

Abstract
Doxorubicin (DOX) is an anthracycline used widely for cancer chemotherapy. Its primary mode of action appears to be topoisomerase II inhibition, DNA cleavage, and free radical generation. However, in non-neuronal cells, DOX also inhibits the expression of dual-specificity phosphatases (also referred to as MAPK phosphatases) and thereby inhibits the dephosphorylation of extracellular signal-regulated kinase (ERK) and p38 mitogen-activated protein kinase (p38 MAPK), two MAPK isoforms important for long-term memory (LTM) formation. Activation of these kinases by DOX in neurons, if present, could have secondary effects on cognitive functions, such as learning and memory. The present study used cultures of rat cortical neurons and sensory neurons (SNs) of Aplysia to examine the effects of DOX on levels of phosphorylated ERK (pERK) and phosphorylated p38 (p-p38) MAPK. In addition, Aplysia neurons were used to examine the effects of DOX on long-term enhanced excitability, long-term synaptic facilitation (LTF), and long-term synaptic depression (LTD). DOX treatment led to elevated levels of pERK and p-p38 MAPK in SNs and cortical neurons. In addition, it increased phosphorylation of the downstream transcriptional repressor cAMP response element-binding protein 2 in SNs. DOX treatment blocked serotonin-induced LTF and enhanced LTD induced by the neuropeptide Phe-Met-Arg-Phe-NH2. The block of LTF appeared to be attributable to overriding inhibitory effects of p-p38 MAPK, because LTF was rescued in the presence of an inhibitor (SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole]) of p38 MAPK. These results suggest that acute application of DOX might impair the formation of LTM via the p38 MAPK pathway.
Keywords: Aplysia, chemotherapy, ERK, p38 MAPK, serotonin, synaptic plasticity
Introduction
Accumulating evidence indicates that chemotherapy is associated with deficits in cognitive functions, including memory (Myers, 2009). However, the mechanisms for these effects are poorly understood. One possibility is that these drugs affect key kinase systems involved in memory. For example, doxorubicin (DOX) is an anthracycline used to treat several forms of cancer (Minotti et al., 2004). Its antitumor actions include oxidative damage to cellular membranes and interference with DNA replication and RNA synthesis (Gewirtz, 1999). DOX also affects multiple isoforms of mitogen-activated protein kinase (MAPK). In heart cells, DOX leads to activation of the p38 isoform of MAPK (Poizat et al., 2005), in mammary epithelial and breast carcinoma cell lines, DOX leads to activation of the extracellular signal-regulated kinase (ERK) isoform of MAPK attributable, at least in part, to the reduced expression of dual-specificity phosphatases (Small et al., 2003), and in lymphoma and lung carcinoma cell lines, DOX leads to activation of both MAPK isoforms (Niiya et al., 2004). It is not known whether DOX affects ERK and p38 MAPK in the same neurons, but such a finding would suggest that DOX treatment affects neuronal plasticity and learning. Because hippocampal long-term potentiation (LTP) is dependent on ERK (English and Sweatt, 1997; Coogan et al., 1999; Bolshakov et al., 2000; Kanterewicz et al., 2000), selective activation of ERK would suggest a potentiation of synaptic transmission. Because p38 MAPK promotes depotentiation of LTP and long-term depression (LTD) (Bolshakov et al., 2000; Murray and O'Connor, 2003; Liang et al., 2008), selective activation of p38 MAPK would suggest an attenuation of synaptic transmission. Therefore, if ERK and p38 MAPK were activated simultaneously, the combined effects would be difficult to predict. Consequently, the effects of DOX on memory are likely to depend on the extent to which DOX leads to the activation of ERK and p38 MAPK and on the relative contribution of these kinases to the particular forms of plasticity within a given memory system.
To gain insight into possible effects of DOX on MAPK in neurons and potential effects on synaptic plasticity, we exploited the technical advantages of the sensorimotor culture system in Aplysia. This system exhibits long-term synaptic plasticity [long-term synaptic facilitation (LTF) and LTD], the mechanisms of which are well understood and analogous to vertebrate LTP and LTD. For example, ERK is essential for LTF (Martin et al., 1997; Michael et al., 1998; Purcell et al., 2003; Ormond et al., 2004; Sharma and Carew, 2004), and activation of p38 MAPK produces LTD (Guan et al., 2002, 2003).
Treatment with DOX produced a rapid (within 45 min) increase in phosphorylation of ERK, p38 MAPK, and cAMP response element-binding protein 2 (CREB2). Moreover, a relatively brief application of DOX (120 min) produced a deficit in serotonin (5-HT)-induced LTF and enhanced Phe-Met-Arg-Phe-NH2 (FMRFa)-mediated LTD. Interestingly, the impairment in LTF was rescued by inhibition of p38 MAPK. These results suggest that DOX leads to activation of both ERK and p38 MAPK in neurons and that impairments in learning and memory produced by DOX chemotherapy may be attributable in part to dominant inhibitory actions of p38 MAPK.
Materials and Methods
Neuronal cultures.
Isolated sensory neurons (SNs) or SN–motor neuron (MN) cocultures from the marine mollusk Aplysia were prepared according to conventional procedures (Zhang et al., 2012). Dishes of SN cultures were plated with 4–10 SNs. Dishes of SN–MN cocultures were plated with a single SN and a single L7 MN.
Cortices from rat embryos (E17–E18) were dissected, dissociated, and plated on glass coverslips coated with poly-d-lysine in four-well tissue-culture plates (3 × 105 cells per well; Tsvetkov et al., 2013). Cortical neuronal cultures were grown in modified neuronal growth medium made from Neurobasal medium, B-27 supplement, GlutaMAX, and penicillin–streptomycin (all from Life Technologies) for 10–18 d before treatment with DOX. In these cultures, serum-free Neurobasal medium supplemented with B-27 limited the growth of glial cells to <5% after 5 d (Boland et al., 2008).
Pharmacological treatment.
The use of DOX in Aplysia has not been reported. Concentrations of DOX used in vitro range from 0.5 to 20 μm (Small et al., 2003; Poizat et al., 2005; Price et al., 2007; Rojo et al., 2009). We found that concentrations of DOX <200 μm were not toxic to Aplysia SNs based on the observations that no characteristic morphologies of cell death (e.g., cell shrinkage, rupture of the plasma membrane, and neurite degeneration) were observed, nor were any changes observed in cellular properties (resting potentials and membrane resistances) in the SNs 24 h after a 2 h DOX exposure at concentrations of 2, 20, and 200 μm. These passive electrical properties are sensitive measures of cell function and integrity (Ehrlich et al., 2007). In addition to examining the potential toxic effects of DOX on the cellular properties of SNs, we examined cellular properties of MNs of the SN–MN cocultures that were used in the physiological studies of the effects of 2.5 μm DOX on the modulation of LTF and LTD. No effects on resting potential or input resistance were observed 24 h after a 2 h DOX exposure.
To examine DOX-induced ERK activation in SNs, we applied DOX at 0, 2.5, 5, and 10 μm for 30 or 120 min. After washout of DOX, SNs were incubated in vehicle solution (Veh; [L15 medium/artificial seawater (ASW)] for 45 min. SNs were then fixed for immunofluorescence analysis. For each experiment, two dishes of SNs from the same animal were treated with either DOX or Veh alone. Therefore, the SNs treated with different concentrations of DOX were compared with their own Veh controls.
To induce LTF, SN–MN cocultures were treated with five 5-min pulses of 1 μm 5-HT (Sigma) with an interpulse interval of 20 min. To induce LTD, SN–MN cocultures were treated with five 5-min pulses of 10 μm FMRFa (Sigma) with an interpulse interval of 20 min (Guan et al., 2003; Fioravante et al., 2008). Control SN–MN cocultures were treated with an equal number of pulses of Veh (L15/ASW). Cocultures were exposed to 2.5 μm DOX (Sigma) for 30 min before and during the 85 min 5-HT or FMRFa treatments (115 min in total). The same procedures were used with isolated SNs to study long-term enhanced excitability (LTEE).
To examine the effect of p38 MAPK inhibition on DOX-impaired LTF, 3 μm SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole (SB); EMD Millipore] was applied to cocultures 30 min before adding DOX and then concurrently with DOX (145 min in total). At this concentration, SB blocks LTD in Aplysia without affecting basal synaptic strength (Guan et al., 2003).
Concentrations of DOX used in cultures of cortical neurons were 2.5 and 0.25 μm. DOX at 2.5 μm was chosen to compare the effects of DOX on ERK and p38 MAPK activation in rat cortical neurons with those examined in Aplysia neurons. The lower concentration of 0.25 μm was investigated because it corresponds to the concentration of DOX detected in rat cerebral hemispheres after intraperitoneal injection of DOX (Sardi et al., 2013).
Immunofluorescence.
