

Abstract
Retinitis pigmentosa (RP) is an inherited neurodegenerative disease involving progressive vision loss, and is often linked to mutations in the rhodopsin gene. Mutations that abolish N-terminal glycosylation of rhodopsin (T4K and T17M) cause sector RP in which the inferior retina preferentially degenerates, possibly due to greater light exposure of this region. Transgenic animal models expressing rhodopsin glycosylation mutants also exhibit light exacerbated retinal degeneration (RD). In this study, we used transgenic Xenopus laevis to investigate the pathogenic mechanism connecting light exposure and RD in photoreceptors expressing T4K or T17M rhodopsin. We demonstrate that increasing the thermal stability of these rhodopsins via a novel disulfide bond resulted in significantly less RD. Furthermore, T4K or T17M rhodopsins that were constitutively inactive (due to lack of the chromophore-binding site or dietary deprivation of the chromophore precursor vitamin A) induced less toxicity. In contrast, variants in the active conformation accumulated in the ER and caused RD even in the absence of light. In vitro, T4K and T17M rhodopsins showed reduced ability to regenerate pigment after light exposure. Finally, although multiple amino acid substitutions of T4 abolished glycosylation at N2 but were not toxic, similar substitutions of T17 were not tolerated, suggesting that the carbohydrate moiety at N15 is critical for cell viability. Our results identify a novel pathogenic mechanism in which the glycosylation-deficient rhodopsins are destabilized by light activation. These results have important implications for proposed RP therapies, such as vitamin A supplementation, which may be ineffective or even detrimental for certain RP genotypes.
Keywords: cell death, glycosylation, instability, misfolding, retinitis pigmentosa, rhodopsin
Introduction
Retinitis pigmentosa (RP) is one of the most common inherited retinopathies and involves progressive loss of rod and cone photoreceptor neurons. Rods are primarily affected, resulting in poor night and peripheral vision, with subsequent cone death and loss of central vision. Mutations of the gene encoding the G-protein-coupled receptor rhodopsin cause ∼27% of autosomal dominant RP (Sullivan et al., 2006). Rhodopsin consists of a protein (rod opsin) and a chromophore ligand (11-cis-retinal) and initiates phototransduction in a specialized organelle called the rod outer segment (OS). Upon photon capture, 11-cis-retinal isomerizes to all-trans resulting in a shift in rhodopsin's conformation and binding to the G-protein transducin (Ridge and Palczewski, 2007). Elucidating the mechanisms underlying the >100 identified RP-causing mutations will increase our understanding of rhodopsin structure and function and facilitate development of therapeutic strategies.
Several mutations located in rhodopsin's N terminus (T4K, T17M and P23H) cause light-exacerbated retinal degeneration (RD) in transgenic animals (Tam and Moritz, 2007, 2009) and sector RP in humans (Heckenlively et al., 1991; Fishman et al., 1992; Sullivan et al., 1993; Li et al., 1994; van den Born et al., 1994). This region is dually glycosylated at N2 and N15 (Hargrave, 1977) and forms part of a multilayer structure that covers the chromophore-binding pocket (Bourne and Meng, 2000; Palczewski, 2000). The structural integrity of the N-terminus is sensitive to changes in amino acid sequence surrounding N2 and possibly dependent on the presence of the carbohydrate moiety itself at N15 (Tam and Moritz, 2009). In transgenic animals, P23H rhodopsin mislocalizes to inner segments (IS) and expresses at low levels (Roof et al., 1994; Tam and Moritz, 2006, 2007; Price et al., 2011; Sakami et al., 2011) and in cultured cells induces ER stress (Chiang et al., 2012), suggesting protein misfolding and degradation. In contrast, T4K and T17M rhodopsins express at high levels, exhibit little or no misfolding and localize primarily to the OS in transgenic frog rods suggesting that ER stress may not play a major role in their pathology. Furthermore, mutations T4K and T17M but not P23H eliminate N-linked glycosylation at either N2 or N15. Thus, the glycosylation-deficient mutants likely differ from P23H in their toxic etiology.
We hypothesize that rod death is associated with light activation of mutant T4K and T17M opsins rather than defective biosynthesis. In this study, we examined transgenic retinas under conditions that promoted either constitutive activation or inactivation to identify aspects of rhodopsin activation relevant to the disease process. Constitutively active or inactive T4K and T17M opsins were produced using a molecular approach in which the ligand-binding site was mutated. Inactive mutant opsins were also generated by dietary depletion of the chromophore. We differentiated between toxicity based on structural instability versus rhodopsin activity by examining the mutants in the context of a thermally stabilized rhodopsin. Structural stability was assessed in vitro by the ability to regenerate photopigment following bleaching. Finally, we further examined the requirements of the peptide sequences surrounding N2 and N15.
