

Abstract
An artificial nucleic acid analogue capable of self-assembly into duplex merely through hydrophobic interactions is presented. The replacement of Watson-Crick hydrogen bonding with strictly hydrophobic interactions has the potential to confer new properties and facilitate the construction of complex DNA nanodevices. To study how the hydrophobic effect works during the self-assembly of nucleic acid bases, we have designed and synthesized a series of fluorinated nucleic acids (FNA) containing 3,5-bis(trifluoromethyl) benzene (F) and nucleic acids incorporating 3,5-dimethylbenzene (M) as hydrophobic base surrogates. Our experiments illustrate that two single-stranded nucleic acid oligomers could spontaneously organize into a duplex entirely by hydrophobic base pairing if the bases were size-complementary and the intermolecular forces were sufficiently strong.
Introduction
Synthetic oligonucleotides, which are programmable biomaterials with recognition and self-assembly properties, have been widely used in medicine,1 biotechnology2 and nanotechnology.3 Assembly of such building blocks creates well-defined structures with applications in highly sensitive and selective sensors or detectors,4 nano-scale electronics,5 and nanomachines.6 The replacement of Watson-Crick hydrogen bonding with strictly hydrophobic interactions confers new properties and facilitates the construction of complex DNA nano-devices, expanding the repertoire of DNA nanotechnology beyond the boundaries of Watson-Crick base pairing.7 Previous studies on DNA structural modifications have revealed that two strands of artificial nucleic acids can be assembled through metal-mediated bonding,8 as well as non-Watson-Crick base pair hydrogen bonding.9
Hydrophobic effects play a dominant role in assembly processes, such as protein folding10 and the construction of liposomes11 and micelles.12 Lipid-oligonucleotide conjugates have also been used in DNA nanotechnology to produce nanostructures with unique properties.13 Hydrophobic “bases” have been extensively investigated in the past two decades.14 It has been demonstrated that hydrogen bonds are not required for base-pair stabilization, since the incorporation of hydrophobic base pairs can also stabilize the duplex if they are size-complementary and provide sufficient π stacking.15 However, hydrogen bonding is not only a factor stabilizing the duplex; it is also the major force driving two complementary strands together. Therefore, despite the existing work, it is not known whether a duplex structure could be constructed using only hydrophobic base pairs. Although interactions between hydrophobic base pairs are less studied,16 they may play a role similar to that of hydrogen bonding between A-T and C-G base pairs during specific recognition and self-assembly.
Therefore, we hypothesized that two single-stranded nucleic acid oligomers could spontaneously organize into a duplex entirely by hydrophobic base pairing, but only if: 1) the hydrophobic bases were complementary in terms of size; and 2) the intermolecular forces were sufficiently strong. Nucleic acids capable of assembly into a duplex merely through hydrophobic interactions would provide a class of unique biomaterials with important applications in biotechnology, essentially because such assembly would not only be orthogonal to the assembly through hydrogen bonding or metal-mediated bonding, but also inert to pH, cation type, and temperature. This new addition to the repertoire of DNA nanotechnology would have unique applications, including, for example, in molecular scale electronics or nanomedicine.
To study how the hydrophobic effect works during the self-assembly of nucleic acid bases, we have designed and synthesized a series of trifluoromethylated nucleic acids (FNA) containing 3,5-bis(trifluoromethyl) benzene (artificial base F), as hydrophobic bases, and nucleic acids incorporating 3,5-dimethylbenzene (artificial base M), as a control. Artificial bases F and M have similar π-systems; thus, they will provide equivalent π-stacking interactions for stabilization of the duplex structure. However, the van der Waals radius of the methyl group is 2.0 Å, while that of trifluoromethyl is 2.2 Å, or more;17 hence, the hydrophobic interactions between F-F and M-M base pairs are quite different. As a result, the comparison of FNA and nucleic acids containing M will directly illuminate the hydrophobic effects in the self-assembly process.
