequencing (of both strands) of exon 13 of MYH7 and exon 3 of JPH2.
Table 1.
Primer Sequences and Corresponding Annealing Temperatures
| Gene/exon | Forward primer | Reverse primer | Anneal temperature |
|---|---|---|---|
| MYH7/exon13 | TGCCACCTCCACATCCTG | CTACCCTGCCCACCCATT | 66°C |
| JPH2/exon2 | CAAGGGAGGTTGGGAAAG | AGACTCACGGGCATCCAG | 64°C |
Function predictions
ClustalX was used to perform multiple sequence alignments to testify the degree of conservation. In silico predictions were performed using Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/). A pdb file was generated by Swiss-model (http://swissmodel.expasy.org/) using the protein sequence of Uniprot (www.uniprot.org/) with an accession number of P12883. Generation of protein plots was performed using Swiss-PdbViewer 4.1.
Results
Clinical data
Clinical data were collected from 12 family members (Table 2). Symptoms were discovered for the first time in most patients at their twenties, while the proband had an early onset of 14 years exhibiting a relatively severe phenotype (Fig. 2). Fatigue, palpitations, angina, dyspnea, swelling of limbs are symptoms mostly seen among the patients and the severity differed from patient to patient. No sudden death was ever reported in this FHC as far as we know.
Table 2.
Clinical and Molecular Genetic Analysis of Familial Hypertrophic Cardiomyopathy Patients
| Family member | Sex/age(years) | IVS, cm | LVOT, m/s | LVEF, % | Symptoms |
|---|---|---|---|---|---|
| Patient | |||||
| 4 | M/51 | 3.84 | 1.4 | 66 | Fatigue, palpitations, angina, dyspnea, swelling of limbs |
| 7 | M/44 | 3.82 | 1.0 | 60 | Fatigue, palpitations, angina, dyspnea |
| 9 | F/25 | 1.62 | 1.4 | 60 | Fatigue, dyspnea |
| 13 | M/37 | 3.85 | 1.0 | 66 | Fatigue, dyspnea |
| 22 | M/14 | 4.19 | 4.05 | 68 | Fatigue, dyspnea |
| Normal | |||||
| 6 | F/46 | 1.26 | 0.8 | 64 | — |
| 8 | F/43 | 1.18 | 0.8 | 66 | |
| 17 | F/25 | 1.0 | 0.8 | 58 | — |
| 18 | F/23 | 1.2 | 1.0 | 62 | — |
| 21 | F/16 | 0.8 | 0.7 | 64 | — |
| 23 | F/7 | 0.5 | 1.0 | 60 | — |
| 24 | M/6 | 0.4 | 1.0 | 59 | — |
M, male; F, female; IVS, interventricular septum diameter; LVOT, left ventricle out tract; LVEF, left ventricle ejection fraction.
FIG. 2.

Echocardiography images of the proband. Color images available online at www.liebertpub.com/dna
Exome analysis and mutation detection
The number of reads produced in the proband was 78,282,594 according to the whole-exome sequencing experiment. The number of reads mapped to the human genome was 56,599,099 (72.3%, out of total reads) with a Mappable yield of 5,615,031,704 bp. When measured at 1x and 10x coverage, 96.1% and 84.9% of the intended target were covered, respectively. Finally, a total of 10,133 exonic variations were found according to the results of exome sequencing. Then, we filter the data by a minimum depth of 20x and a frequency lower than 5% to get 61 variants (59 nonsynonymous single nucleotide variant [SNV] and 2 stopgain SNV) in 46 genes, of which no one showed any relationship with HCM.
To own an informative analysis of the exome sequencing, we then made a list of genes seen mostly, often resulting in FHC on the basis of OMIM (www.ncbi.nlm.nih.gov/omim), which includes MYH7, ACTC, CSRP3, TNNC1, MYH6, VCL, MYOZ2, JPH2, PLN, CALR2, and NEXN. Finally, two mutations of MYH7 p.G407C and JPH2 p.A396T were detected when we compared all of the aforementioned FHC genes to the results of the exome sequencing of the proband (Fig. 3). One hundred normal DNA samples were sequenced to confirm that JPH2 p.A396T was a polymorphism, while MYH7 p.G407C was not (Table 1). Then, exon 13 of MYH7 was amplified, and direct DNA sequencing was utilized for confirmation using samples from the family. In a word, the proband with the number of 22 in the family pedigree turned out to own a pathogenic mutation of MYH7 p.G407C. Then, family members of 4, 7, 9, and 13 were found to harbor the mutation, while 6, 8, 17, 18, 21, 23, and 24 did not have the mutation. These results are consistent with the ECG and clinical symptoms.