Materials and Methods
Molecular biology.
Xenopus laevis transgene constructs were based on XOP0.8-eGFP-N1 (Tam et al., 2006) in which the green fluorescent protein cDNA was replaced with wild-type (WT) or mutant human rhodopsin cDNAs. Expression constructs for transfection of cultured cells were based on the pMT3 plasmid (Franke et al., 1988). WT and mutant cDNAs were cloned into the EcoRI and NotI sites of both expression plasmids. T4I and N2C mutations were introduced into oligonucleotides used for PCR amplification of rhodopsin cDNAs. All other point mutations (T17V, K296E, K296R, and N282C) were introduced via PCR mutagenesis (Nelson and Long, 1989) or the Quikchange Site Directed Mutagenesis Kit (Agilent). Human T4K and T17M rhodopsin expression vectors were previously described (Tam and Moritz, 2009). Double- and triple-mutant constructs were produced by ligating together fragments containing the individual mutations. All mutant cDNAs were verified by DNA sequencing. Transgene constructs were linearized with FseI (New England Biolabs) and purified using the QIAquick Gel Extraction Kit (Qiagen) for integration into sperm nuclei.
Generation and rearing of primary transgenic X. laevis.
Transgenic X. laevis tadpoles were generated by the methods previously described (Kroll and Amaya, 1996; Tam et al., 2006). Briefly, linearized expression plasmids are incubated with permeabilized sperm. This mixture was then treated with X. laevis egg extract and restriction enzyme and subsequently injected into unfertilized eggs. Resulting normally developed embryos of either sex were housed in 4 L tanks in an 18°C incubator either in complete darkness or on a 12 h light cycle. When lit, the average light intensity inside the incubator was 1700 lux. Unless otherwise stated, embryos were exposed to 20 μg/ml G418 (∼24 h after fertilization) for 4–5 d to select for transgenic tadpoles (Moritz et al., 2002). Tadpoles were fed ground frog brittle (Nasco) when they reached swimming stages. At 14 d postfertilization (dpf) corresponding to developmental stage 48, normally developed X. laevis were killed and one eye was fixed in 4% paraformaldehyde buffered with 0.1 m sodium phosphate pH7.4, while the contralateral eye was solubilized in 100 μl of a 1:1 mixture of PBS and SDS-PAGE loading buffer containing 1 mm EDTA and 100 μg/ml PMSF as previously described. All experiments were conducted in adherence to the Statement for the Use of Animals in Ophthalmic and Visual Research.
Vitamin A deprivation.
Animals used in vitamin A deprivation experiments were f1 offspring of mature transgenic founders (either line T4KF1 or T4KM3). Rearing conditions are as described above except for feeding. Tadpoles were fed food either containing (5LP3) or lacking (5B8V) vitamin A acetate and carotene (TestDiet).
Dot-blot analysis.
Dot blots of X. laevis eye extracts were performed as previously described (Tam et al., 2006) with slight modifications. Aliquots of solubilized eyes were diluted 1:300 in 20 mm NaPO4 buffer, pH7.4, and drawn through Immobilon P membrane. After washing and drying the membrane, duplicate dot blots were probed with primary antibodies mAb B630N (Adamus et al., 1991) at 1:10 dilution of tissue culture supernatant and mAb A5-3 (Adamus et al., 1991) at 1:5 dilution of tissue culture supernatant or mAb 1D4 (MacKenzie et al., 1984; UBC-UILO) at 1:750 dilution of 1 mg/ml solution followed by IR-dye800-conjugated goat anti-mouse antibody at 1:10,000 of 1 mg/ml solution (LI-COR Biosciences). Blots were imaged and quantified using the LI-COR Odyssey imaging system. Each dot blot included standards containing 100% X. laevis rhodopsin (from WT retinas) and 100% human WT rhodopsin (from transfected, cultured cells). The relative affinities of mAb B630N (recognizes both endogenous X. laevis and transgenic human rhodopsin) and mAb A5-3 or mAb1D4 label (recognizes transgenic human rhodopsin but not X. laevis rhodopsin) were determined from the control samples and used to calculate the percentage transgenic rhodopsin expression levels. Standard curves derived from serial dilutions of control samples were used to determine the linear range of both antibodies. Plots of expression level versus total rhodopsin were fit to dose–response curves using SigmaPlot (Systat Software). Nonparametric statistical methods were used for data analysis of experiments using primary transgenic animals due to non-normal (i.e., skewed and bimodal) data distributions. Nonparametric multiple-comparisons tests were performed as described by Conover (1999). Parametric statistical methods were used for data analysis of experiments using f1 animals.
