

Abstract
Upon glucose elevation, pancreatic beta-cells secrete insulin in a Ca2+-dependent manner. In diabetic animal models, different aspects of the calcium signaling pathway in beta-cells are altered, but there is no consensus regarding their relative contributions to the development of beta-cell dysfunction. In this study, we compared the increase in cytosolic Ca2+ ([Ca2+]i) via Ca2+ influx, Ca2+ mobilization from endoplasmic reticulum (ER) calcium stores, and the removal of Ca2+ via multiple mechanisms in beta-cells from both diabetic db/db mice and non-diabetic C57BL/6J mice. We refined our previous quantitative model to describe the slow [Ca2+]i recovery after depolarization in beta-cells from db/db mice. According to the model, the activity levels of the two subtypes of the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) pump, SERCA2 and SERCA3, were severely down-regulated in diabetic cells to 65% and 0% of the levels in normal cells. This down-regulation may lead to a reduction in the Ca2+ concentration in the ER, a compensatory up-regulation of the plasma membrane Na+/Ca2+ exchanger (NCX) and a reduction in depolarization-evoked Ca2+ influx. As a result, the patterns of glucose-stimulated calcium oscillations were significantly different in db/db diabetic beta-cells compared with normal cells. Overall, quantifying the changes in the calcium signaling pathway in db/db diabetic beta-cells will aid in the development of a disease model that could provide insight into the adaptive transformations of beta-cell function during diabetes development.
Electronic supplementary material
The online version of this article (doi:10.1007/s13238-014-0075-7) contains supplementary material, which is available to authorized users.
Keywords: diabetic beta-cells, calcium signaling alterations, SERCA pump, db/db mice
INTRODUCTION
Diabetes affects millions of people and exacts a significant toll on both individual health and society as a whole. Because diabetes is associated with both genetic and environmental factors, the etiology of the disease is complicated and unclear. Interestingly, different Ca2+ signaling components are disturbed in a wide range of organelles in diabetic animals and patients, suggesting a pivotal role for the dysregulation of Ca2+ signaling in the development of diabetes (Bergsten 2000).
Upon blood glucose elevation, pancreatic beta-cells secrete insulin in an intracellular Ca2+ concentration ([Ca2+]i)-dependent manner, which acts on downstream target tissues to facilitate glucose uptake. This process involves different components of the Ca2+ signaling pathways. For example, depolarization triggered by glucose metabolism opens L-type voltage-gated Ca2+ channels, leading to an initial extracellular Ca2+ influx (Rorsman 1997) and the subsequent mobilization of Ca2+ stores via pathways such as the IP3 and ryanodine receptors (Islam 2002; Duman et al. 2006). Simultaneously, clearance mechanisms such as the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), the plasma membrane Ca2+-ATPase (PMCA) and the sodium calcium exchanger (NCX) are activated to reduce elevated [Ca2+]i to the physiological resting level (Chen et al. 2003; Hughes et al. 2006). Altered Ca2+ signaling is consistently observed in pancreatic beta-cells in diabetic animal models. For example, the L-type calcium channel was up-regulated in both neonatally streptozocin-induced and Goto-Kakizaki (GK) diabetic rats (Kato et al. 1996; Kato et al. 1994) but was down-regulated in islets from other rat models of type II diabetes (Iwashima et al. 2001; Roe et al. 1996). An altered release of endoplasmic reticulum (ER) Ca2+ stores via the ryanodine receptor has also been hypothesized to occur during diabetes development (Islam 2002).
SERCA is the major Ca2+ extrusion mechanism in mouse and rat pancreatic beta-cells (Chen et al. 2003; Hughes et al. 2006). SERCA pump activity could not be detected in db/db islets (Roe et al. 1994), whereas a selective down-regulation of SERCA3 sub-type mRNA has been reported in GK rats (Varadi et al. 1996). Missense mutations in human SERCA3 have been associated with type II diabetes (Varadi et al. 1999), suggesting a crucial role for SERCA3 in the etiology of diabetes. However, SERCA3-specific knockout (KO) mice are normoglycemic and have normal insulinemia (Arredouani et al. 2002). PMCA activity is down-regulated in certain types of diabetic islets (Roe et al. 1994; Hoenig et al. 1990; Levy et al. 1998), in contrast to beta-cells cultured under high glucose conditions, which exhibited enhanced NCX transcription (Ximenes et al. 2003). These controversies can only be resolved if the functions of different Ca2+ signaling proteins in normal and diabetic beta-cells are systematically compared under identical experimental conditions.
To address these questions, we compared calcium clearance in age-matched pancreatic beta-cells that were isolated from both C57BL/6J control mice and db/db mice, a widely used type II diabetic mouse model. The changes can be quantitatively modeled as a 35% reduction in SERCA2 activity, a full inhibition of the SERCA3 pump, a 30% increase in the NCX capacity and a 27% reduction in Ca2+ influx. Despite the severely compromised SERCA function, the Ca2+ concentration in the ER ([Ca2+]ER) was reduced only slightly, to 89% of the control, while the ER mobilization pathways remained unchanged. Overall, these changes led to significant alterations in the glucose-induced calcium oscillations in the beta-cells of the db/db mice relative to the control.