Results and Discussion
As seen from the molecular model, two 3,5-bis(trifluoromethyl) benzenes are structurally self-complementary, and their aggregation forms a homogeneous hydrophobic phase bridging the two strands (Figure 1). Although 3,5-dimethylbenzene is self-complementary, the hydrophobic phase formed is less continuous and smaller in volume, indicating that the intermolecular base-pair force between 3,5-bis(trifluoromethyl)benzenes is stronger than that of 3,5-dimethylbenzenes.
Figure 1.

Chemical structure, molecular model and the Connolly surface models of hydrophobic base pairs.
When oligonucleotides are incorporated with F as the base, the resulting FNA strands can orthogonally recognize each other and assemble into a duplex with the hydrophobic phase in the central zone. The duplex structure of FNAs may be quite different from the helix duplex of natural DNAs. Our preliminary simulation results showed that the duplex assembled through hydrophobic effects could be linear instead of helical (Figure 2; Ball-and-Stick model of FNA is also illustrated in Figure S3).
Figure 2.

Linear Duplex model of FNA with hydrophobic phase formed in the central zone.
Details of the synthesis of diol 1 are described in the experimental section. Briefly, commercially available (S)-3-amino-1,2-propanediol was benzoylated to afford compound 1, and the corresponding phosphoramidite 4 (Scheme 1) was prepared for DNA solid phase synthesis by standard methods 18. Following the protocols, phosphoramidites 5 and 6 (see Supporting Information) were also synthesized to prepare corresponding oligonucleotides for comparison. All the phosphoramidites were synthesized from inexpensive commercially available materials in three steps in 52%-57% yields, demonstrating that the simplicity of their preparation would easily meet the requirement of industrial production.
Scheme 1.
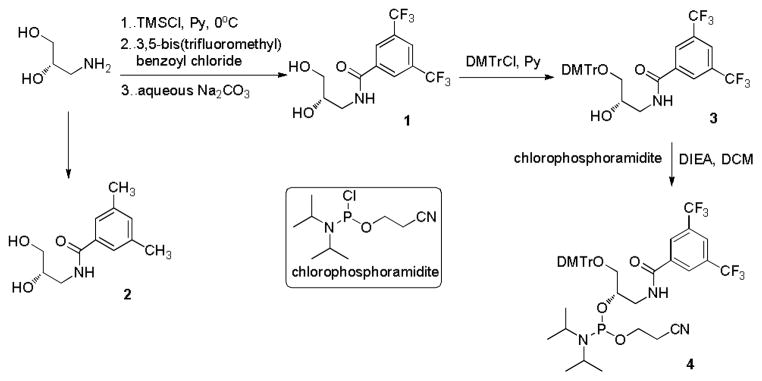
Synthesis of compounds 1, 2 and 4.
The thermal melting properties of the unnatural base pairs were evaluated by determining the melting temperature (Tm) of duplexes containing F in the center (Table 1). The F-F-containing duplex (entry 3) is significantly less stable than the duplex containing A-T (entry 1) or C-G (entry 2) base pairs, but still more stable than the corresponding duplexes containing mismatched base pairs F-A, F-G, F-C, and F-T. We also investigated duplexes with 2 or 3 consecutive F bases in each strand (see Supporting Information), and we found a negative correlation between increasing incorporation of F units and thermal stability of the duplex. When two complementary strands were modified with three F bases each, they could not form a stable duplex.
Table 1.
Melting temperature (Tm) for DNA duplexes containing F base.a
| Entry | 5′-d(GCGTACXCATGCG) 3′-d(CGCATGYGTACGC) |
|
|---|---|---|
| X-Y | Tm (°C) | |
| 1 | A-T | 61.5 |
| 2 | C-G | 62.4 |
| 3 | F-F | 51.5 |
| 4 | F-A | 50.1 |
| 5 | F-G | 48.8 |
| 6 | F-C | 47.2 |
| 7 | F-T | 49.1 |
| 8 | A-F | 50.4 |
| 9 | G-F | 45.7 |
Conditions: 1 μM DNA, 100 mM NaCl, 10 mM MgCl2, 10 mM PIPES, pH 7.0. The heating rates were 0.5 °C/min.