Immunofluorescence procedures for SNs and rat cortical neurons followed those of Chin et al. (1999). Briefly, cells were fixed in a solution of 4% paraformaldehyde in PBS containing 20% sucrose. After three rinses in PBS, fixed cells were blocked for 30 min at room temperature in Superblock buffer (Pierce), 0.2% Triton X-100, and 3% normal goat serum and subsequently incubated overnight at 4°C with anti-phosphorylated ERK (pERK) antibody (1:500), anti-phosphorylated p38 (p-p38) MAPK antibody (1:200), anti-MAPK phosphatase 1 (MKP1) antibody (1:500), or anti-pCREB2 antibody (1:500). Anti-pERK antibody was purchased from Cell Signaling Technology. Anti-p-p38 MAPK and anti-MKP1 antibodies were purchased from Santa Cruz Biotechnology. The anti-pCREB2 antibody was raised by a commercial vendor (Genemed Biotechnologies) against the phosphorylated version of a CREB2 hybrid peptide (SPPDSPEQGPSSPET) constructed to juxtapose the sequences immediately surrounding two putative MAPK phosphorylation sites with phosphate groups chemically added on Ser152 (first underline in the sequence) and Ser237 (second underline) for the phospho-peptide (Mohamed et al., 2005). Binding specificity was characterized in previous studies (Mohamed et al., 2005; Fioravante et al., 2008). Aplysia MKP1 has not been cloned, and an Aplysia-specific antibody is not available. Consequently, we used a rabbit polyclonal antibody raised against a peptide of human origin with a molecular weight of ∼40 kDa from Santa Cruz Biotechnology. In a Western blot analysis of Aplysia nervous tissue (see below and Fig. 4), the antibody recognized a single band at ∼40 kDa. To test the specificity of anti-MKP1 antibody, a blocking peptide specific to anti-MKP1 antibody was purchased from Santa Cruz Biotechnology. The antibody was mixed with a fivefold (by weight) excess of blocking peptide in 500 μl of PBS and incubated for 2 h at room temperature. For each immunofluorescence procedure, two dishes of SNs from the same animal were treated with either anti-MKP1 antibody or antibody mixed with blocking peptide. As an additional negative control, a third dish of SNs from the same animal was treated with Veh. Fluorescence intensity was compared among these three dishes.
Figure 4.

DOX decreased MKP1 levels. A1, Western blot analysis of pleural-pedal ganglia revealed a single band with a molecular weight appropriate for MKP1. A2, Preabsorption analysis of cultured SNs revealed the specificity of anti-MKP1 antibody. BP, Blocking peptide. B, Effect of DOX on MKP1 levels. B1, Representative confocal images of MKP1 immunofluorescence in SNs treated with DOX. Scale bar, 20 μm. B2, Summary data. Treatment for 120 min with 2.5 μm DOX significantly reduced MKP1 levels, whereas a 30-min 10 μm DOX treatment did not.
Secondary antibody (goat anti-rabbit secondary antibody conjugated to Cy-3; 1:200 dilution; The Jackson Laboratory) was applied for 1 h at room temperature. Cells were then mounted using Fluoromount-G (Southern Biotechnology). The intensity of staining in SNs was quantified in images obtained with a Zeiss LSM510 confocal microscope using a 63× oil-immersion lens as described previously (Liu et al., 2011). A z-series of optical sections through the cell body (0.5 μm increments) were taken, and the section through the middle of the nucleus was used for analysis of mean fluorescence intensity with MetaMorph Offline software (Universal Imaging Corporation). Approximately five neurons on each coverslip were analyzed, and measurements from neurons on the same coverslip were averaged. To test the specificity of secondary antibody, two dishes of SNs from the same animal were treated with either anti-MKP1 antibody followed by secondary antibody or anti-MKP1 antibody followed by Veh. Fluorescence intensity was compared between these two dishes.
The intensity of staining in cortical neurons was quantified in images obtained with a Zeiss LSM510 confocal microscope using a 63× oil-immersion lens as described previously (Fukushima et al., 2007). Cortical neurons were randomly chosen for analysis by capturing five images observed during scanning of coverslips from each experimental condition. A z-series of optical sections through the cell body (0.2 μm increments through 2 μm) was taken, and the section through the middle of the nucleus was analyzed using MetaMorph Offline software. These experiments were performed in a blind manner so that the investigator analyzing the images was unaware of the treatment.
Western blot analysis.
Western blot analysis was performed following established procedures (Liu et al., 2008). Pleural-pedal ganglia were lysed 45 min after the end of treatment with Veh or DOX in ice-cold lysis buffer and stored at −20°C until use. Extracts were resolved using SDS-PAGE. Protein samples were transferred to a nitrocellulose membrane. The membrane was probed with anti-pERK antibody (1:200) or anti-MKP1 antibody (1:1000), followed by incubation with peroxidase-conjugated secondary antibody (1:500; Thermo Fisher Scientific). Immunoreactive bands were visualized by ECL (GE Healthcare) and analyzed with ImageQuant 5.0 software.
Electrophysiology.
For LTF and LTD experiments, stimulation of presynaptic SNs from SN–MN cocultures was performed extracellularly using a blunt patch electrode filled with L15/ASW. Intracellular recordings from MNs were made with 10–20 MΩ sharp electrodes filled with 3 m potassium acetate connected to an Axoclamp 2-B amplifier (Molecular Devices). Data acquisition and analyses of resting potential, input resistance, and EPSP amplitude were performed with pClamp 8 software (Molecular Devices). Before measurement of EPSPs, MNs were held at −90 mV by passing constant hyperpolarizing current into the MN. Several measurements were made, including resting potential of the SN and MN, input resistance of the MN, and EPSP amplitude. Cultures were excluded from additional use if pretreatment measurements of EPSP amplitudes were <5 mV, >35 mV, or sufficiently large to trigger an action potential. MNs with resting potentials more positive than −40 mV or input resistances <10 MΩ were also excluded. These measurements were repeated 24 h after treatment. In cases in which the post-test EPSP amplitude was accompanied by an action potential, the EPSP amplitude was assigned a value of 45 mV, which was the largest synaptic potential obtained in our culture system that did not trigger an action potential. For statistical analysis, the amplitudes of the EPSPs 24 h after treatment (post-test) were normalized to the EPSPs measured before treatment (pre-test).
Excitability of SNs was measured using isolated SNs following established protocols (Liu et al., 2011). SNs were impaled with a single microelectrode (10–20 MΩ resistance) and held at −55 mV. Input resistance was measured by applying 0.3 nA of hyperpolarizing current for 1 s. Firing threshold was measured by applying 1 s of depolarizing current in increasing increments of 0.1 nA until an action potential was triggered. The lowest current intensity necessary to trigger a single action potential was considered the firing threshold. When the firing threshold was <0.5 nA, excitability was measured by counting the number of action potentials triggered by applying 0.5 nA of depolarizing current for 1 s. When the firing threshold of SNs was equal to or >0.5 nA but <1.0 nA, 1.0 nA of depolarizing current was used to measure excitability. When the firing threshold was equal to or >1.0 nA but <2.0 nA, 2.0 nA of depolarizing current was used. SNs were excluded from additional use if cells had resting potentials more positive than −40 mV, failed to respond to depolarizing current up to 2.0 nA, or exhibited a drop in input resistance of ≥50% between the pre-test and post-test. Measurements from two to five viable cells in each dish were averaged. The number of samples (n) reported in Results indicate number of dishes. All the above experiments were performed in a blind manner so that the investigator performing the electrophysiology was unaware of the treatment the neurons received.
Statistical analyses.
SigmaPlot version 11 (Systat Software) was used to perform all statistical analyses. Data are presented as means ± SEMs or medians (interquartile range), and p < 0.05 was considered to represent statistical significance.
Results
DOX induced ERK and p38 MAPK activation in cultured Aplysia SNs
In breast epithelial and breast carcinoma cells, DOX represses MKP1 transcription and thereby indirectly increases phosphorylation of ERK (Small et al., 2003; Rojo et al., 2009). As a first step to examine the effects of DOX on isolated nerve cells, immunofluorescence techniques were used to measure pERK levels in isolated Aplysia SNs at several different concentrations and exposure times. SNs were fixed for immunofluorescence 45 min after the start of treatment, a time when a transient increase in pERK is observed after a 5 min pulse of 5-HT (Philips et al., 2013). A 30 min treatment with 10 μm DOX led to a 113 ± 29% (n = 5) increase in levels of pERK compared with Veh controls. This difference was statistically significant (Fig. 1A; paired t test, t(4) = 4.23, p = 0.01). Compared with the corresponding Veh controls, treatment with lower concentrations (2.5 and 5 μm) were without effect. However, treatment with 2.5 μm DOX for a longer period (120 min) increased pERK (Fig. 1A; paired t test, t(9) = 3.91, p = 0.004). We also applied 2.5 μm DOX to pleural-pedal ganglia for 2 h and used Western blot analysis to measure pERK levels. Compared with Veh-treated ganglia, 2.5 μm DOX did not increase pERK in ganglia. However, a 2 h treatment with 10 μm DOX produced a 36 ± 18% increase in pERK (n = 5, paired t test, t(4) = 3.00, p = 0.04). The need for a higher DOX concentration to increase pERK in ganglia may be attributable to the presence of multiple cell types. SNs are only a small portion of the neurons in pleural-pedal ganglia, so the changes occurring specifically in SNs may be obscured in Western blots that use the whole pleural-pedal ganglion. Nonetheless these results indicate that DOX is an effective activator of the ERK cascade.