RESULTS
Ca2+ clearance after depolarization stimulated Ca2+ influx in normal and db/db beta-cells
We have previously dissected the contributions of multiple clearance mechanisms in pancreatic beta-cells isolated from Balb/c mice through pharmacological manipulation (Chen et al. 2003). In this work, we used this method to compare the clearance mechanisms in beta-cells isolated from age-matched db/db and C57BL/6J mice. To quantitatively evaluate Ca2+ clearance after depolarization, control and db/db cells were stimulated with 70 mmol/L KCl for 3 s and then switched to normal extracellular solution (Fig. 1A and 1B). Compared with normal beta-cells, the db/db cells exhibited a reduced depolarization-triggered [Ca2+]i elevation, and [Ca2+]i returned to the basal level in normal extracellular solution at a significantly slower rate. In cells that had been pre-treated with thapsigargin (TG), an irreversible inhibitor of the SERCA pump, the increase in [Ca2+]i remained smaller, but the clearance was faster in db/db cells than it was in the control cells (Fig. 1C and 1D). This result suggested not only an impairment of the SERCA pump but also a possible up-regulation of clearance mechanisms other than the SERCA pump in db/db beta-cells. To dissect the contributions of NCX and PMCA in clearance, we switched to a Na+-free solution (Li7.4) or a high pH extracellular solution (Na8.8) after depolarization in cells pretreated with TG (Chen et al. 2003). Compared with the control cells, Ca2+ clearance in db/db cells was much faster in the presence of SERCA and PMCA inhibition (TG + Na8.8). In contrast, [Ca2+]i returned to the basal level with the same kinetics in db/db and control cells when only the PMCA pump was functioning (TG + Li7.4). Taken together, these results indicate the selective down-regulation of depolarization-induced Ca2+ influx and the SERCA pump and the up-regulation of NCX in db/db cells.
Figure 1.

Ca 2+ recoveries after short depolarizations in normal and db/db diabetic beta-cells. (A) Averaged clearances in normal beta-cells pretreated with 1 μmol/L TG in control solution (n = 42, red), Li7.4 solution (n = 21, blue) and Na8.8 solution (n = 22, purple) and in cells that were not pretreated with TG (n = 25, dark). (B) Averaged clearances in db/db beta-cells pretreated with 1 μmol/L TG in control solution (n = 32, red), Li7.4 solution (n = 16, blue) and Na8.8 solution (n = 16, purple) and in cells that were not pretreated with TG (n = 34, dark). (C) Summary of the recovery time constants in different solutions from normal and db/db beta-cells. Individual calcium recovery traces were fitted with single-exponential functions. (D) Summary of KCl-triggered [Ca2+]i elevations ([Ca2+]i) in different solutions from normal and db/db beta-cells
Reduced function of the SERCA pump in db/db beta-cells compared with the control
The relative expression levels of the SERCA pump in normal and db/db islets were studied using a SERCA-specific antibody that recognizes all three isoforms of SERCA. In agreement with previous experiments (Roe et al. 1994; Varadi et al. 1996), the SERCA protein levels were severely reduced in db/db islets compared with normal islets (Fig. 2A). Because the Western blotting experiments were performed using whole islets that contain alpha- and beta-cells, it is difficult to determine the specific reduction in SERCA in beta-cells. Therefore, we took a direct approach to measure SERCA function in live beta-cells (Duman et al. 2006; Albrecht et al. 2002). The application of a high concentration of a rapid inhibitor of the SERCA pump, BHQ (100 μmol/L), in resting cells (Fig. 2B) or in cells that were stimulated with a short (3 s, Fig. 2C) or long (30 s, Fig. 2D) depolarization, produced an upstroke in the [Ca2+]i trace. The difference between the BHQ-induced rate of change in [Ca2+]i (J2) and the initial slope of the [Ca2+]i trace immediately before BHQ application (J1) represented the SERCA-dependent uptake of cytosolic Ca2+, which was correlated with different [Ca2+]i levels (Duman et al. 2006; Albrecht et al. 2002). SERCA activity was inhibited at [Ca2+]i ranging from 200 nm to 1000 nm, with increased inhibition at higher [Ca2+]i (Fig. 2D).
Figure 2.
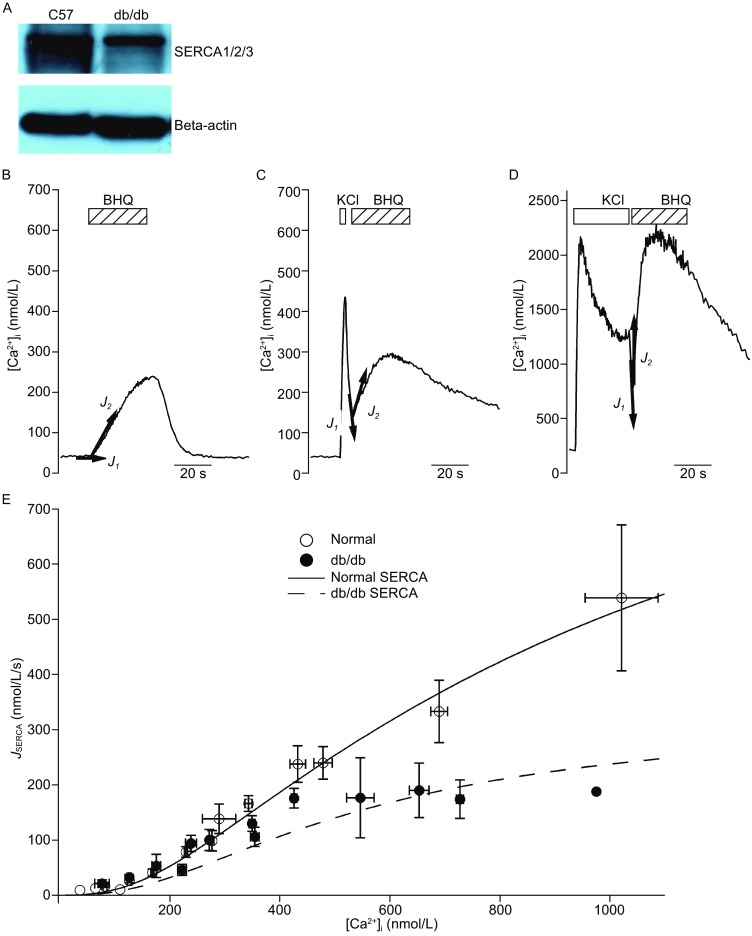
Impaired SERCA function in beta-cells from db/db mice. (A) Reduced total SERCA protein levels in islets from db/db mice compared to those from normal mice. Beta-actin was used as an internal control, and the figure is representative of three independent repeats. (B–D) BHQ (100 μmol/L) was acutely applied to resting cells (B) and cells that had been previously stimulated with KCl for 3 s (C) or 30 s (D). The total cellular Ca2+ flux (defined as –d[Ca2+]i/dt) before (J 1) and after (J 2) BHQ application was plotted on each figure. (E) J SERCA from normal beta-cells (open circle, n = 256) and from db/db diabetic beta-cells (filled circle, n = 119) as a function of [Ca2+]i. The line indicates the total SERCA activity in normal beta-cells (including both SERCA2 and SERCA3 activity) according to the model, whereas the dashed line represents the activity in db/db cells