The thermal melting results may indicate that the base-stacking interactions of F-F base pairs are orthogonal to those of natural A-T and C-G base pairs; accordingly, the incorporation of such artificial base pairs does not cumulatively contribute to the thermal stability of the duplexes.19 However, two strands of nucleic acids modified with successive F bases may be capable of self-assembly into a duplex merely through hydrophobic F-F base pairing if the number of F-F pairs is sufficient.
We performed calculations of binding free energies of the two strands for FNAs with 4, 6 and 8 F-F base pairs (BP) based on the trajectories obtained from molecular dynamics (MD) simulation using the popular molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA)20 and molecular mechanics/generalized Born surface area (MM/GBSA) methods.21 According to the results, the duplex structure of 6 BP is more stable than that of 4 BP, but less stable than that of 8 BP (Table 2). The results also indicate that the base-stacking between F-F is a positive cumulative interaction.
Table 2.
Binding free energies of FNAs (kcal/mol)
| 4 BP FNA | 6 BP FNA | 8 BP FNA | |
|---|---|---|---|
| MM/PBSA | −11.75±2.31 | −18.35±2.26 | −21.58±3.45 |
| MM/GBSA | −12.61±2.05 | −20.27±2.01 | −27.64±2.89 |
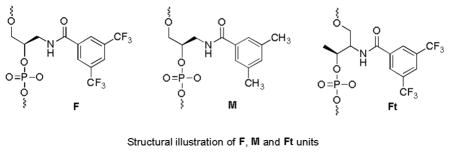
Based on the calculation results, we prepared FNA F4 and F6 to verify that a duplex structure can be constructed merely through F-F base pair interactions (Table 3).
Table 3.
Sequence information of FNAs
| FNA | Sequence |
|---|---|
| F6 | 5′ FAM-FFF FFF TCT AAA TCA CTA TGG TCG C FFF FFF-Dabcyl 3′ |
| F4 | 5′ FAM-FFFF TCT AAA TCA CTA TGG TCG C FFFF-Dabcyl 3′ |
| FM6 | 5′ FAM-FFM MFM TCT AAA TCA CTA TGG TCG C FMF FMM-Dabcyl 3′ |
| M6 | 5′ FAM-MMM MMM TCT AAA TCA CTA TGG TCG C MMM MMM-Dabcyl 3′ |
| Ft6 | 5′ FAM-FtFtFt FtFtFt TCT AAA TCA CTA TGG TCG C FtFtFt FtFtFt-Dabcyl 3′ |
| Ft8 | 5′ FAM-FtFtFt FtFtFt FtFt TCT AAA TCA CTA TGG TCG C FtFtFt FtFtFt FtFt-Dabcyl 3′ |
If hydrophobic bases spontaneously aggregate and form an intramolecular duplex, this self-assembly, through hydrophobic interactions, would place fluorescein (FAM) at the 5′-end and the Dabcyl quencher on the 3′-end in close proximity, thereby yielding a weak fluorescence signal. However, the addition of complementary DNA (cDNA) of the loop to the buffer solution will result in an open state with a fully extended structure (see Scheme 2, using F6 to demonstrate a typical procedure). Consequently, the fluorescence intensity will increase dramatically, and the self-assembly property of FNA can be characterized by fluorescence variations.
Scheme 2.

Hairpin-structured F6 and its hybridization with cDNA, which results in the open state.
Fluorescence variation induced by cDNA is a direct way to determine the assembly properties of FNAs with hairpin structure.22 To determine the importance of hydrophobic interactions in self-assembly of the duplex, we also synthesized nucleic acid analogues M6 and FM6 (Table 3) modified with artificial base M for comparison and studied their fluorescence variation.
Figure 3 shows the signal enhancement after hybridization of M6 (Figure 3a), FM6 (Figure 3b) and F6 (Figure 3c) with cDNA in 20 mM Tris. The fluorescence intensities of M6 and FM6 (background) are quite high, and their signal-to-background (S/B) ratios are less than 2, indicating that the duplex structure may be very unstable. In comparison with M6 and FM6, the S/B ratios of F6 are approximately 9-fold, which is comparable to the S/B ratio of natural base pairs.23 These results illustrate that hydrophobic interaction between base pairs is critical in the self-assembly of such nucleic acid analogues. The fluorescence variation result showed that 4 base-pair nucleic acid analogue F4 does not provide a thermodynamically stable duplex. The observations for F6 and F4 are consistent with our MD simulation. Our gel experiment also confirmed the hairpin structure of F6, in which 31mer F6 migrated faster than single-stranded 27mer F4 (Figure S2).