Figure 1.
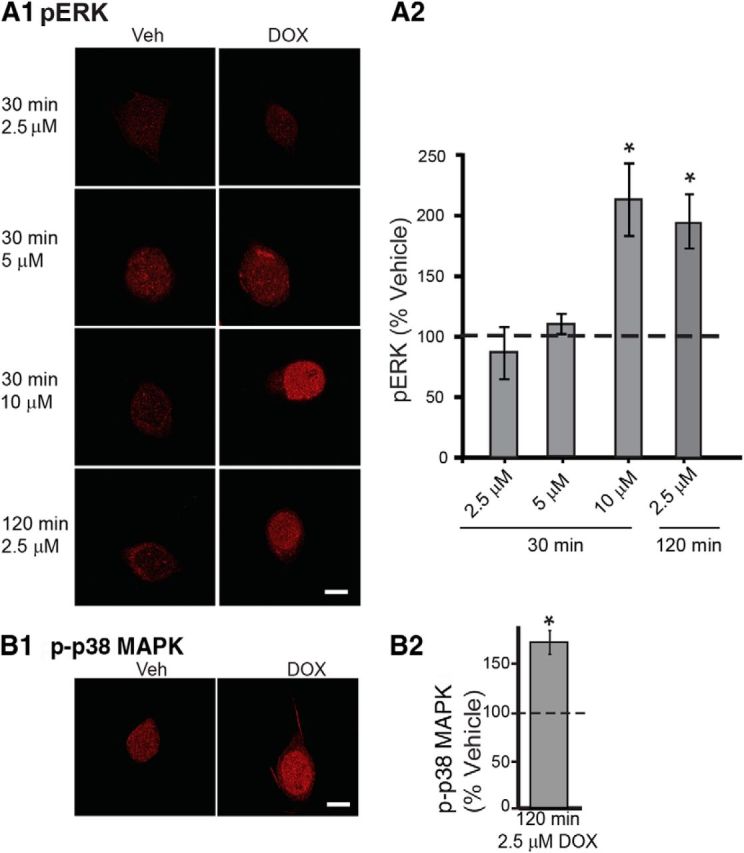
DOX activated ERK and p38 MAPK in cultured SNs. A, ERK activation by DOX is concentration and time dependent. A1, Representative confocal images of pERK immunofluorescence in Aplysia SNs treated with DOX at different concentrations and times. A2, Summary data. Only the 30 min treatment with 10 μm DOX or 120 min treatment with 2.5 μm DOX significantly increased pERK levels. B, p38 MAPK activation by 2.5 μm DOX treatment for 120 min. B1, Representative confocal images of p-p38 MAPK immunofluorescence. B2, Summary data. Treatment for 120 min with 2.5 μm DOX induced a significant increase in p-p38 MAPK. Scale bar, 20 μm. In this and subsequent illustrations, significant differences are indicated by *p < 0.05.
As noted above, DOX represses MKP1 transcription in some types of cells. Both ERK and p38 MAPK are substrates for MKP1 (Jeffrey et al., 2007). To examine whether DOX affects levels of p-p38 MAPK, isolated Aplysia SNs were treated for 120 min with 2.5 μm DOX, a treatment that significantly induced ERK activation (Fig. 1A). At 45 min after treatment, SNs were fixed for immunofluorescence (Fig. 1B). Example responses are illustrated in Figure 1B1, and summary data are presented in Figure 1B2. DOX led to a 73 ± 12% (n = 4) increase in p-p38 MAPK compared with Veh controls (t(3) = 3.81, p = 0.03). These results indicate that DOX is an effective activator of p38 MAPK and ERK in SNs.
DOX induced ERK and p38 MAPK activation in rat cultured cortical neurons
The generality of the finding that DOX leads to the activation of both ERK and p38 MAPK was examined. Rat cortical neurons were exposed to 2.5 μm DOX for 120 min, the same concentration that induced phosphorylation of ERK and p38 MAPK in Aplysia SNs (Fig. 1A). Consistent with previous findings (Lopes et al., 2008), a 120 min treatment with 2.5 μm DOX induced cell death that was detected by light microscopy, including cell shrinkage, nuclear condensation, and fragmentation (data not shown). To avoid DOX-elicited cell death, cortical neurons were exposed to 2.5 μm DOX for only 30 min and fixed for immunofluorescence 45 min after treatment. The immunoreactivity to both pERK and p-p38 MAPK was specifically localized in the nucleus (Fig. 2A). In agreement with the results from Aplysia SNs, a 30 min 2.5 μm DOX exposure significantly increased the immunoreactivity to both pERK [Fig. 2C; Veh, 10.7 ± 0.5 A.U.; DOX, 52.9 ± 1.6 A.U.; paired t test, t(2) = 37.42, p = 0.001) and p-p38 MAPK (Veh, 5.3 ± 1.3 A.U.; DOX, 55.7 ± 2.8 A.U.; paired t test, t(2) = 16.7, p = 0.004). Thus, the effects of DOX generalize to rat cortical neurons, wherein the drug appears more potent than in SNs.
Figure 2.

DOX activated both ERK and p38 MAPK in cultured cortical neurons from rat. A, B, Representative confocal images of pERK and p-p38 MAPK immunofluorescence in cortical neurons treated with 2.5 (A) or 0.25 (B) μm DOX for 30 min. Scale bar, 20 μm. C, Summary data. Treatment for 30 min with 0.25 or 2.5 μm DOX induced a significant increase in both pERK and p-p38 MAPK at 45 min after treatment.
We examined whether DOX is effective at lower concentrations as well. When administered to rats through intraperitoneal injection at a dose of 10 mg/kg, the effective concentration detected in rat cerebral hemispheres is 0.25 μm because of the limited penetration through the blood–brain barrier (Sardi et al., 2013). Therefore, we examined whether ERK and p38 MAPK are activated by 0.25 μm DOX. Despite the 10-fold lower concentration, a 30 min 0.25 μm DOX treatment significantly increased the immunoreactivity to both pERK (Fig. 2B,C; Veh, 13 ± 0.8 A.U.; DOX, 48 ± 8.0 A.U.; paired t test, t(2) = 5.01, p = 0.04) and p-p38 MAPK (Veh, 14 ± 1.4 A.U.; DOX, 43 ± 8.0 A.U.; paired t test, t(2) = 4.41, p = 0.048).
DOX induced CREB2 phosphorylation in cultured Aplysia SNs
Given that DOX leads to activation of both ERK and p38 MAPK (Figs. 1, 2) and that these two MAPKs regulate phosphorylation of CREB2 (Bartsch et al., 1995; Guan et al., 2003; Fioravante et al., 2008), we next examined changes in pCREB2 levels after DOX treatment using immunofluorescence. The anti-pCREB2 antibody was raised against the phosphorylated form of a CREB2 hybrid peptide containing two putative MAPK phosphorylation sites. To determine whether detected changes in pCREB2 are mediated by ERK or p38 MAPK, we also included the specific p38 MAPK inhibitor SB. SNs were treated for 120 min with 2.5 μm DOX or Veh. In a third group, 3 μm SB was applied to cultures, beginning 30 min before adding DOX and continuing through the duration of DOX treatment. A fourth group was treated with SB alone for 150 min. SNs were fixed 45 min after the end of treatment for immunofluorescence. Example responses are illustrated in Figure 3A, and summary data are presented in Figure 3B. DOX alone led to a 30.8 ± 11.8% (n = 9) increase in pCREB2, whereas the SB-alone group showed no change in pCREB2 (−1.7 ± 11.2%; n = 6). The DOX + SB group showed a slight decrease in pCREB2 (−8.8 ± 7.4%; n = 7). A one-way ANOVA revealed a significant overall effect of the treatments (F(3,27) = 3.34, p = 0.03). Subsequent pairwise comparisons revealed a significant difference between the DOX-alone group and the Veh group (q = 3.33, p = 0.03), but no difference was observed between the SB + DOX group and the Veh group (q = 0.89, p = 0.81). Moreover, a significant difference was observed between the DOX-alone group and the DOX + SB group (q = 4.0, p = 0.04), which indicates that the effect of DOX on pCREB2 levels is dependent on activation of p38 MAPK.
Figure 3.

DOX-mediated increases in phosphorylation of CREB2 in cultured SNs. A, Representative confocal images of pCREB2 immunofluorescence in SNs treated with 2.5 μm DOX for 120 min, in the absence or presence of the p38 MAPK inhibitor SB. Scale bar, 20 μm. B, Summary data. Treatment with 2.5 μm DOX for 120 min induced a significant increase in pCREB2, and this increase was blocked by SB.