Quantitative modeling of the alterations in Ca2+ influx and clearance in db/db beta-cells
Previously, we built a mathematical model to simulate calcium clearance in normal beta-cells that used the Kd for SERCA2 for SERCA in the simulation (Chen et al. 2003). However, pancreatic beta-cells also express the SERCA3 subtype (Varadi et al. 1996), which has a much lower affinity for Ca2+ (1100 nmol/L) than does SERCA2 (270 nmol/L) (Lytton et al. 1992). We therefore revised our model to accommodate two SERCA subtypes and manipulated the maximal flux rates through the influx and clearance mechanisms to best simulate the peaks of transient [Ca2+]i and the time constants following depolarization in normal cells under different conditions (Fig. 3A). This simulation yielded a VmaxSERCA2:VmaxSERCA3 ratio of approximately 1.2:1, and the total SERCA activity correlated well with the experimental data throughout a wide range of [Ca2+]i (Fig. 2D, dark dashed lines). By comparing the experimental depolarization-evoked peak [Ca2+]i elevations in the control and db/db beta-cells pretreated with TG, we determined that a 27% decrease in Ca2+ influx from db/db cells was required to account for the decrease in amplitude triggered by depolarization. Based on the clearance dynamics in normal and db/db cells treated with TG + Li7.4 and TG + Na8.8, we determined that the NCX activity in db/db cells was up-regulated 130% relative to control cells. The residual activity levels of SERCA2 and SERCA3 in db/db beta-cells were 65% and 0% of their respective levels in the control cells, which approximated the experimental data nicely (Fig. 2D, red dashed line). With these altered parameters, the simulated [Ca2+]i elevation and the clearance dynamics after depolarization fit the experimental data relatively well (Fig. 3B–D). With the parameters chosen, we reproduced the relative flux rates of the model for normal and beta-cells from db/db mice (Fig. 3E and 3F). The total Ca2+ extrusion rate in db/db beta-cells was ~66% of the control, and the relative role of SERCA was also reduced (from ~66%–71% of the total flux to ~41%–56%). In contrast, the relative contribution of the NCX increased, especially at high [Ca2+]i (>900 nm), indicating that NCX hyper-activity may compensate for the loss of the SERCA3 subtype.
Figure 3.

Simulated Ca 2+ recoveries after short depolarizations in normal and db/db diabetic beta-cells. (A and B) Simulated clearances under different conditions obtained from normal beta-cells (A) and db/db diabetic beta-cells (B). (C and D) Simulated recovery time constants (C) and KCl-triggered [Ca2+]i elevations (D) in beta-cells from normal and db/db mice. (E and F) Simulated kinetic model for Ca2+ transport in normal (E) and db/db (F) beta-cells. The flux rates were much higher than the rates of [Ca2+]i change shown in Fig. 2E because the model includes the effects of strong Ca2+ binding in the cytoplasm by endogenous buffers and fluorescent indicators
No alterations in ER Ca2+ permeability in db/db beta-cells compared to the control
SERCA pump defects combined with an up-regulation of NCX function often result in reduced uptake of Ca2+ to ER calcium stores (Ximenes et al. 2003), and alterations in ER permeability during diabetes have been suggested (Islam 2002). To quantitatively evaluate the Ca2+ content in the ER and its permeability, we modified a protocol from a previous study conducted in sympathetic neurons (Albrecht et al. 2002). Cells were initially bathed in an extracellular solution that did not contain Ca2+ or Na+ (Li7.4) to block the NCX and Ca2+ influx and were then perfused with BHQ to trigger Ca2+ release from the ER calcium stores (Fig. 4A). After [Ca2+]i returned to the basal level, the cells were depolarized for 3 s in the presence of 4 mmol/L Ca2+ to boost the Ca2+ influx. Finally, the extracellular solution was changed back to the Ca2+- and Na+-free solution with BHQ. The rate of Ca2+ extrusion after KCl depolarization was therefore due solely to the PMCA (J PMCA) and could be described by a Hill function that correlated with the different [Ca2+]i levels (Fig. 4B). Because the calcium-flux changes induced by the first BHQ application (J BHQ(t)) were a result of both the release from the ER (J release) and Ca2+ extrusion via the PMCA, the J release(t) was calculated as the difference between J BHQ and J PMCA at each time point (Fig. 4C). The drop in the ER Ca2+ concentration ([Ca2+]ER) at different time points was calculated based on the following equation:
Figure 4.