Figure 3.

S/B fluorescence of M6 (a), FM6 (b), F6 (c) and F4 (d) in 20 mM Tris buffer. Final concentration ratio of FNA:cDNA=1:10.
The influence of the backbone on the stability of nucleic acids is complicated. Previous studies showed that threose nucleic acid (TNA)24 and glycol nucleic acid (GNA)25 can hybridize with DNA and RNA to form stable duplex structures. To study the effect of the backbone on the stability of FNA duplex, we synthesized Ft6 and Ft8 (Table 3) using phosphoramidite 6. FNA Ft6 and Ft8 are composed of threoninol, instead of glycidol, as the backbone, the repeat unit of which is one atom longer than that of F6 in the stem region. The S/B ratio of Ft6 in Tris buffer ranges from 3- to 4-fold higher, which indicates that the 6 base-pair duplex of Ft6 is less stable than that of F6. When base pairs are increased to 8, a more stable duplex is formed in Ft8 (Figure S1). This phenomenon is consistent with the simulation results.
The closed and opened states of FNA can also be characterized by 19F NMR. In the closed state, CF3 groups are aggregated into a hydrophobic phase, but they are dispersed into the aqueous phase when cDNA is added to open the hydrophobic duplex. 19F NMR signals of CF3 are quite different from each other in these two environments.
The 19F spectrum of F6 in Tris buffer (Figure 4a) shows three peaks at δ −58.81 ppm, −59.34 ppm and −59.57ppm. We assume that the signal at −58.81 ppm, as a sharp peak, could originate from the CF3 group in the aqueous phase. The signal at −59.34 ppm, which is partially merged with the signal at −59.57ppm, could arise from the CF3 group between the aqueous and hydrophobic phases. The strongest signal at −59.57 ppm belongs to the CF3 group in the hydrophobic phase. When cDNA was added to the solution of F6 (Figure 4b and 4c), the signals at −59.57 ppm shifted to the position at −58.81 ppm, indicating that hairpin-structured F6 was eventually opened and that the assembled hydrophobic phase was dispersed.
Figure 4.

19F NMR spectrum of F6 (a), F6 /cDNA (3:1) (b) and F6 /cDNA (1:3) (c) in 20 mM Tris buffer.
The thermodynamic behavior of the FNA duplex was studied by monitoring the fluorescence variation of F6. When the temperature was increased from 10°C to 95°C, no obvious change in fluorescence was observed. Nor were differences in 19F NMR spectra observed between samples run at 25°C and 65°C. Quantitative calorimetric data are needed to determine whether the self-assembly process is governed by the classical vs non-classical hydrophobic effect.26
Initial experiments of FNA in a living system showed that incorporation of artificial base F increases the biostability, cellular binding and internalization of nucleic acids,27 factors which are important for biomedical applications, such as DNA-based therapy.28
Conclusions
To study how the hydrophobic effect works during the self-assembly of nucleic acid bases and to determine if a duplex structure could be constructed using only hydrophobic base pairs, we had developed three phosphoramidite reagents for incorporation of hydrophobic base surrogates F and M into nucleic acids. Based on our preliminary simulation results, a series of trifluoromethylated nucleic acids (FNA) containing 3,5-bis(trifluoromethyl) benzene (F) and 3,5-dimethylbenzene (M) were synthesized and characterized by fluorescence and NMR studies. The thermal melting results may indicate that the base-stacking interactions of F-F base pairs are orthogonal to those of natural A-T and C-G base pairs. Our experiments illustrate that two single-stranded nucleic acid oligomers could spontaneously organize into a duplex by hydrophobicity if the bases were size-complementary and the intermolecular forces were sufficiently strong. This type of biomaterial may have unique applications in biological probes, such as molecular beacons, and in nanotechnology.