FIG. 3.

(a) Shows mutation of MYH7 p.G407C of the proband; (b) shows the wild type of MYH7. (c) Shows mutation of JPH2 p.A396T of the proband; (d) shows the wild type of JPH2. Color images available online at www.liebertpub.com/dna
Importance of the mutation
The p.G407C mutation corresponds to a one-base substitution in codon 407 (GGG to GTG), resulting in an amino acid change (glycine to cysteine). To make an assessment of the effects of the mutation p.G407C on protein, multiple sequence alignments by ClustalX were used to testify that the position of this mutation is highly conserved among different species (Fig. 4). In addition, the p.G407C mutation is predicted to be probably damaging to the function of the protein with a score of 1.000 by Polyphen-2 and −7.956 by SIFT. The visualization of crystallographic structure of the protein with Swiss-PdbViewer 4.1 gave some clues on how the specific mutation p.G407C might affect the function of the protein (Fig. 5). The amino Gly407 is located in an inverse gamma turn as well as a beta turn, which has no hydrogen bond, so the amino acid change can damage the second structure of the protein. We could also tell the formation of an extra hydrogen bond between the sulfur atom of the mutant Cys407 and the nitrogen atom of Asn408.
FIG. 4.

Alignment of this region of the cardiac MYH7 amino acid sequence from multiple species demonstrating the conservation of the Pro 407.
FIG. 5.
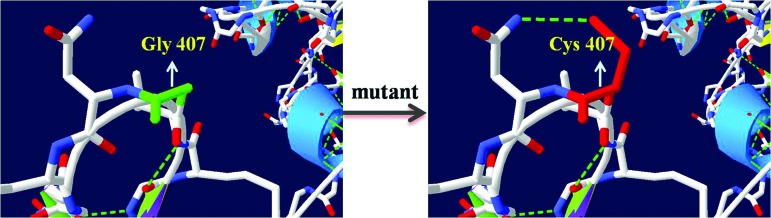
Three-dimensional structures of protein bearing wild type or the mutation of MYH7 p.G407C. Color images available online at www.liebertpub.com/dna
Discussion
Only a 24-year-old patient in Italy harboring the p.G407C mutation has been reported, who suffered from SCA with little detailed clinical or functional analysis provided (Smaniotto et al., 2009). To our knowledge, this is the first study to report the occurrence of the p.G407C mutation in a large family.
Exome sequencing was used for mutation screening in the proband. And after a reasonable analysis of genes responsible for FHC, the p.G407C mutation tended to be the pathogenic mutation in this family. A nonpolar, hydrophobic glycine residue is replaced by a polar, hydrophilic cysteine residue. The wild-type and mutant amino acids differ in size. The glycine residue is the most flexible residue. With the location of an inverse gamma turn as well as a beta turn, it is possible that the residue is needed at this position to make a special backbone conformation or to facilitate movement of the protein (Venselaar et al., 2010). An extra hydrogen bond between the sulfur atom of the mutant Cys407 and the nitrogen atom of Asn408 that can be seen after the transition can be a disruption for the formation of second structure of the protein. In this way, the specific mutation in a highly conserved position may end up damaging the function of the protein of MYH7 and causing myocardial hypertrophy in the whole family.
A genotype–phenotype correlation was also determined in our work. All affected members of the family harbored a heterozygous mutation. The echocardiography of every patient showed an obstruction in the outflow tract. The affected family members shared a number of symptoms such as palpitations, fatigue, and low levels of physical activity to varying degrees (Table 2). A sudden death has not occurred in this family until now, which is different from the previous report in Italy. Although certain mutations of MYH7 have been associated with a significantly shorter lifespan in patients with HCM (Watkins et al., 1992), the fact that most of the affected members were not diagnosed until adulthood shows that the p.G407C mutation tends to be associated with a relatively benign prognosis in the heterozygous state. However, the existence of the 14-year-old proband suggests that early onset of the disease is also possible. This clinical heterogeneity indicates that there are numerous factors, both genetic and environmental that underlie the evolution of FHC caused by the p.G407C mutation.
It is not difficult to find differences in the interventricular septal diameters of family members who harbor the p.G407C mutation (Table 2). Interestingly, the interventricular septal diameter of the female patients was smaller than the male patients, irrespective of age. Although we are not able to make any definite conclusions due to the small number of patients with accurate echocardiography data, this phenomenon is consistent with earlier observations involving other mutations.
Based on this study, genetic counseling can be made available for young people, especially newborns in this family, which will guarantee a better quality of life for them. Furthermore, a population study for the incidence of the p.G407C mutation is currently still needed.