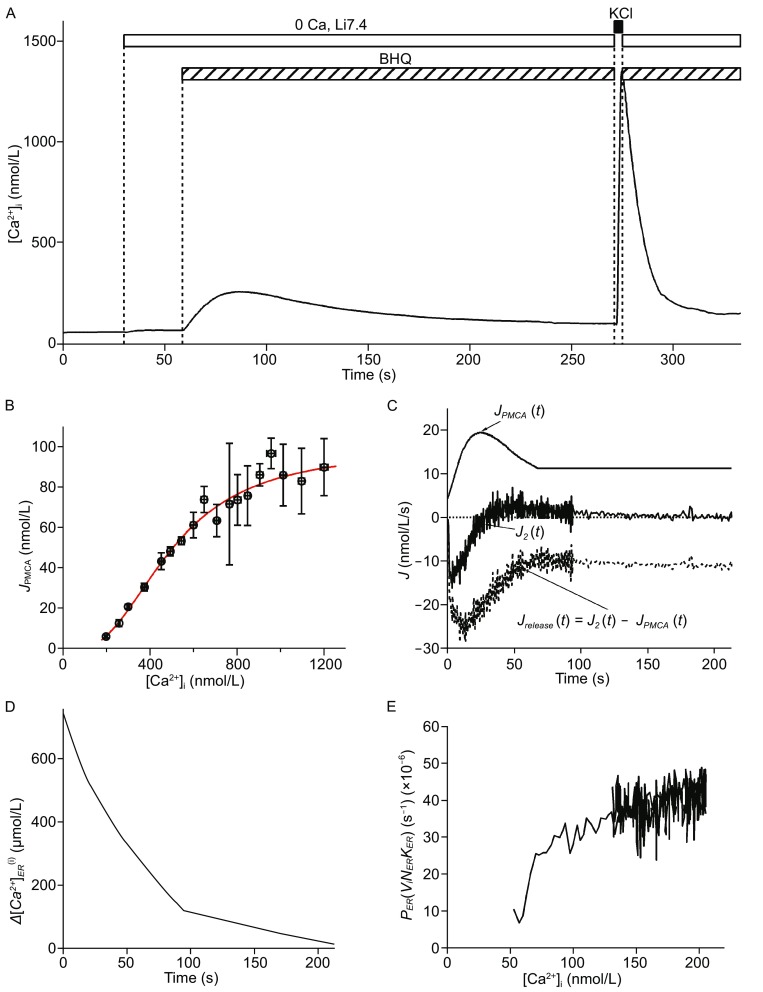
Detection of Ca 2+ content in the ER and ER Ca 2+ permeability in single beta-cells. (A) A cell was initially bathed in Ca2+-free and Na+-free solution, then switched to a solution containing 100 μmol/L BHQ to block the SERCA pump. After depletion of the Ca2+ in the ER, KCl (70 mmol/L) with Ca2+ (4 mmol/L) was applied to the cell for 3 s to induce a transient increase in [Ca2+]i. The clearance dynamics thereafter were mainly due to the removal of Ca2+ by the PMCA (J PMCA). (B) The correlation of J PMCA with [Ca2+]i obtained from (A) can be fitted with a Hill function. (C) Time course of JPMCA, total cytoplasmic Ca2+ flux (J 2) and J release after BHQ application. J release was calculated as the difference between the J 2 and the simulated J PMCA at different time points after BHQ application. (D) Plot of time course of after BHQ application, which was calculated as stated in the main text (Albrecht et al. 2002). (E) Relationship of P ER [v i /(v ER k ER )] to different [Ca2+]i obtained during the BHQ-stimulated ER calcium release
(Albrecht et al. 2002), in which v i and v ER are the volumes of the cytoplasm and the ER, respectively, and κi and κER are the calcium buffering ratios of the cytoplasm and the ER, respectively. The minus of the integral () was the change in total cytoplasmic Ca2+ concentration that would occur if from time t onward, J release were deposited into a closed compartment that has the same volume as the cytoplasm. Assuming that [Ca2+]ER equilibrates with [Ca2+]i after BHQ application, we calculated the initial content in the ER according to the following equation:
Therefore, the time course of was shown in Fig. 4D, and the relative permeability of the ER was estimated according to the equation
(Fig. 4E) (Albrecht et al. 2002).
In beta-cells from db/db mice, the BHQ-triggered [Ca2+]i transient was significantly lower than in the control cells (Fig. 5A). Based on the calculations, the average in the db/db beta-cells was approximately 89% of the value in the normal cells (Fig. 5C, P < 0.05). Although the time courses of J release were apparently different in different cell types (Fig. 5B), the relative permeabilities of the ER were approximately the same (Fig. 5D), suggesting that the difference in ER release was due to a difference in the initial [Ca2+]ER.
Figure 5.
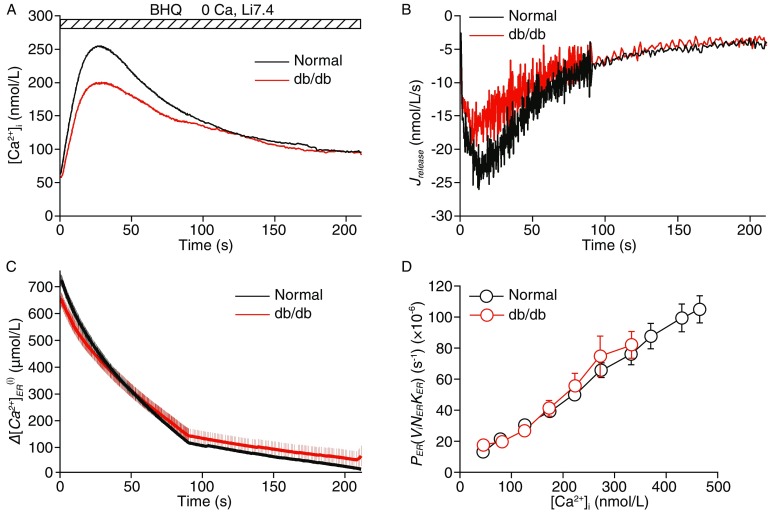
Reduced and unaltered ER Ca 2+ permeability in db/db beta-cells. (A) Average BHQ-triggered [Ca2+]i transients in Ca2+-free and Na+-free solutions from normal beta-cells (n = 43, dark) and db/db diabetic beta-cells (n = 31, red). (B) Time courses of average J release in normal and db/db cells. (C) Reduced initial in db/db diabetic beta-cells relative to normal cells (P < 0.05). (D) The average relative ER permeability during the BHQ stimulation was approximately the same in normal and db/db diabetic beta-cells
Glucose-induced calcium oscillations in normal and db/db beta-cells
Finally, we directly monitored the calcium oscillations induced by 15 mmol/L glucose in normal and db/db beta-cells (Fig. 6A). Upon the application of glucose, [Ca2+]i often decreased slightly before being elevated, which was suggested to be due to the ATP-activated SERCA-dependent sequestration of [Ca2+]i in the ER (Marie et al. 2001). Consistent with a significant inhibition of SERCA2 activity (Fig. 3), the initial decrease in [Ca2+]i was reduced significantly in db/db cells (Fig. 6B). The maximal elevation in [Ca2+]i triggered by glucose was also reduced in db/db cells compared with the control cells. However, the glucose-stimulated [Ca2+]i increase was accelerated in the db/db cells, and the percentage of time the cells spent over the [Ca2+]i plateau was slightly, but not significantly, different in the db/db cells. Overall, our data suggest that multiple characteristics of the glucose-triggered calcium transient are altered in db/db beta-cells.