Experimental Section
All DNA synthesis reagents were purchased from Glen Research. FNA F6, F4, FM6 and M6 were synthesized and purified by Sangon Biotech (Shanghai). FNA Ft6, Ft8 and other oligonucleotides were synthesized on an ABI 3400 synthesizer (Applied Biosystems). Dabcyl CPG was used for all FAM-labeled FNA. The completed sequences were then deprotected in AMA (ammonium hydroxide/40% aqueous methylamine, 1:1) at 65 °C for 30 min and further purified by reversed-phase HPLC (ProStar; Varian) on a C-18 column using 0.1 M triethylamine acetate(TEAA) buffer (Glen Research) and acetonitrile (SigmaAldrich) as the eluents. The collected DNA products were dried and detritylated by dissolving and incubating DNA products in 200 μL of 80% acetic acid for 20 min. The detritylated DNA product was precipitated with NaCl (3 M, 25 μL) and ethanol (600 μL).
Unless otherwise noted below, all commercially available reagents and solvents were purchased from Sigma Aldrich and used without further purification. 1H NMR (TMS as the internal standard) and 19F NMR spectra (CFCl3 as the outside standard and low field positive) were recorded on a Bruker AM300 or Bruker AM400 spectrometer. 13C NMR was recorded on a Bruker AM400 spectrometer. Chemical shifts (δ) are reported in ppm, and coupling constants (J) are in Hertz (Hz).
Synthesis of compound 1
To a solution of (S)-3-amino-1,2-propanediol (1.01g, 11 mmol) in anhydrous pyridine (55 mL) was added chlorotrimethylsilane (5.97 g, 55 mmol) dropwise at 0°C. After 30 min, 3,5-bis(trifluoromethyl)benzoyl chloride (3.34 g, 12 mmol) was added at 0°C, and the mixture was allowed to warm to RT and was stirred for an additional 2 h. Then a saturated NaHCO3 solution was added to terminate the reaction. The resulting reaction mixture was concentrated in vacuo and subjected to a flash silica gel column (CH2Cl2/MeOH, 15:1). A white solid (S)-N-(2,3-dihydroxypropyl)-3,5-bis(trifluoromethyl)benzamide 1 (3.20 g, 88 % yield) was obtained after flash chromatography: 1H NMR (400 MHz, acetone-d6) δ 8.51 (s, 2H), 8.41 (s, 1H), 8.20 (s, 1H), 4.12 (br, 2H), 3.82–3.88 (m, 1H), 3.60–3.66 (m, 1H), 3.47–3.54 (m, 3H); 13C NMR (100 MHz, acetone-d6) δ 164.65, 137.10, 131.31 (q, JC-F= 35.8 Hz), 127.98, 124.59, 121.97, 70.77, 63.84, 43.22; 19F NMR (282 MHz, acetone-d6) δ −63.43; MS (ESI-): m/z 330.0578 (Calculated M-H: 330.0570).
Synthesis of compound 3
To a solution of compound 1 (1.66g, 5 mmol) in anhydrous pyridine (50 mL) was added DMTrCl (1.86 g, 5.5 mmol), and the reaction was stirred overnight. The reaction mixture was concentrated in vacuo, and the residue was subjected to a flash silica gel column (ethyl acetate/hexane, 1:3). Compound 3 (2.25 g, 71 % yield) was obtained as a white solid after flash chromatography: 1H NMR (300 MHz, acetone-d6) δ 8.47 (s, 2H), 8.27 (s, 1H), 8.22 (s, 1H), 7.49 (d, J=5.7 Hz, 2H), 7.36 (d, J=6.6 Hz, 4H), 7.27 (t, J=5.7 Hz, 2H), 7.18 (t, J=5.4 Hz, 1H), 6.83 (d, J=6.6 Hz, 4H),4.38 (d, J=3.6 Hz, 1H), 4.05–4.72 (m, 1H), 3.72–3.78 (m, 7H), 3.46–3.51 (m, 1H), 3.10–3.20 (m, 2H); 13C NMR (100 MHz, acetone-d6) δ 164.21, 158.63, 145.38, 137.26, 136.12, 131.30 (q, JC-F = 33.7 Hz), 130.05, 128.12, 127.96, 127.59, 126.54, 124.71, 124.52, 122.00, 112.88, 85.86, 69.49, 65.77, 54.53, 44.02; 19F NMR (282 MHz, acetone-d6) δ −62.68; MS (ESI+): m/z 656.1825 (Calculated M+Na: 656.1842).