DOX decreased levels of MKP1
DOX-induced ERK activation could be the result of downregulation of MKP1 expression (Small et al., 2003; Rojo et al., 2009). Therefore, we examined protein levels of MKP1 after DOX treatment using immunofluorescence techniques. To assess the specificity of the MKP1 antibody, Western blot analysis was performed using pleural-pedal ganglia lysate. This antibody recognized a single band (Fig. 4A1) with a molecular weight (∼40 kDa) appropriate for MKP1 (Bokemeyer et al., 1998). A preabsorption test also confirmed the specificity of the MKP1 antibody (Fig. 4A2). In the absence of MKP1 antibody or when MKP1 antibody was added in the presence of blocking peptide, the immunofluorescence intensities of SNs were extremely low. Also in the absence of secondary antibody, the immunofluorescence intensities of SNs were extremely low. In subsequent immunofluorescence analyses, SNs were exposed to 10 μm DOX for 30 min or 2.5 μm DOX for 120 min, treatments that significantly enhanced ERK phosphorylation in previous experiments (Fig. 1). At 45 min after treatment, SNs were fixed for immunofluorescence. As shown in Figure 4, B1 and B2, the 30 min treatment with 10 μm DOX produced a 9 ± 7% (n = 4) increase in MKP1, but this change was not statistically significant (paired t test, t(3) = 1.32, p = 0.28), whereas the 120 min treatment of 2.5 μm DOX induced a modest (−13 ± 9%, n = 8) but significant decrease in MKP1 levels (paired t test, t(7) = 2.66, p = 0.03). Western blot analysis of whole pedal-pleural ganglia revealed that a 2 h treatment with 10 μm DOX produced a weak 9 ± 12% decrease in MKP1 in pleural-pedal ganglia (n = 5), but this effect was not statistically significant (paired t test, t(4) = 1.34, p = 0.25). The increase in 30 min, 10 μm DOX-induced activation of ERK (Fig. 1) in the absence of any detectable effect on MKP1 protein levels indicates that downregulation of MKP1 expression is unlikely to be involved in short-term (30 min) DOX-induced activation of ERK. However, downregulation of MKP1 expression, albeit small, may at least in part contribute to the more prolonged (120 min) DOX-induced ERK activation. The reason for the difference between the effects of the 30 and 120 min treatments may be the time required for MKP1 protein levels to adjust to the decreased MKP1 mRNA levels (Small et al., 2003; Rojo et al., 2009).
DOX reduced 5-HT-induced LTF
Given that DOX leads to activation of both ERK and p38 MAPK and that these kinases have effects on long-term plasticity in Aplysia and other species, we examined the effect of DOX on 5-HT-induced LTF. Presumably, LTF would be attenuated if the effects of p38 MAPK predominate but would be enhanced if the effects of ERK predominate. We first performed an analysis of the relationship between the magnitude of LTF produced by five pulses of 5-HT at different concentrations. This was accomplished to ensure that LTF was in a range that would be most sensitive to possible effects of DOX. Concentrations of 5-HT of 0, 1, 5, 10, and 50 μm were examined (while keeping the duration and interstimulus interval the same; Fig. 5A). Confirming previous results (Alberini et al., 1994; Ghirardi et al., 1995; Sharma et al., 2003a; Hu et al., 2004; Liu et al., 2008; Weatherill et al., 2010), 5-HT produced LTF at all concentrations.
Figure 5.
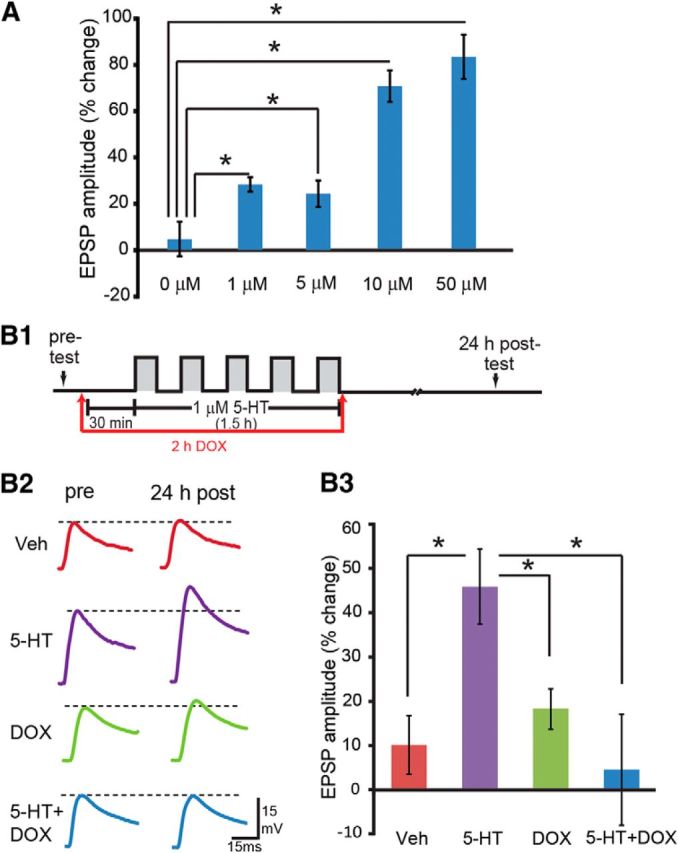
5-HT dose effects for LTF and inhibition of 5-HT-induced LTF by DOX. A, The changes in EPSP amplitude at different concentrations of 5-HT. One, 5, 10, and 50 μm 5-HT-induced LTF compared with Veh (0 μm) control. Significant differences are indicated by *p < 0.05. B, DOX inhibited 5-HT-induced LTF. B1, Protocol for DOX application with 1 μm 5-HT or Veh. B2, Representative EPSPs recorded immediately before (pre-test) and 24 h after (post-test) treatment with 5-HT or Veh. Dashed lines represent the amplitude of the pre-test EPSP. B3, Summary data.
A concentration of 1 μm 5-HT was selected for subsequent experiments. EPSPs were measured immediately before five pulses of 5-HT treatment (pre-test) and 24 h after treatment (post-test) in four groups: (1) Veh; (2) 1 μm 5-HT; (3) 2.5 μm DOX alone; and (4) 5-HT + DOX. As shown in Figure 5B, 5-HT led to a 44.6 ± 8.7% (n = 8) increase in EPSP amplitude. However, the increase in the 5-HT + DOX group was only 4.5 ± 12.2% (n = 9). The increases in the Veh group and the DOX-alone group were 10.0 ± 6.2% (n = 8) and 18.4 ± 4.2% (n = 9), respectively. A one-way ANOVA indicated significant overall differences among the groups (F(3,33) = 4.29, p = 0.01). Post hoc comparisons revealed significant 5-HT-induced LTF (5-HT vs Veh, q = 4.05, p = 0.02). DOX alone appeared to produce a small facilitation compared with the Veh group, but this effect was not statistically significant (Veh vs DOX, q = 1.08, p = 0.45). Importantly, a significant difference was observed between the 5-HT-alone and 5-HT + DOX groups (5-HT vs 5-HT + DOX, q = 4.71, p = 0.01). These results suggest that DOX inhibits LTF.
The attenuation of LTF by DOX could be attributable to a secondary neurotoxic effect (Lopes et al., 2008). This is unlikely, because EPSPs from the DOX-alone group were not significantly different from the Veh group (Fig. 5B). Indeed, they were slightly enhanced. Moreover, DOX (2.5 μm) had no effect on resting potential (percentage change as post/pre: Veh group, 1.33 ± 4.5%, n = 8; DOX group, 1.40 ± 4.5%, n = 9; t test, t(15) = 0.01, p = 0.99) or input resistance (Veh group, median of 16.45 MΩ, n = 8; DOX group, median of 16.7 MΩ, n = 9; Mann–Whitney rank-sum test, U = 29.5, p = 0.56), suggesting that the impairment of LTF by DOX was not attributable to a neurotoxic effect of DOX.
DOX enhanced basal long-term excitability but did not affect 5-HT-induced long-term enhanced excitability (LTEE)
In addition to changes in synaptic strength, 5-HT also produces long-term changes in SN excitability (Dale et al., 1987; Cleary et al., 1998; Liu et al., 2008), which is an independent mechanism for memory storage (Mozzachiodi and Byrne, 2010). Given that ERK is involved in the regulation of neuronal excitability in both vertebrates and invertebrates (Sung et al., 2001; Chin et al., 2006; Rosenkranz et al., 2009; Stamboulian et al., 2010) and that DOX enhances ERK activation, DOX may increase long-term changes in neuronal excitability. To test this hypothesis, we investigated the effect of DOX on five 5-min pulses of 1 μm 5-HT-induced LTEE using isolated SNs. As described in the above LTF experiment, cell excitability was measured immediately before treatment (pre-test) and 24 after treatment (post-test) in four groups: (1) Veh; (2) 1 μm 5-HT; (3) 2.5 μm DOX alone; and (4) 5-HT + DOX (for protocol and representative traces, see Fig. 6A,B1). 5-HT alone led to a 70.9% (54.2–75%) [median (interquartile range)] (n = 6) increase in excitability at 24 h compared with the pre-test. DOX alone increased excitability by 22.9% (3.1–64.6%) (n = 6). The combination of 5-HT + DOX led to a 43.8% (18.8–78.1%) (n = 6) increase in excitability. Veh treatment led to no apparent (0–9.4%) (n = 6) change in excitability (Fig. 6B2). A Kruskal–Wallis one-way ANOVA on ranks revealed a significant difference among the four groups (H = 9.16, df = 3, p = 0.03). Post hoc comparisons revealed that DOX alone produced a significant increase in cell excitability 24 h after treatment compared with Veh treatment (Fig. 6; DOX vs Veh: difference of ranks = 37; q = 4.189, p < 0.05). 5-HT alone also significantly increased excitability 24 h after treatment compared with Veh treatment (5-HT vs Veh: difference of ranks = 71; q = 4.099, p < 0.05). 5-HT-induced LTEE appeared attenuated by DOX, but this effect was not statistically significant (5-HT + DOX vs 5-HT: difference of ranks = 19; q = 2.151, p > 0.05). These results demonstrate that, unlike the findings for LTF, DOX alone induces LTEE but does not significantly affect 5-HT-induced LTEE.