Figure 6.

Altered glucose-triggered calcium oscillations in db/db beta-cells. (A) Representative examples of calcium oscillations triggered by glucose (15 mmol/L) in one beta-cell from normal mice (dark) and in one beta-cell from db/db mice (red). The initial decrease in [Ca2+]i was quantified as the difference between the resting [Ca2+]i and the minimal [Ca2+]i reached within 120 s after glucose perfusion. The maximal increase in [Ca2+]i was calculated as the difference between the resting [Ca2+]i and the maximal [Ca2+]i reached within the period of glucose perfusion. The time to rise was defined as the time delay between the perfusion of glucose and the time when [Ca2+]i reached 50% of the maximal increase in [Ca2+]i. Finally, the percentage over plateau of each trace was defined as the time during which the [Ca2+]i was greater than 50% of the maximal increase divided by the total time of glucose stimulation. (B, C, D, E) Compared to normal cells (n = 23), pancreatic beta-cells from db/db mice (n = 21) exhibited a reduction in the average initial decrease in [Ca2+]i (B), a reduction in the average maximal increase in [Ca2+]i (C), and an accelerated response to glucose (D). Although db/db cells spent a slightly greater percentage of time at [Ca2+]i greater than 50% of the maximal increase in [Ca2+]i during glucose stimulation, the average difference compared to normal cells was not significant (E)
DISCUSSION
In the current study, we showed that the PMCA activity in islets from db/db mice was not significantly different compared with that observed in normal mice, in contrast to the previously reported down-regulation of PMCA function in db/db islets and islets with insulin resistance (Roe et al. 1994; Hoenig et al. 1990; Levy et al. 1998; Alzugaray et al. 2009). The difference may be due to the different experimental conditions tested and the different cell models used. However, because PMCA plays a minor role in calcium clearance after depolarization in both mouse and rat pancreatic beta-cells (Chen et al. 2003; Hughes et al. 2006), we believe that PMCA may not be the primary target in the reshaping of beta-cell calcium signaling pathways during the development of diabetes. The functions of the SERCA2 and SERCA3 subtypes were both impaired in db/db beta-cells, as opposed to the selective impairment of SERCA3 function in GK rats. This result partially explains the normoglycemic phenotype of SERCA3 KO mice (Arredouani et al. 2002). Because the high-affinity SERCA2 subtype functions in basal [Ca2+]i regulation in beta-cells (Arredouani et al. 2002), its down-regulation correlates with the reduction in the glucose-stimulated initial decrease in [Ca2+]i in db/db beta-cells (Fig. 6B), which may perturb glucose-stimulated insulin secretion (Roe et al. 1994; Marie et al. 2001). In addition, we report for the first time a 30% increase in NCX activity in db/db beta-cells, which is consistent with the enhancement of NCX transcription observed in islets cultured in high glucose (Ximenes et al. 2003). Thus our data generally agree with previous studies conducted in this field. In addition, by fitting the experimental data to a modified mathematical model, we obtained a quantitative description of all alterations in the calcium signaling pathways in db/db mice, which has not been previously reported.
Perturbed Ca2+ homeostasis has long been proposed as a hallmark of diabetes (Bergsten 2000). Despite the many alterations in the calcium signaling pathway in beta-cells from db/db mice, we propose that the down-regulation of SERCA is the earliest event. SERCA2 and SERCA3 expression levels are reduced in islets from both type I and type II diabetic mice at earlier stages (Varadi et al. 1996; Liang et al. 2011), which may be due to increased levels of glucose (Levy et al. 1998), saturated fatty acids (Cunha et al. 2008), and cytokines (Dula et al. 2010; Cardozo et al. 2005) in the blood vessels and enhanced insulin resistance in beta-cells (Borge et al. 2002). Because the SERCA pump is the dominant clearance mechanism in beta-cells that limits the amplitude of [Ca2+]i transients after depolarization, SERCA pump inhibition dramatically enhances depolarization-induced insulin secretion (Chen et al. 2003; Hughes et al. 2006). Therefore, the down-regulation of SERCA is a beneficial adaptation mechanism that enables beta-cells to secrete more insulin to compensate for the loss of beta-cell mass in type I diabetes or to overcome insulin resistance in type II diabetes. This explanation is supported by the accelerated onset of the glucose-stimulated Ca2+ transient in db/db diabetic beta-cells relative to the control cells (Fig. 6B). Along with the reduction in SERCA expression, fine-tuning of the voltage-gated calcium channel currents is needed to generate suitable [Ca2+]i transients that maximize the secretion response and minimize the apoptosis triggered by high [Ca2+]i. Because the NCX contributes more to the total clearance mechanism of beta-cells at relatively high [Ca2+]i (Chen et al. 2003), the loss of low-affinity SERCA3 may lead to the adaptive up-regulation of NCX observed here, which assists in calcium clearance from the cytosol during the stimulation and shaping of glucose-triggered calcium transients. Despite the significant reduction in SERCA activity and the enhanced NCX activity, the decrease in [Ca2+]ER (11%) in db/db beta-cells was relatively small, in contrast to the severe reduction in [Ca2+]ER from insulin-secreting cell lines treated acutely or chronically with saturated fatty acids (Cunha et al. 2008; Gwiazda et al. 2009). These results highlight the importance of systematic investigations of calcium signaling pathways alterations in primary beta-cells isolated from diabetic animals, rather than solely detecting alterations in beta-cells cultured in conditions that mimic diabetes. Decreased [Ca2+]ER is a susceptibility factor for ER stress (Cunha et al. 2008; Cardozo et al. 2005). Therefore, in the long run, a prolonged small decrease in [Ca2+]ER in combination with other ER stress susceptibility factors, such as excessive insulin biosynthesis (Grill and Bjorklund 2001), may ultimately result in beta-cell failure, apoptosis, and severe glucose intolerance.