Synthesis of phosphoramidite 4
To a solution of compound 3 (2.11g, 3.33 mmol) in anhydrous DCM (35 mL) was added DIEA, followed by chlorophosphoramidite (920 mg, 3.90 mmol) at 0°C. The mixture was allowed to warm to RT and stirred for 1 h. Then the reaction mixture was diluted with 50 mL of DCM and washed with saturated NaHCO3 solution and saturated saline solution. The organic phase was dried over Na2SO4 and then concentrated in vacuo. A white solid foam 4 (2.31 g, 85 % yield) was obtained as a mixture of diastereomers after flash chromatography: 1H NMR (300 MHz, acetone-d6) δ 8.43 (s, 1H), 8.41 (s, 1H), 8.23 (s, 1H), 8.17 (s, 1H), 7.51 (d, J=6.0 Hz, 2H), 7.37 (d, J=6.6 Hz, 4H), 7.26–7.30 (m, 2H), 7.187.21 (m, 1H), 6.83–6.87 (m, 4H),4.29–4.35 (m, 1H), 3.62–3.93 (m, 12H), 3.17–3.34 (m, 2H), 2.70–2.74 (m, 1H), 2.60–2.63 (m, 1H), 1.10–1.25 (m, 12H); 19F NMR (282 MHz, acetone-d6) δ: −63.33, −63.35; 31P NMR( acetone-d6) δ: 148.79, 148.61.
Supplementary Material
Acknowledgments
We acknowledge the National Basic Research Program of China (2012CB21600) for support. This work is supported by grants awarded by the National Key Scientific Program of China (2011CB911000), the Foundation for Innovative Research Groups of NSFC (Grant 21221003), the China National Instrumentation Program (2011YQ03012412) and by the National Institutes of Health (GM079359 and CA133086). We thank Prof. Barry Gold at the University of Pittsburgh for helpful discussion.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
Footnotes should appear here. These might include comments relevant to but not central to the matter under discussion, limited experimental and spectral data, and crystallographic data.
Contributor Information
Feng-Ling Qing, Email: flq@mail.sioc.ac.cn.
Weihong Tan, Email: tan@chem.ufl.edu.
Notes and references
- 1.(a) Merki E, Graham MJ, Mullick AE, Miller ER, Crooke RM, Pitas RE, Witztum JL, Tsimikas SA. Circulation. 2008;118:743–753. doi: 10.1161/CIRCULATIONAHA.108.786822. [DOI] [PubMed] [Google Scholar]; (b) Bennett FC, Swayze EE. Annu Rev Pharmacol Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]; (c) Eschgfaller B, Konig M, Boess F, Boelsterli UA, Benner SA. J Med Chem. 1998;41:276–283. doi: 10.1021/jm970538o. [DOI] [PubMed] [Google Scholar]; (d) Ni X, Castanares M, Mukherjee A, Lupold SE. Curr Med Chem. 2011;18:4206–4214. doi: 10.2174/092986711797189600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Shangguan D, Li Y, Tang Z, Cao ZC, Chen HW, Mallikaratchy P, Sefah K, Yang CJ, Tan W. PNAS. 2006;103:11838–11843. doi: 10.1073/pnas.0602615103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schena M, Shalon D, Davis RW, Brown PO. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]; (c) Tyaqi S, Kramer FR. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]; (d) You M, Chen Y, Peng L, Han D, Yin B, Ye B, Tan W. Chem Sci. 2011;2:1003–1010. [Google Scholar]