Figure 6.

DOX induced basal LTEE but had no effect on 5-HT-induced LTEE. A, Protocol for DOX application with 5-HT or Veh. B1, Representative action potentials were recorded immediately before (pre-test) and 24 h after (post-test) treatment with 5-HT or Veh. B2, Summary data. DOX induced LTEE but did not affect 5-HT-induced LTEE. Diamond markers represent means.
Effects of DOX on 5-HT-induced activation of ERK and p38 MAPK
The attenuation of LTF by DOX could be attributable to an attenuation of the normal 5-HT-induced activation of ERK necessary for LTF (Martin et al., 1997; Michael et al., 1998; Purcell et al., 2003; Sharma and Carew, 2004) or to p38 MAPK-induced LTD (Guan et al., 2002, 2003) that overrides 5-HT-induced LTF. To examine whether the effect of DOX on pERK levels are affected by 5-HT, SNs were pretreated for 120 min with 2.5 μm DOX, followed by a 5 min treatment with 1 μm 5-HT in the presence of DOX. Cells were fixed for immunofluorescence 45 min after the onset of 5-HT treatment, a time when a transient increase in pERK is observed after a 5 min pulse of 5-HT (Philips et al., 2013). Four groups of experiments were performed: (1) 1 μm 5-HT alone; (2) 2.5 μm DOX; (3) 5-HT + DOX; and (4) Veh alone. Example responses are illustrated in Figure 7A1, and summary data are presented in Figure 7A2. DOX led to a 139 ± 38% (n = 6) increase in pERK, whereas the increase by 5-HT + DOX was 161 ± 41% (n = 6). The 5-HT-alone group showed a slight increase in pERK (15 ± 18%; n = 6). A one-way ANOVA revealed a significant overall effect of the treatments (F(5,18) = 10.79, p = 0.001). Subsequent pairwise comparisons revealed that, as expected, the DOX-alone group was significantly different from the Veh group (q = 5.51, p = 0.004) and the 5-HT + DOX group was significantly different from the Veh group (q = 6.38, p = 0.002). However, there was no significant difference between the DOX-alone group and the 5-HT + DOX group (q = 0.87, p = 0.55), which indicates that the effect of DOX on ERK activation is not affected by 5-HT.
Figure 7.

DOX- and 5-HT-induced changes in pERK and p-p38 MAPK levels. A1, B1, Representative confocal images of pERK immunofluorescence in SNs treated with DOX and/or 5-HT at 1 μm (A1) and 50 μm (B1). Scale bar, 20 μm. C1, Representative confocal images of p-p38 MAPK immunofluorescence in SNs treated with DOX and/or 5-HT at 1 μm. A2, B2, C2, Summary data. Summary data for Veh and DOX treatment in B2 are duplicated from Figure 1A.
Because a single 5 min pulse of 1 μm 5-HT did not significantly increase the phosphorylation of ERK in the preceding experiment, we increased the concentration of 5-HT to 50 μm to test whether the ability of DOX to activate ERK would be attenuated by 5-HT at a higher concentration. Example responses are illustrated in Figure 7B1, and summary data are presented in Figure 7B2. DOX led to a 41 ± 14% (n = 4) increase in pERK, whereas the increase by 5-HT + DOX was 76 ± 16% (n = 4). Consistent with previous results (Philips et al., 2013), the 50 μm 5-HT-alone group showed a significant increase (37 ± 12%; n = 4) in pERK. A one-way ANOVA revealed a significant overall effect of the treatments (F(3,12) = 33.13, p = 0.001). Subsequent pairwise comparisons revealed that, as expected, the DOX-alone group was significantly different from the Veh group (q = 7.60, p = 0.001) and the 5-HT + DOX group was significantly different from the Veh group (q = 14.08, p = 0.001). The difference between the DOX-alone group and the 5-HT + DOX group was statistically significant (q = 6.47, p = 0.001). These results indicate that the effects of DOX and 50 μm 5-HT on ERK are additive. The ability of DOX to activate ERK is not attenuated by 5-HT, regardless of the concentration of 5-HT used, suggesting that the impairment of LTF by DOX is not attributable to an attenuation of the normal 5-HT-induced activation of ERK necessary for LTF.
In Aplysia SNs, 5-HT leads to a decrease of p38 MAPK phosphorylation (Guan et al., 2003), whereas DOX leads to an increase of p38 MAPK phosphorylation (Fig. 1B). To examine whether the effect of DOX on p-p38 MAPK levels is affected by 5-HT, SNs were treated for 120 min with 2.5 μm DOX, followed by a 5 min treatment with 1 μm 5-HT in the presence of DOX. Cells were fixed for immunofluorescence immediately after 5-HT treatment, a time when a transient decrease in p-p38 MAPK is observed after a 10 min pulse of 5-HT (Guan et al., 2003). Four groups of experiments were performed: (1) 1 μm 5-HT alone; (2) 2.5 μm DOX; (3) 5-HT + DOX; and (4) Veh alone. As shown in Figure 7C, DOX led to a 66.9 ± 7.7% (n = 4) increase in p-p38 MAPK, whereas the 5-HT-alone group showed a slight decrease in p-p38 MAPK (−12 ± 1.9%; n = 4). The combined effect of 5-HT + DOX was a 69.8 ± 9.7% increase in p-p38 MAPK (n = 3). A one-way ANOVA revealed a significant overall effect of the treatments (F(3,11) = 55.69, p = 0.001). Subsequent pairwise comparisons revealed that, as expected, the DOX-alone group was significantly different from the Veh group (q = 11.98, p = 0.001) and the 5-HT + DOX group was significantly different from the Veh group (q = 11.56, p = 0.001). Moreover, no significant difference was observed between the DOX-alone group and the 5-HT + DOX group (q = 0.48, p = 0.74), indicating that DOX plays the predominant role in p38 MAPK activity.
Rescue of DOX-induced impairment of LTF
Activation of p38 MAPK alone induces LTD (Guan et al., 2002; Guan et al., 2003). Therefore, we next examined whether the DOX-induced activation of p38 MAPK was responsible for the impairment of LTF by DOX. The experiment of Figure 5 was repeated in the presence of SB. Three groups of SN–MN cocultures were treated with 5-HT. In one group, cocultures were treated with 1 μm 5-HT alone (5-HT, n = 9). In the second group, cocultures were exposed to 2.5 μm DOX 30 min before and during the entire 5-HT treatment (5-HT + DOX, n = 10) to replicate the inhibition of LTF by DOX. In the third group, cocultures were preincubated with 3 μm SB for 30 min before DOX and concurrently with DOX treatment 1 h before and during the entire 5-HT treatment (5-HT + DOX + SB, n = 10). EPSPs were measured immediately before (pre-test) 5-HT treatment and 24 h later (post-test; Fig. 8). Confirming the experiment of Figure 5, LTF was attenuated as a result of pretreatment with DOX. In contrast, LTF was rescued by preexposure to SB. Indeed, although not statistically significant, LTF produced by 5-HT + DOX + SB was somewhat greater than the LTF produced by 5-HT alone. A one-way ANOVA indicated significant overall differences among the treatment groups (F(2,28) = 10.69, p = 0.001). LTF in the 5-HT + DOX group was significantly less than that in the 5-HT group (q = 4.00, p = 0.01). Importantly, LTF in the 5-HT + DOX + SB group was significantly greater than in the 5-HT + DOX group (q = 6.41, p < 0.001). No significant difference in LTF was observed between the 5-HT and 5-HT + DOX + SB groups (q = 1.99, p = 0.17). Therefore, inhibition of p38 MAPK rescued the deficit in LTF induced by DOX.
Figure 8.

SB prevented DOX-induced inhibition of LTF. A, Protocol for SB application. B1, Representative EPSPs recorded immediately before (pre-test) and 24 h after (post-test) treatment with 5-HT. Dashed lines represent the amplitude of the pre-test EPSP. B2, Summary data.