In conclusion, we characterize changes in multiple aspects of the calcium signaling pathway in beta-cells isolated from db/db mice compared with the control. These quantitative studies will help build better models to describe glucose-induced Ca2+ oscillations in diabetic beta-cells (Bertram et al. 2004), which will provide insights into the development of insulin secretion dysfunction and beta-cell failure in the development of diabetes.
MATERIALS AND METHODS
Cell culture and Western blotting experiments
Primary islets and beta-cells were isolated from age-matched C57BL/6J and db/db mice (7–8 weeks) as previously described (Chen et al. 2003). Mouse handling and experimental procedures were conducted in accordance with the Committee for the Use of Live Animals in Teaching and Research at Institute of Molecular Medicine, Peking University. The protocol was approved by the Committee on the Ethics of Animal Experiments of Peking University (Permit Number: IMM-ChenLY-1). All surgery was performed under chloral hydrate anesthesia, and all efforts were made to minimize suffering. At this age, db/db mice were overweight and glucose intolerant, as previously reported (Kobayashi et al. 2000). The isolated cells were plated onto polyornithine-coated glass coverslips and cultured at 37°C in 5% CO2 in RPMI 1640 culture medium containing 10 mmol/L glucose, 10% fetal bovine serum, 100 µg/mL streptomycin and 100 IU/mL penicillin. Pancreatic beta-cells were identified by size (Cho et al. 2010) and generally used on culture day 2 or 3. For the Western blotting experiments (Fig. 2), isolated islets were homogenized using homogenate buffer to obtain total proteins. Equal amounts of proteins (approximately 30 µg/lane) were loaded to SDS-PAGE and analyzed by Western blotting using anti-SERCA1/2/3 (SANTA CRUZ, 1:800) and anti-beta actin antibody (Sigma, St. Louis, MO, 1:2000). The incubation with the antibodies was followed by the application of rabbit anti-mouse IgG peroxidase conjugate (Sigma, St. Louis, MO, 1:5000) and goat anti-rabbit IgG (Perkin Elmer, 1:5000). The blots were then probed with Western Lighting plus-ECL (Perkin Elmer, Inc.) (He et al. 2008).
Experimental set-up and Ca2+ photometry
Cells were loaded with fura-2-AM (10 µmol/L) in modified Ringer’s solution (130 mmol/L NaCl, 2.5 mmol/L KCl, 1 mmol/L MgCl2, 2 mmol/L CaCl2, 4 mmol/L glucose, and 10 mmol/L HEPES, pH 7.3) at room temperature for 20–25 min. The modified Ringer’s solution was also used as the extracellular solution for basal recording in the glucose-induced calcium oscillation experiments, in which 15 mmol/L glucose replaced 4 mmol/L glucose as the stimulus. For the Ca2+ clearance/[Ca2+]ER evaluation experiment, the cells were perfused with the modified Ringer’s solution containing 15 mmol/L (instead of 4 mmol/L) glucose and 250 µmol/L diazoxide, and various reagents were added to this solution to inhibit clearance mechanisms, as described in the individual experiments. The depolarization solution (KCl) consisted of 70 mmol/L KCl, 67 mmol/L NaCl, 4 mmol/L CaCl2, 1 mmol/L MgCl2, 15 mmol/L glucose, 250 µmol/L diazoxide, and 10 mmol/L HEPES, pH 7.3. To inhibit the NCX, we used a Na+-free solution in which Li+ replaced Na+ (Li7.4) and raised the pH of the solution to 8.8 (Na8.8) to slow the PMCA pump (Chen et al. 2003). Rapid solution changes (<500 ms) were digitally controlled by a fast local perfusion system (Chen et al. 2003).
For photometry, the cells were excited by sequential 340 and 380 nm light generated by a computer-controlled PolyChrome IV (TILL Photonics) light source, and the emission at 505 nm was collected by a photodiode. The PULSE software was used to coordinate protocols and collect data, which were analyzed using IGOR Pro. The standard calibration parameters Rmin, Rmax and K* were determined as previously described (Chen et al. 2003).
Measurement of JSERCA and Jrelease activity at different [Ca2+]i in live beta-cells
Following previously published reports (Duman et al. 2006; Albrecht et al. 2002), we applied a high dose of tert-butylhydroquinone (BHQ, 100 μmol/L) to abruptly stop the uptake of Ca2+ by the ER via the SERCA pump. The total cellular Ca2+ flux can be calculated as the rate of change of [Ca2+]i (defined as –d[Ca2+]i/dt). Prior to the application of BHQ, the total cellular Ca2+ flux (J 1) can be described by the equation
| Eq. 1 |
J SERCA is the Ca2+ flux due to the BHQ-sensitive pumping of Ca2+ into the ER; J release is the flux into the cytosol from intracellular stores; and J PM is the flux across the plasma membrane. The acute application of BHQ changes the new total cellular Ca2+ flux (J 1) according to
| Eq. 2 |
Therefore, the value of J SERCA was calculated as the difference between J 1 and J 2. In Fig. 4, we briefly treated the beta-cells with High K+ solution containing BHQ to obtain Ca2+ clearance by the PMCA pump alone, and J PMCA was fitted to a Hill equation to describe its relationship to different [Ca2+]i levels. Therefore, a different J release(t) was calculated based on Eq. 2, and the appropriate value of J PMCA was inserted.