- 3.(a) Seeman NC. Nature. 2003;421:427–431. doi: 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]; (b) Lin C, Liu Y, Rinker S, Yan H. ChemPhysChem. 2006;7:1641–1647. doi: 10.1002/cphc.200600260. [DOI] [PubMed] [Google Scholar]; (c) Storhoff JJ, Mirkin CA. Chem Rev. 1999;99:1849–1862. doi: 10.1021/cr970071p. [DOI] [PubMed] [Google Scholar]; (d) Mclaughlin CK, Hamblin GD, Sleiman HF. Chem Soc Rev. 2011;40:5647–5656. doi: 10.1039/c1cs15253j. [DOI] [PubMed] [Google Scholar]; (e) Benhaj AB, Smith BD, Liu J. Chem Sci. 2012;3:3216–3220. [Google Scholar]
- 4.(a) Xiang Y, Lu Y. Nature Chem. 2011;3:697–703. doi: 10.1038/nchem.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ma DL, He HZ, Chan DSH, Leung CH. Chem Sci. 2013;4:3366–3380. [Google Scholar]
- 5.Park S-J, Taton TA, Mirkin CA. Science. 2002;295:1503–1506. doi: 10.1126/science.1067003. [DOI] [PubMed] [Google Scholar]
- 6.Han D, Pal S, Nangreave J, Deng Z, Liu Y, Yan H. Science. 2011;332:342–346. doi: 10.1126/science.1202998. [DOI] [PubMed] [Google Scholar]
- 7.(a) Tolle F, Mayer G. Chem Sci. 2013;4:60–67. [Google Scholar]; (b) Voth AR, Khuu P, Ho KO. Nature Chem. 2009;1:74–79. doi: 10.1038/nchem.112. [DOI] [PubMed] [Google Scholar]
- 8.(a) Tanaka K, Tengeiji A, Kato T, Toyama N, Shionoya M. Science. 2003;299:1212–1213. doi: 10.1126/science.1080587. [DOI] [PubMed] [Google Scholar]; (b) Schlegel MK, Zhang L, Pagano N, Meggers E. Org Bio Chem. 2009;7:476–482. doi: 10.1039/b816142a. [DOI] [PubMed] [Google Scholar]; (c) Johannsen S, Megger N, Bohme D, Sigel RO, Muller J. Nature Chem. 2010;2:229–234. doi: 10.1038/nchem.512. [DOI] [PubMed] [Google Scholar]
- 9.(a) Piccirilli JA, Krauch T, Moroney SE, Benner SA. Nature. 1990;343:33–37. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]; (b) Liu H, Gao J, Lynch S, Saito D, Maynard L, Kool ET. Science. 2003;302:868–871. doi: 10.1126/science.1088334. [DOI] [PubMed] [Google Scholar]; (c) Lu H, He K, Kool ET. Angew Chem Int Ed. 2004;43:5834–583. doi: 10.1002/anie.200461036. [DOI] [PubMed] [Google Scholar]
- 10.Lins L, Brasseur R. FASEB J. 1995;9:535–540. doi: 10.1096/fasebj.9.7.7737462. [DOI] [PubMed] [Google Scholar]
- 11.Antonietti M, Forster S. Adv Mater. 2003;15:1323–1333. [Google Scholar]
- 12.Israelachvili JN, Mitchell DJ, Ninham BW. J Chem Soc, Faraday Trans 2. 1976;72:1525–1568. [Google Scholar]
- 13.(a) Edwardson TGW, Carneiro KMM, Mclaughlin CK, Serpell CJ, Sleiman HF. Nature Chem. 2013;5:868–875. doi: 10.1038/nchem.1745. [DOI] [PubMed] [Google Scholar]; (b) Wu Y, Kwame S, Liu H, Wang R, Tan W. PNAS. 2010;107:5–10. doi: 10.1073/pnas.0909611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For representative researches, see Schweltzer BA, Kool ET. J Am Chem Soc. 1995;117:1863–1872. doi: 10.1021/ja00112a001.McMinn DL, Ogawa AK, Wu Y, Liu J, Schultz PG, Romesberg FE. J Am Chem Soc. 1999;121:11585–11586.Berger M, Ogawa AK, McMinn DL, Wu Y, Schultz PG, Romesberg FE. Angew Chem Int Ed. 2000;39:2940–2943. doi: 10.1002/1521-3773(20000818)39:16<2940::aid-anie2940>3.0.co;2-#.Matray TJ, Kool ET. Nature. 