Enhancement by DOX of FMRFa-induced LTD
Aplysia SN–MN synapses undergo bidirectional plasticity. The facilitatory neurotransmitter 5-HT produces LTF, which requires ERK activity, whereas the neuropeptide FMRFa leads to LTD, which requires p38 MAPK activity. Because DOX activates p38 MAPK and the downstream transcriptional inhibitor CREB2 and inhibits LTF, we examined whether DOX would potentiate FMRFa-induced LTD. EPSPs were measured immediately before treatment with five pulses of 10 μm FMRFa (pre-test) and 24 h after treatment (post-test) in three groups: (1) Veh; (2) 10 μm FMRFa; and (3) 10 μm FMRFa + 2.5 μm DOX. FMRFa led to an 8 ± 3% (n = 5) decrease in EPSP amplitude, whereas EPSPs were decreased by 24 ± 5% (n = 5) in the FMRFa + DOX group (Fig. 9). A one-way ANOVA indicated significant overall differences among the three groups (F(2,12) = 11.82, p = 0.001). Post hoc comparisons revealed that FMRFa reduced amplitudes of EPSPs (FMRFa vs Veh, q = 3.02, p = 0.05; FMRFa + DOX vs Veh, q = 6.86, p = 0.001). Importantly, a significant difference was observed between the FMRFa-alone and FMRFa + DOX groups (FMRFa + DOX vs FMRFa, q = 3.84, p = 0.02). These results suggest that DOX enhanced FMRFa-induced LTD.
Figure 9.

DOX enhanced FMRFa-induced LTD. A, Protocol for DOX application with 10 μm FMRFa or Veh. B1, Representative EPSPs recorded immediately before (pre-test) and 24 h after (post-test) treatment with FMRFa or Veh. Dashed lines represent the amplitude of the pre-test EPSPs. B2, Summary data.
Discussion
Activation of ERK and p38 MAPK by DOX in both invertebrate and vertebrate neurons
Treatment with DOX produced an increase in phosphorylation of both ERK and p38 MAPK in isolated SNs of Aplysia and in rat cortical neurons. To our knowledge, this is the first report of DOX affecting these kinases in isolated neurons, although effects have been reported in other types of cells, including neuroblastoma cells (Guise et al., 2001), cardiac cells (Kang et al., 2000; Poizat et al., 2005), lymphoma and lung carcinoma cells (Niiya et al., 2004; Elsea et al., 2008), and breast cancer cells (Small et al., 2003; Rojo et al., 2009). The mechanisms underlying enhanced ERK and p38 MAPK activation are not completely clear. One possibility suggested by the actions of DOX in human cell lines (Small et al., 2003; Rojo et al., 2009) is that activation is attributable to decreased expression of MKP1, thereby leading to a reduction in basal phosphatase activity and an increase in basal phosphorylation of ERK. Indeed, activation of ERK was accompanied by decreased levels of MKP1 in SNs after a 120 min exposure to DOX (Fig. 4B), but this effect was relatively small. In addition, this mechanism does not appear to be sufficient for increasing levels of pERK, because a 30 min treatment of DOX was sufficient to increase pERK (Fig. 1) in the absence of a reduction in levels of MKP1 (Fig. 4B). This conclusion is consistent with the idea that the degradation of MKP1 is relatively slow (Small et al., 2003; Rojo et al., 2009). The relatively weak effects of DOX after a 120 min exposure and the lack of an effect of DOX on MKP1 levels at 30 min may suggest that DOX affects pERK through other mechanisms. For example, DOX might stimulate the phosphorylation of ERK by directly inhibiting MKP1 or other MKPs. Additional studies including the cloning and characterization of the Aplysia MKP1 gene will be required to delineate the mechanism(s) by which DOX affects the ERK and p38 MAPK pathways in the SNs.
ERK and p38 MAPK determine the direction of synaptic plasticity and mediate the effects of DOX on plasticity
MAPK pathways are pivotal for mediating synaptic plasticity by conveying extracellular signals to the nucleus (Adams and Sweatt, 2002). ERK and p38 MAPK mediate opposing forms of synaptic plasticity. In vertebrates and invertebrates, ERK activation is required for LTP and LTF, respectively (English and Sweatt, 1997; Martin et al., 1997; Michael et al., 1998; Coogan et al., 1999; Bolshakov et al., 2000; Kanterewicz et al., 2000; Purcell et al., 2003; Sharma and Carew, 2004). In contrast, p38 MAPK mediates both LTD and short-term synaptic depression (Bolshakov et al., 2000; Guan et al., 2002, 2003; Zhong et al., 2008). Consequently, it appears that the balance of these two MAPKs determines the direction of synaptic plasticity. Because DOX clearly leads to significant activation of ERK and p38 MAPK in the same neuron (Fig. 1), it serves as a useful tool for exploring the consequences of jointly activating these MAPK isoforms for synaptic plasticity.
DOX alone has no effect on synaptic strength because it induces neither LTF nor LTD by itself (Fig. 5). This result may be attributable to simultaneous activation of both ERK and p38 MAPK by DOX with these two MAPKs counteracting each other. However, when DOX is combined with excitatory (e.g., 5-HT) or inhibitory (e.g., FMRFa) neurotransmitters, synaptic plasticity is modulated by DOX. As illustrated in the model scheme of Figure 10, a key site of convergence of ERK and p38 MAPK is the transcriptional repressor CREB2. During 5-HT-induced LTF, protein kinase A (PKA) phosphorylates and activates the transcriptional activator CREB1, whereas ERK phosphorylates CREB2, removing its inhibitory effect. Activation of CREB1 and inactivation of CREB2 together induce the expression of CCAAT/enhancer-binding protein (C/EBP) that is critical for the induction of LTF (Alberini et al., 1994). In contrast, p38 MAPK activates CREB2 by phosphorylation at a different site, thereby inhibiting the transcription of C/EBP (Guan et al., 2002, 2003). 5-HT enhances pERK (Sharma et al., 2003b; Fioravante et al., 2006; Fig. 7B) but inhibits p38 MAPK activity (Guan et al., 2003). However, the application of DOX leads to an increase in both pERK and p-p38 MAPK levels, as well as p38 MAPK-mediated CREB2 phosphorylation (Figs. 1, 3). In the presence of both 5-HT and DOX, it is possible that this increase in CREB2 activity cannot be sufficiently suppressed by the enhanced activation of ERK. Thus, the activation of CREB2 by p38 MAPK dominated any suppression of CREB2 caused by ERK. Therefore, the expression of C/EBP is suppressed and LTF is impaired. When the suppressive effect of p38 MAPK is prevented by an inhibitor of p38 MAPK, LTF is restored (Fig. 8). This model is consistent with previous work by Guan et al. (2002, 2003), who observed that simultaneous application of 5-HT and FMRFa results in LTD.
Figure 10.

Schematic of a model for action of DOX within key signaling pathways necessary for LTF. The model is adapted and simplified from the previous model of Liu et al. (2013) with the addition of the p38 MAPK pathway and the effects of DOX. p38 MAPK suppresses LTF via CREB2 activation. ERK plays a key role in the induction of LTF via inactivation of CREB2. 5-HT activates PKA and ERK and inhibits p38 MAPK, which in turn favors the expression of C/EBP and induction of LTF. DOX has dual effects on synaptic plasticity. It leads to activation of both p38 MAPK and ERK, at least in part, by inhibiting the expression of MKP1. The balance between p38 MAPK and ERK activation determines whether CREB2 is sufficiently inactivated for the expression of C/EBP. Arrows represent activation; balls represent inhibition.
Although DOX alone has no significant effects on basal synaptic strength, it enhances FMRFa-induced LTD (Fig. 9). As shown in the model in Figure 10, FMRFa can enhance p38 MAPK activation (Guan et al., 2003). In addition, it reduces the phosphorylation of ERK via p38 MAPK (Fioravante et al., 2006). When FMRFa is applied to neurons, activation of p38 MAPK inhibits ERK phosphorylation and activates CREB2, leading to LTD. When FMRFa and DOX are applied to neurons, the combined effects of these treatments might induce a synergistic activation of p38 MAPK. The highly activated p38 MAPK may not only suppress ERK activation induced by DOX but also reduce pERK to a level even lower than when FMRFa alone is applied. Therefore, LTD is enhanced.
DOX has no effect on 5-HT-induced LTEE
LTEE is a form of plasticity that is also induced by 5-HT (Dale et al., 1987; Cleary et al., 1998; Liu et al., 2008; Mozzachiodi and Byrne, 2010). DOX alone produced a long-term change in excitability (Fig. 6) but had no effect on synaptic strength (Fig. 5). This observation is in agreement with the finding that changes in short-term excitability have a lower threshold than changes in short-term facilitation (Emptage and Carew, 1993). In addition, although DOX significantly attenuated LTF, the attenuation of 5-HT-induced LTEE in the presence of DOX was not statistically significant (Fig. 6). These differences could be attributable to divergent roles of ERK and p38 MAPK, including different downstream effectors, in LTF and LTEE.
DOX-induced activation of p38 MAPK could contribute to chemotherapy-associated cognitive dysfunction
Although our study focused on LTF in Aplysia, the results may provide some insights into the deleterious side effects on learning and memory associated with the use of DOX in cancer chemotherapy (Wefel et al., 2010; Jansen et al., 2011). DOX also leads to memory deficits in animals (Seigers and Fardell, 2011). For example, rats show impairments on an inhibitory avoidance task 24 h after a single dose of DOX administration (Liedke et al., 2009) and have significantly impaired performance on novel place recognition and contextual fear conditioning tasks, suggesting disrupted hippocampal-based memory function (Christie et al., 2012).