Data analysis
All data were analyzed using the Igor Pro software (Wavemetrics, Lake Oswego, OR). The averaged results are presented as the mean value ± SEM of the number of experiments indicated. The statistical significance was evaluated using either Student’s t-test for single Gaussian distributed datasets or the Mann-Whitney rank sum test for non-single Gaussian-distributed datasets. The asterisks *, **, and *** denote statistical significances with P values less than 0.05, 0.01, and 0.001, respectively.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ACKNOWLEDGEMENTS
The work was supported by grants from the National Basic Research Program (973 Program) (No. 2013CB531200), the National Key Technology R&D Program (No. SQ2011SF11B01041), National Natural Science Foundation of China (Grant Nos. 31221002 and 81222020), the Beijing Natural Science Foundation (Nos. 7121008 and 7142071), the Basic and Clinical Research Fund of Capital Medical University (No. 13JL19) and the Talents Project of Department of Beijing Municipal Party Committee Organization of the Communist Party of China (No. 2011D005018000004). This work is also supported by the grant GM83913 from the U.S. National Institutes of Health.
COMPLIANCE WITH ETHICS GUIDELINES
Kuo Liang, Wen Du, Jingze Lu, Fei Li, Lu Yang, Yanhong Xue, Bertil Hille and Liangyi Chen declare that they have no conflict of interest.
All animal experimental procedures were conducted in accordance with the Committee for the Use of Live Animals in Teaching and Research at Institute of Molecular Medicine, Peking University.
Footnotes
Kuo Liang, Wen Du, Jingze Lu have contributed equally to this work.
Contributor Information
Fei Li, Email: feili36@ccmu.edu.cn.
Liangyi Chen, Email: lychen@pku.edu.cn.
References
- Albrecht MA, Colegrove SL, Friel DD. Differential regulation of ER Ca2+ uptake and release rates accounts for multiple modes of Ca2+-induced Ca2+ release. J Gen Physiol. 2002;119:211–233. doi: 10.1085/jgp.20028484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzugaray ME, Garcia ME, Del Zotto HH, Raschia MA, Palomeque J, et al. Changes in islet plasma membrane calcium-ATPase activity and isoform expression induced by insulin resistance. Arch Biochem Biophys. 2009;490:17–23. doi: 10.1016/j.abb.2009.07.022. [DOI] [PubMed] [Google Scholar]
- Arredouani A, Guiot Y, Jonas JC, Liu LH, Nenquin M, et al. SERCA3 ablation does not impair insulin secretion but suggests distinct roles of different sarcoendoplasmic reticulum Ca(2+) pumps for Ca(2+) homeostasis in pancreatic beta-cells. Diabetes. 2002;51:3245–3253. doi: 10.2337/diabetes.51.11.3245. [DOI] [PubMed] [Google Scholar]
- Bergsten P. Pathophysiology of impaired pulsatile insulin release. Diabetes Metab Res Rev. 2000;16:179–191. doi: 10.1002/1520-7560(200005/06)16:3<179::AID-DMRR115>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Bertram R, Satin L, Zhang M, Smolen P, Sherman A. Calcium and glycolysis mediate multiple bursting modes in pancreatic islets. Biophys J. 2004;87:3074–3087. doi: 10.1529/biophysj.104.049262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borge PD, Moibi J, Greene SR, Trucco M, Young RA, et al. Insulin receptor signaling and sarco/endoplasmic reticulum calcium ATPase in beta-cells. Diabetes. 2002;51(Suppl 3):S427–S433. doi: 10.2337/diabetes.51.2007.S427. [DOI] [PubMed] [Google Scholar]
- Cardozo AK, Ortis F, Storling J, Feng YM, Rasschaert J, et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes. 2005;54:452–461. doi: 10.2337/diabetes.54.2.452. [DOI] [PubMed] [Google Scholar]
- Chen LY, Koh DS, Hille B. Dynamics of calcium clearance in mouse pancreatic beta-cells. Diabetes. 2003;52:1723–1731. doi: 10.2337/diabetes.52.7.1723. [DOI] [PubMed] [Google Scholar]
- Cho JH, Chen L, Kim MH, Chow RH, Hille B, et al. Characteristics and functions of {alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors expressed in mouse pancreatic {alpha}-cells. Endocrinology. 2010;151:1541–1550. doi: 10.1210/en.2009-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha DA, Hekerman P, Ladriere L, Bazarra-Castro A, Ortis F, et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci. 2008;121:2308–2318. doi: 10.1242/jcs.026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dula SB, Jecmenica M, Wu R, Jahanshahi P, Verrilli GM, et al. Evidence that low-grade systemic inflammation can induce islet dysfunction as measured by impaired calcium handling. Cell Calcium. 2010;48:133–142. doi: 10.1016/j.ceca.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman JG, Chen LY, Palmer AE, Hille B. Contributions of intracellular compartments to calcium dynamics: implicating an acidic store. Traffic. 2006;7:859–872. doi: 10.1111/j.1600-0854.2006.00432.x. [DOI] [PubMed] [Google Scholar]
- Grill V, Bjorklund A. Overstimulation and beta-cell function. Diabetes. 2001;50(Suppl 1):S122–S124. doi: 10.2337/diabetes.50.2007.S122. [DOI] [PubMed] [Google Scholar]