Acknowledgments
The study is supported, in part, by a project supported by the Shanghai Committee of Science and Technology, China (124119a3900), grants from the National Basic Research Program of China (2010CB529500), a project supported by the Shanghai Committee of Science and Technology, China (13JC1401705), the Joint health research program for major diseases in Shanghai (2013ZYJB0016), and the National Natural Science Foundation of China (81070134/30772349).
Disclosure Statement
The authors declare they have no conflicts of interest.
References
- Arad M., Seidman J.G., and Seidman C.E. (2002). Phenotypic diversity in hypertrophic cardiomyopathy. Hum Mol Genet 11,2499–2506 [DOI] [PubMed] [Google Scholar]
- Bashyam M.D., Bashyam L., Savithri G.R., Gopikrishna M., Sangal V., Devi A.R. (2004). Molecular genetic analyses of beta-thalassemia in South India reveals rare mutations in the beta-globin gene. J Hum Genet 49,408–413 [DOI] [PubMed] [Google Scholar]
- Bashyam M.D., Savithri G.R., Kumar M.S., Narasimhan C., and Nallari P. (2003). Molecular genetics of familial hypertrophic cardiomyopathy (FHC). J Hum Genet 48,55–64 [DOI] [PubMed] [Google Scholar]
- Ho C.Y., Lever H.M., DeSanctis R., Farver C.F., Seidman J.G., and Seidman C.E. (2000). Homozygous mutation in cardiac troponin T:Implications for hypertrophic cardiomyopathy. Circulation 102,1950–1955 [DOI] [PubMed] [Google Scholar]
- Mamanova L., Coffey A.J., Scott C.E., Kozarewa I., Turner E.H., et al. (2010). Target-enrichment strategies for next-generation sequencing. Nat Methods 7,111–118 [DOI] [PubMed] [Google Scholar]
- Maron B.J. (2002). Hypertrophic cardiomyopathy: a systematic review. JAMA 287,1308–1320 [DOI] [PubMed] [Google Scholar]
- Maron B.J., Carney K.P., Lever H.M., et al. (2003). Relationship of race to sudden cardiac death in competitive athletes with hypertrophic cardiomyopathy. J Am Coll Cardiol 41,974–980 [DOI] [PubMed] [Google Scholar]
- Maron B.J., Epstein S.E., and Roberts W.C. (1986). Causes of sudden death in competitive athletes. J Am Coll Cardiol 7,204–214 [DOI] [PubMed] [Google Scholar]
- Maron B.J., Gardin J.M., Flack J.M., Gidding S.S., Kurosaki T.T., and Bild D.E. (1995). Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary artery risk development in (Young) adults. Circulation 92,785–789 [DOI] [PubMed] [Google Scholar]
- Metzker M.L. (2009). Sequencing technologies—the next generation. Nat Rev Genet 11,31–46 [DOI] [PubMed] [Google Scholar]
- Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., et al. (2009). Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42,30–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment I., Holden H.M., Sellers J.R., Fananapazir L., and Epstein N.D. (1995). Structural interpretation of the mutations in the beta-cardiac myosin that have been implicated in familial hypertrophic cardiomyopathy. Proc Natl Acad Sci U S A 92,3864–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P., Charron P., Carrier L., Ledeuil C., Cheav T., Pichereau C., Benaiche A., Isnard R., Dubourg O., Burban M., Gueffet J.P., Millaire A., Desnos M., Schwartz K., Hainque B., and Komajda M. (2003). Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107,2227–2232 [DOI] [PubMed] [Google Scholar]
- Seidman J.G., and Seidman C. (2001). The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 104,557–567 [DOI] [PubMed] [Google Scholar]
- Smaniotto G., Melacini P., Calore C., et al. (2009). Screening of the four genes most commonly involved in hypertrophic cardiomyopathy. Eur Heart J 30 (Abstract Suppl), 543 [Google Scholar]
- Spirito P., Seidman C.E., McKenna W.J., and Maron B.J. (1997). The management of hypertrophic cardiomyopathy. N Engl J Med 336,775–785 [DOI] [PubMed] [Google Scholar]
- Venselaar H., Te Beek T.A., Kuipers R.K., Hekkelman M.L., and Vriend G. (2010). Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 11, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins H., Rosenzweig A., Hwang D.S., Levi T., McKenna W., et al. (1992). Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med 326,1108–1114 [DOI] [PubMed] [Google Scholar]
- Xu Q., Dewey S., Nguyen S., and Gomes A.V. (2010). Malignant and benign mutations in familial cardiomyopathies: insights into mutations linked to complex cardiovascular phenotypes. J Mol Cell Cardiol 48,899–909 [DOI] [PubMed] [Google Scholar]