A variety of mechanisms have been suggested to account for the effects of DOX on cognitive function, often referred to as chemobrain. They include neurotoxicity, decreased neurogenesis, cytokine-induced inflammatory responses, and oxidative stress (Jansen et al., 2005; Ahles and Saykin, 2007). Our findings that DOX activates both ERK and p38 MAPK pathways in Aplysia neurons and rat cortical neurons and that acute exposure to DOX leads to inhibition of LTF and enhancement of LTD provide support for an alternative, although not necessarily mutually exclusive, hypothesis to explain long-term cognitive deficits associated with DOX treatment. Moreover, our finding that LTF can be restored by p38 MAPK inhibition suggests a pharmacological approach that may ameliorate those cognitive deficits.
Footnotes
The research was supported by National Institutes of Health Grant NS019895 and the Zilkha Family Discovery Fellowship. We thank D. Baxter and P. Smolen for comments on previous drafts of the manuscript, E. Kartikaningrum and J. Liu for preparing the Aplysia cultures, and A. Tsvetkov for preparing the cultured cortical neurons.
References
- Adams JP, Sweatt JD. Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol. 2002;42:135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401. [DOI] [PubMed] [Google Scholar]
- Ahles TA, Saykin AJ. Candidate mechanisms for chemotherapy-induced cognitive changes. Nat Rev Cancer. 2007;7:192–201. doi: 10.1038/nrc2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini CM, Ghirardi M, Metz R, Kandel ER. C/EBP is an immediate-early gene required for the consolidation of long-term facilitation in Aplysia. Cell. 1994;76:1099–1114. doi: 10.1016/0092-8674(94)90386-7. [DOI] [PubMed] [Google Scholar]
- Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, Kandel ER. Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell. 1995;83:979–992. doi: 10.1016/0092-8674(95)90213-9. [DOI] [PubMed] [Google Scholar]
- Bokemeyer D, Lindemann M, Kramer HJ. Regulation of mitogen-activated protein kinase phosphatase-1 in vascular smooth muscle cells. Hypertension. 1998;32:661–667. doi: 10.1161/01.HYP.32.4.661. [DOI] [PubMed] [Google Scholar]
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Carboni L, Cobb MH, Siegelbaum SA, Belardetti F. Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nat Neurosci. 2000;3:1107–1112. doi: 10.1038/80624. [DOI] [PubMed] [Google Scholar]
- Chin J, Angers A, Cleary LJ, Eskin A, Byrne JH. TGF-beta1 in Aplysia: role in long-term changes in the excitability of sensory neurons and distribution of TbetaR-II-like immunoreactivity. Learn Mem. 1999;6:317–330. [PMC free article] [PubMed] [Google Scholar]
- Chin J, Liu RY, Cleary LJ, Eskin A, Byrne JH. TGF-beta1-induced long-term changes in neuronal excitability in Aplysia sensory neurons depend on MAPK. J Neurophysiol. 2006;95:3286–3290. doi: 10.1152/jn.00770.2005. [DOI] [PubMed] [Google Scholar]
- Christie LA, Acharya MM, Parihar VK, Nguyen A, Martirosian V, Limoli CL. Impaired cognitive function and hippocampal neurogenesis following cancer chemotherapy. Clin Cancer Res. 2012;18:1954–1965. doi: 10.1158/1078-0432.CCR-11-2000. [DOI] [PubMed] [Google Scholar]
- Cleary LJ, Lee WL, Byrne JH. Cellular correlates of long-term sensitization in Aplysia. J Neurosci. 1998;18:5988–5998. doi: 10.1523/JNEUROSCI.18-15-05988.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coogan AN, O'Leary DM, O'Connor JJ. P42/44 MAP kinase inhibitor PD98059 attenuates multiple forms of synaptic plasticity in rat dentate gyrus in vitro. J Neurophysiol. 1999;81:103–110. doi: 10.1152/jn.1999.81.1.103. [DOI] [PubMed] [Google Scholar]
- Dale N, Kandel ER, Schacher S. Serotonin produces long-term changes in the excitability of Aplysia sensory neurons in culture that depend on new protein synthesis. J Neurosci. 1987;7:2232–2238. doi: 10.1523/JNEUROSCI.07-07-02232.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci U S A. 2007;104:4176–4181. doi: 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsea CR, Roberts DA, Druker BJ, Wood LJ. Inhibition of p38 MAPK suppresses inflammatory cytokine induction by etoposide, 5-fluorouracil, and doxorubicin without affecting tumoricidal activity. PLoS One. 2008;3:e2355. doi: 10.1371/journal.pone.0002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emptage NJ, Carew TJ. Long-term synaptic facilitation in the absence of short-term facilitation in Aplysia neurons. Science. 1993;262:253–256. doi: 10.1126/science.8211146. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Fioravante D, Smolen PD, Byrne JH. The 5-HT- and FMRFa-activated signaling pathways interact at the level of the Erk MAPK cascade: potential inhibitory constraints on memory formation. Neurosci Lett. 2006;396:235–240. doi: 10.1016/j.neulet.2005.11.036. [DOI] [PubMed] [Google Scholar]
- Fioravante D, Liu RY, Byrne JH. The ubiquitin-proteasome system is necessary for long-term synaptic depression in Aplysia. J Neurosci. 2008;28:10245–10256. doi: 10.1523/JNEUROSCI.2139-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima T, Liu RY, Byrne JH. Transforming growth factor-beta2 modulates synaptic efficacy and plasticity and induces phosphorylation of CREB in hippocampal neurons. Hippocampus. 2007;17:5–9. doi: 10.1002/hipo.20243. [DOI] [PubMed] [Google Scholar]
- Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57:727–741. doi: 10.1016/S0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- Ghirardi M, Montarolo PG, Kandel ER. A novel intermediate stage in the transition between short- and long-term facilitation in the sensory to motor neuron synapse of Aplysia. Neuron. 1995;14:413–420. doi: 10.1016/0896-6273(95)90297-X. [DOI] [PubMed] [Google Scholar]
- Guan Z, Giustetto M, Lomvardas S, Kim JH, Miniaci MC, Schwartz JH, Thanos D, Kandel ER. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/S0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Guan Z, Kim JH, Lomvardas S, Holick K, Xu S, Kandel ER, Schwartz JH. p38 MAP kinase mediates both short-term and long-term synaptic depression in Aplysia. J Neurosci. 2003;23:7317–7325. doi: 10.1523/JNEUROSCI.23-19-07317.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guise S, Braguer D, Carles G, Delacourte A, Briand C. Hyperphosphorylation of tau is mediated by ERK activation during anticancer drug-induced apoptosis in neuroblastoma cells. J Neurosci Res. 2001;63:257–267. doi: 10.1002/1097-4547(20010201)63:3<257::AID-JNR1019>3.0.CO%3B2-T. [DOI] [PubMed] [Google Scholar]
- Hu JY, Glickman L, Wu F, Schacher S. Serotonin regulates the secretion and autocrine action of a neuropeptide to activate MAPK required for long-term facilitation in Aplysia. Neuron. 2004;43:373–385. doi: 10.1016/j.neuron.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Jansen CE, Cooper BA, Dodd MJ, Miaskowski CA. A prospective longitudinal study of chemotherapy-induced cognitive changes in breast cancer patients. Supportive Care Cancer. 2011;19:1647–1656. doi: 10.1007/s00520-010-0997-4. [DOI] [PubMed] [Google Scholar]
- Jansen C, Miaskowski C, Dodd M, Dowling G, Kramer J. Potential mechanisms for chemotherapy-induced impairments in cognitive function. Oncol Nurs Forum. 2005;32:1151–1163. doi: 10.1188/05.ONF.1151-1163. [DOI] [PubMed] [Google Scholar]
- Jeffrey KL, Camps M, Rommel C, Mackay CR. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov. 2007;6:391–403. doi: 10.1038/nrd2289. [DOI] [PubMed] [Google Scholar]
- Kang YJ, Zhou ZX, Wang GW, Buridi A, Klein JB. Suppression by metallothionein of doxorubicin-induced cardiomyocyte apoptosis through inhibition of p38 mitogen-activated protein kinases. J Biol Chem. 2000;275:13690–13698. doi: 10.1074/jbc.275.18.13690. [DOI] [PubMed] [Google Scholar]