- Gwiazda KS, Yang TL, Lin Y, Johnson JD. Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am J Physiol Endocrinol Metab. 2009;296:E690–E701. doi: 10.1152/ajpendo.90525.2008. [DOI] [PubMed] [Google Scholar]
- He ZX, Fan JM, Kang LJ, Lu JZ, Xue YH, et al. Ca2+ triggers a novel clathrin-independent but actin-dependent fast endocytosis in pancreatic beta cells. Traffic. 2008;9:910–923. doi: 10.1111/j.1600-0854.2008.00730.x. [DOI] [PubMed] [Google Scholar]
- Hoenig M, Lee RJ, Ferguson DC. Glucose inhibits the high-affinity (Ca2+ + Mg2+)-ATPase in the plasma membrane of a glucose-responsive insulinoma. Biochim Biophys Acta. 1990;1022:333–338. doi: 10.1016/0005-2736(90)90282-S. [DOI] [PubMed] [Google Scholar]
- Hughes E, Lee AK, Tse A. Dominant role of sarcoendoplasmic reticulum Ca2+-ATPase pump in Ca2+ homeostasis and exocytosis in rat pancreatic beta-cells. Endocrinology. 2006;147:1396–1407. doi: 10.1210/en.2005-1023. [DOI] [PubMed] [Google Scholar]
- Islam MS. The ryanodine receptor calcium channel of beta-cells: molecular regulation and physiological significance. Diabetes. 2002;51:1299–1309. doi: 10.2337/diabetes.51.5.1299. [DOI] [PubMed] [Google Scholar]
- Iwashima Y, Abiko A, Ushikubi F, Hata A, Kaku K, et al. Downregulation of the voltage-dependent calcium channel (VDCC) beta-subunit mRNAs in pancreatic islets of type 2 diabetic rats. Biochem Biophys Res Commun. 2001;280:923–932. doi: 10.1006/bbrc.2000.4122. [DOI] [PubMed] [Google Scholar]
- Kato S, Ishida H, Tsuura Y, Okamoto Y, Tsuji K, et al. Increased calcium-channel currents of pancreatic beta cells in neonatally streptozocin-induced diabetic rats. Metabolism. 1994;43:1395–1400. doi: 10.1016/0026-0495(94)90034-5. [DOI] [PubMed] [Google Scholar]
- Kato S, Ishida H, Tsuura Y, Tsuji K, Nishimura M, et al. Alterations in basal and glucose-stimulated voltage-dependent Ca2+ channel activities in pancreatic beta cells of non-insulin-dependent diabetes mellitus GK rats. J Clin Invest. 1996;97:2417–2425. doi: 10.1172/JCI118688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Forte TM, Taniguchi S, Ishida BY, Oka K, et al. The db/db mouse, a model for diabetic dyslipidemia: molecular characterization and effects of Western diet feeding. Metabolism. 2000;49:22–31. doi: 10.1016/S0026-0495(00)90588-2. [DOI] [PubMed] [Google Scholar]
- Levy J, Zhu Z, Dunbar JC. The effect of glucose and calcium on Ca2+-adenosine triphosphatase in pancreatic islets isolated from a normal and a non-insulin-dependent diabetes mellitus rat model. Metabolism. 1998;47:185–189. doi: 10.1016/S0026-0495(98)90218-9. [DOI] [PubMed] [Google Scholar]
- Liang K, Du W, Zhu W, Liu S, Cui Y, et al. Contribution of different mechanisms to pancreatic beta-cell hyper-secretion in non-obese diabetic (NOD) mice during pre-diabetes. J Biol Chem. 2011;286:39537–39545. doi: 10.1074/jbc.M111.295931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton J, Westlin M, Burk SE, Shull GE, MacLennan DH. Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J Biol Chem. 1992;267:14483–14489. [PubMed] [Google Scholar]
- Marie JC, Bailbe D, Gylfe E, Portha B. Defective glucose-dependent cytosolic Ca2+ handling in islets of GK and nSTZ rat models of type 2 diabetes. J Endocrinol. 2001;169:169–176. doi: 10.1677/joe.0.1690169. [DOI] [PubMed] [Google Scholar]
- Roe MW, Philipson LH, Frangakis CJ, Kuznetsov A, Mertz RJ, et al. Defective glucose-dependent endoplasmic reticulum Ca2+ sequestration in diabetic mouse islets of Langerhans. J Biol Chem. 1994;269:18279–18282. [PubMed] [Google Scholar]
- Roe MW, Worley JF, 3rd, Tokuyama Y, Philipson LH, Sturis J, et al. NIDDM is associated with loss of pancreatic beta-cell L-type Ca2+ channel activity. Am J Physiol. 1996;270:E133–E140. doi: 10.1152/ajpendo.1996.270.1.E133. [DOI] [PubMed] [Google Scholar]
- Rorsman P. The pancreatic beta-cell as a fuel sensor: an electrophysiologist’s viewpoint. Diabetologia. 1997;40:487–495. doi: 10.1007/s001250050706. [DOI] [PubMed] [Google Scholar]
- Varadi A, Molnar E, Ostenson CG, Ashcroft SJ. Isoforms of endoplasmic reticulum Ca(2+)-ATPase are differentially expressed in normal and diabetic islets of Langerhans. Biochem J. 1996;319(Pt 2):521–527. doi: 10.1042/bj3190521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi A, Lebel L, Hashim Y, Mehta Z, Ashcroft SJ, et al. Sequence variants of the sarco(endo)plasmic reticulum Ca(2+)-transport ATPase 3 gene (SERCA3) in Caucasian type II diabetic patients (UK Prospective Diabetes Study 48) Diabetologia. 1999;42:1240–1243. doi: 10.1007/s001250051298. [DOI] [PubMed] [Google Scholar]
- Ximenes HM, Kamagate A, Van Eylen F, Carpinelli A, Herchuelz A. Opposite effects of glucose on plasma membrane Ca2+-ATPase and Na/Ca exchanger transcription, expression, and activity in rat pancreatic beta-cells. J Biol Chem. 2003;278:22956–22963. doi: 10.1074/jbc.M212339200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.