1999;399:704–708. doi: 10.1038/21453.Ogawa AK, Wu Y, Berger M, Schultz PG, Romesberg FE. J Am Chem Soc. 2000;122:8803–8804.Lai JS, Qu J, Kool ET. Angew Chem Int Ed. 2003;42:5973–5977. doi: 10.1002/anie.200352531.Lai JS, Kool ET. J Am Chem Soc. 2004;126:3040–3041. doi: 10.1021/ja039571s.Kimoto M, Yamashige R, Matsunaga KI, Yokoyama S, Hirao I. Nat Biotech. 2013;31:453–457. doi: 10.1038/nbt.2556.For reviews, see Kool ET, Morales J, Guckian KM. Angew Chem Int Ed. 2000;39:990–1009. doi: 10.1002/(sici)1521-3773(20000317)39:6<990::aid-anie990>3.0.co;2-0.Henry AA, Romesberg FE. Curr Opin Chem Biol. 2003;7:727–733. doi: 10.1016/j.cbpa.2003.10.011.Kool ET. Acc Chem Res. 2002;35:936–943. doi: 10.1021/ar000183u.Wojciechowski F, Leumann CJ. Chem Soc Rev. 2011;40:5669–5679. doi: 10.1039/c1cs15027h.
- 15.(a) Matray TJ, Kool ET. J Am Chem Soc. 1998;120:6191–6192. doi: 10.1021/ja9803310. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu Y, Ogawa AK, Berger M, McMinn DL, Schultz PG, Romesberg FE. J Am Chem Soc. 2000;122:7621–7632. [Google Scholar]
- 16.Kaufmann M, Gisler M, Leumann C. Angew Chem Int Ed. 2009;48:3810–3813. doi: 10.1002/anie.200900153. [DOI] [PubMed] [Google Scholar]
- 17.Terazono Y, Dolphin D. J Org Chem. 2003;68:1892–1900. doi: 10.1021/jo020562w. [DOI] [PubMed] [Google Scholar]
- 18.Wang R-W, Gold B. Org Lett. 2009;11:2465–2468. doi: 10.1021/ol9007537. [DOI] [PubMed] [Google Scholar]
- 19.Yakovchuk P, Protozanova E, Frank-Kamenetski MD. Nucleic Acids Res. 2006;34:564–574. doi: 10.1093/nar/gkj454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 21.Gohlke H, Kiel C, Case DA. J Mol Biol. 2003;330:891–913. doi: 10.1016/s0022-2836(03)00610-7. [DOI] [PubMed] [Google Scholar]
- 22.Tan W, Wang K, Drake TJ. Curr Opin Chem Biol. 2004;8:547–553. doi: 10.1016/j.cbpa.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 23.Yang CJ, Medley CD, Tan W. Curr Pharm Biotechnol. 2005;6:445–452. doi: 10.2174/138920105775159322. [DOI] [PubMed] [Google Scholar]
- 24.Schoning K-U, Scholz P, Guntha S, Wu X, Krishnamurthy R, Eschenmoser A. Science. 2000;290:1347–1351. doi: 10.1126/science.290.5495.1347. [DOI] [PubMed] [Google Scholar]
- 25.Zhang L, Peritz A, Meggers E. J Am Chem Soc. 2005;127:4174–4175. doi: 10.1021/ja042564z. [DOI] [PubMed] [Google Scholar]
- 26.Meyer EA, Castellano RK, Diederich F. Angew Chem Int Ed. 2003;42:1210–1250. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- 27.Preliminary results, to be published.
- 28.(a) Li W, Szoka FC., Jr Pharm Res. 2007;24:438–449. doi: 10.1007/s11095-006-9180-5. [DOI] [PubMed] [Google Scholar]; (b) Guo X, Huang L. Acc Chem Res. 2012;45:971–979. doi: 10.1021/ar200151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.