- Kanterewicz BI, Urban NN, McMahon DB, Norman ED, Giffen LJ, Favata MF, Scherle PA, Trzskos JM, Barrionuevo G, Klann E. The extracellular signal-regulated kinase cascade is required for NMDA receptor-independent LTP in area CA1 but not area CA3 of the hippocampus. J Neurosci. 2000;20:3057–3066. doi: 10.1523/JNEUROSCI.20-09-03057.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YC, Huang CC, Hsu KS. A role of p38 mitogen-activated protein kinase in adenosine A(1) receptor-mediated synaptic depotentiation in area CA1 of the rat hippocampus. Mol Brain. 2008;1:13. doi: 10.1186/1756-6606-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedke PE, Reolon GK, Kilpp B, Brunetto AL, Roesler R, Schwartsmann G. Systemic administration of doxorubicin impairs aversively motivated memory in rats. Pharmacol Biochem Behav. 2009;94:239–243. doi: 10.1016/j.pbb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Liu RY, Fioravante D, Shah S, Byrne JH. cAMP response element-binding protein 1 feedback loop is necessary for consolidation of long-term synaptic facilitation in Aplysia. J Neurosci. 2008;28:1970–1976. doi: 10.1523/JNEUROSCI.3848-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RY, Cleary LJ, Byrne JH. The requirement for enhanced CREB1 expression in consolidation of long-term synaptic facilitation and long-term excitability in sensory neurons of Aplysia. J Neurosci. 2011;31:6871–6879. doi: 10.1523/JNEUROSCI.5071-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RY, Zhang Y, Baxter DA, Smolen P, Cleary LJ, Byrne JH. Deficit in long-term synaptic plasticity is rescued by a computationally predicted stimulus protocol. J Neurosci. 2013;33:6944–6949. doi: 10.1523/JNEUROSCI.0643-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes MA, Meisel A, Dirnagl U, Carvalho FD, Bastos Mde L. Doxorubicin induces biphasic neurotoxicity to rat cortical neurons. Neurotoxicology. 2008;29:286–293. doi: 10.1016/j.neuro.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/S0896-6273(00)80330-X. [DOI] [PubMed] [Google Scholar]
- Michael D, Martin KC, Seger R, Ning MM, Baston R, Kandel ER. Repeated pulses of serotonin required for long-term facilitation activate mitogen-activated protein kinase in sensory neurons of Aplysia. Proc Natl Acad Sci U S A. 1998;95:1864–1869. doi: 10.1073/pnas.95.4.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- Mohamed HA, Yao W, Fioravante D, Smolen PD, Byrne JH. cAMP-response elements in Aplysia creb1, creb2, and Ap-uch promoters: implications for feedback loops modulating long term memory. J Biol Chem. 2005;280:27035–27043. doi: 10.1074/jbc.M502541200. [DOI] [PubMed] [Google Scholar]
- Mozzachiodi R, Byrne JH. More than synaptic plasticity: role of nonsynaptic plasticity in learning and memory. Trends Neurosci. 2010;33:17–26. doi: 10.1016/j.tins.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray HJ, O'Connor JJ. A role for COX-2 and p38 mitogen activated protein kinase in long-term depression in the rat dentate gyrus in vitro. Neuropharmacology. 2003;44:374–380. doi: 10.1016/S0028-3908(02)00375-1. [DOI] [PubMed] [Google Scholar]
- Myers JS. Chemotherapy-related cognitive impairment. Clin J Oncol Nurs. 2009;13:413–421. doi: 10.1188/09.CJON.413-421. [DOI] [PubMed] [Google Scholar]
- Niiya M, Niiya K, Shibakura M, Asaumi N, Yoshida C, Shinagawa K, Teshima T, Ishimaru F, Ikeda K, Tanimoto M. Involvement of ERK1/2 and p38 MAP kinase in doxorubicin-induced uPA expression in human RC-K8 lymphoma and NCI-H69 small cell lung carcinoma cells. Oncology. 2004;67:310–319. doi: 10.1159/000081332. [DOI] [PubMed] [Google Scholar]
- Ormond J, Hislop J, Zhao Y, Webb N, Vaillaincourt F, Dyer JR, Ferraro G, Barker P, Martin KC, Sossin WS. ApTrkl, a Trk-like receptor, mediates serotonin- dependent ERK activation and long-term facilitation in Aplysia sensory neurons. Neuron. 2004;44:715–728. doi: 10.1016/j.neuron.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Philips GT, Ye X, Kopec AM, Carew TJ. MAPK establishes a molecular context that defines effective training patterns for long-term memory formation. J Neurosci. 2013;33:7565–7573. doi: 10.1523/JNEUROSCI.5561-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poizat C, Puri PL, Bai Y, Kedes L. Phosphorylation-dependent degradation of p300 by doxorubicin-activated p38 mitogen-activated protein kinase in cardiac cells. Mol Cell Biol. 2005;25:2673–2687. doi: 10.1128/MCB.25.7.2673-2687.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DM, Wloka MT, Chik CL, Ho AK. Mitogen-activated protein kinase phosphatase-1 (MKP-1) preferentially dephosphorylates p42/44MAPK but not p38MAPK in rat pinealocytes. J Neurochem. 2007;101:1685–1693. doi: 10.1111/j.1471-4159.2007.04557.x. [DOI] [PubMed] [Google Scholar]
- Purcell AL, Sharma SK, Bagnall MW, Sutton MA, Carew TJ. Activation of a tyrosine kinase-MAPK cascade enhances the induction of long-term synaptic facilitation and long-term memory in Aplysia. Neuron. 2003;37:473–484. doi: 10.1016/S0896-6273(03)00030-8. [DOI] [PubMed] [Google Scholar]
- Rojo F, González-Navarrete I, Bragado R, Dalmases A, Menéndez S, Cortes-Sempere M, Suárez C, Oliva C, Servitja S, Rodriguez-Fanjul V, Sánchez-Pérez I, Campas C, Corominas JM, Tusquets I, Bellosillo B, Serrano S, Perona R, Rovira A, Albanell J. Mitogen-activated protein kinase phosphatase-1 in human breast cancer independently predicts prognosis and is repressed by doxorubicin. Clin Cancer Res. 2009;15:3530–3539. doi: 10.1158/1078-0432.CCR-08-2070. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Frick A, Johnston D. Kinase-dependent modification of dendritic excitability after long-term potentiation. J Physiol. 2009;587:115–125. doi: 10.1113/jphysiol.2008.158816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardi I, la Marca G, Cardellicchio S, Giunti L, Malvagia S, Genitori L, Massimino M, de Martino M, Giovannini MG. Pharmacological modulation of blood-brain barrier increases permeability of doxorubicin into the rat brain. Am J Cancer Res. 2013;3:424–432. [PMC free article] [PubMed] [Google Scholar]
- Seigers R, Fardell JE. Neurobiological basis of chemotherapy-induced cognitive impairment: a review of rodent research. Neuroscience and biobehavioral reviews. 2011;35:729–741. doi: 10.1016/j.neubiorev.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Carew TJ. The roles of MAPK cascades in synaptic plasticity and memory in Aplysia: facilitatory effects and inhibitory constraints. Learning and memory. 2004;11:373–378. doi: 10.1101/lm.81104. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Bagnall MW, Sutton MA, Carew TJ. Inhibition of calcineurin facilitates the induction of memory for sensitization in Aplysia: requirement of mitogen-activated protein kinase. Proc Natl Acad Sci U S A. 2003a;100:4861–4866. doi: 10.1073/pnas.0830994100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SK, Sherff CM, Shobe J, Bagnall MW, Sutton MA, Carew TJ. Differential role of mitogen-activated protein kinase in three distinct phases of memory for sensitization in Aplysia. J Neurosci. 2003b;23:3899–3907. doi: 10.1523/JNEUROSCI.23-09-03899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small GW, Somasundaram S, Moore DT, Shi YY, Orlowski RZ. Repression of mitogen-activated protein kinase (MAPK) phosphatase-1 by anthracyclines contributes to their antiapoptotic activation of p44/42-MAPK. J Pharmacol Exp Ther. 2003;307:861–869. doi: 10.1124/jpet.103.055806. [DOI] [PubMed] [Google Scholar]
- Stamboulian S, Choi JS, Ahn HS, Chang YW, Tyrrell L, Black JA, Waxman SG, Dib-Hajj SD. ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel Na(v)1.7 and alters its gating properties. J Neurosci. 2010;30:1637–1647. doi: 10.1523/JNEUROSCI.4872-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung YJ, Povelones M, Ambron RT. RISK-1: a novel MAPK homologue in axoplasm that is activated and retrogradely transported after nerve injury. J Neurobiol. 2001;47:67–79. doi: 10.1002/neu.1016. [DOI] [PubMed] [Google Scholar]
- Tsvetkov AS, Arrasate M, Barmada S, Ando DM, Sharma P, Shaby BA, Finkbeiner S. Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nature chemical biology. 2013;9:586–592. doi: 10.1038/nchembio.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherill DB, Dyer J, Sossin WS. Ribosomal protein S6 kinase is a critical downstream effector of the target of rapamycin complex 1 for long-term facilitation in Aplysia. J Biol Chem. 2010;285:12255–12267. doi: 10.1074/jbc.M109.071142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wefel JS, Saleeba AK, Buzdar AU, Meyers CA. Acute and late onset cognitive dysfunction associated with chemotherapy in women with breast cancer. Cancer. 2010;116:3348–3356. doi: 10.1002/cncr.25098. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu RY, Heberton GA, Smolen P, Baxter DA, Cleary LJ, Byrne JH. Computational design of enhanced learning protocols. Nat Neurosci. 2012;15:294–297. doi: 10.1038/nn.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong P, Liu W, Gu Z, Yan Z. Serotonin facilitates long-term depression induction in prefrontal cortex via p38 MAPK/Rab5-mediated enhancement of AMPA receptor internalization. J Physiol. 2008;586:4465–4479. doi: 10.1113/jphysiol.2008.155143. [DOI] [PMC free article] [PubMed] [Google Scholar]