Immunohistochemistry and confocal microscopy.
Within 48 h, fixed eyes were infiltrated in 20% sucrose and embedded in OCT compound (Sakura Finetek). For anti-rhodopsin labeling, 12 μm frozen sections were labeled overnight with mAb 2B2 (MacKenzie et al., 1984) cell culture supernatant at 1:10 dilution, followed by 1:750 dilution of Cy3-conjugated secondary antibody (Jackson Immunoresearch) and counterstained with AlexaFluor 488-conjugated wheat germ agglutinin (WGA; Life Technologies) and Hoechst 33342 (Sigma-Aldrich) as previously described (Moritz et al., 1999). MAb 2B2 recognizes the N-terminus of mammalian but not frog rhodopsin. For anti-BiP labeling, the same protocol was used except Ab32618 (Abcam) was used at 1:200 dilution. Labeled sections were mounted with MOWIOL 4-88 Reagent (EMD Millipore). Sections were imaged using a Zeiss 510 laser scanning confocal microscope using either LSM510 or Zen 2009 software and 10× (Zeiss, plan-neofluar, NA 0.3) or 40× (Zeiss, C-apochromat, NA 1.2) objectives. Postmicroscopy image processing was performed using Adobe Photoshop. The brightness and contrast of fluorescent signals derived from antibody labeling were adjusted linearly (i.e., no changes to gamma settings) to preserve the relative intensities from different parts of the cell. However, signals derived from WGA and Hoechst 33342 staining were adjusted in a nonlinear manner to best represent the architecture of the retina. For quantitative BiP imaging, all microscope settings were held constant and all images were collected in a single session. BiP signal was quantified using ImageJ software in 1.5 × 2.0 μm regions of rod ISs immediately adjacent to the nucleus (consistent with the location of ER in X. laevis rod photoreceptors) selected in unprocessed images using the WGA and DAPI channels as guides (Schneider et al., 2012). Animals derived from the F1 T4K rhodopsin line (transgenic or their WT siblings) were used for each condition and measurements from five rods in two labeled sections were combined to give a single measurement. Results were analyzed by two-way ANOVA using n = 4 per condition.
In vitro rhodopsin regeneration.
pMT3 expression vectors containing human WT or mutant rhodopsin cDNAs were transiently transfected into COS-1 cells using the DEAE-dextran method (Oprian, 1993; McKee et al., 2007). On the third day after transfection, cells expressing WT or T4K opsin were harvested, centrifuged, and stored at −80°C until used. All subsequent steps were conducted under dim red light conditions. Pigments were generated by resuspending the cell pellets in 5 ml 10 mm sodium phosphate, 150 mm NaCl, pH 7.0, containing 20 μm 11-cis-retinal (provided by Dr R. Crouch, MUSC) and incubated at least 1 h at 4°C. The cells were then solubilized in 1% dodecyl maltoside by adding an equal volume of 2% dodecyl maltoside in the resuspension buffer. After 1 h, samples were centrifuged and the supernatant incubated with 1D4-Sepharose immunoaffinity beads. This was extensively washed with 0.1% dodecyl maltoside, 10 mm sodium phosphate, 150 mm NaCl, and the pigment was eluted with a peptide corresponding to the last 9 aa residues of rhodopsin dissolved in the wash buffer. Cells expressing T17M opsin were cultured in the presence of 10 μm 11-cis-retinal to increase the yield of folded pigment (Li et al., 1998). All steps were performed under dim red light conditions. Twenty-four and 48 h after transfection the media was replaced with media containing 10 μm 11-cis-retinal. As before, cells were harvested on the third day after transfection and the pigment was purified using the same protocol as for WT and T4K rhodopsins. Absorption spectra of purified rhodopsins were acquired on a Varian-Cary 300 spectrophotometer modified by the manufacturer for dark room use. Sample temperature was regulated by a temperature controlled cuvette holder from Quantum Northwest. A 5 to tenfold molar excess of 11-cis-retinal was added to the sample before bleaching. After recording an initial spectrum, samples were then exposed to >495 nm light for 10 s and several spectra were recorded.
Rhodopsin thermal stability assay.
pMT3 plasmids containing human WT or N2C/N282C opsin cDNAs were transiently transfected into HEK293 cells using Lipofectamine (Life Technologies). After 12 h, cell culture medium was changed. All subsequent steps were performed in a dark room under red light. Twenty-micromoles of 11-cis-retinal were added to the cells and incubated at 37°C for 48 h. Cells were then harvested, washed, and resuspended in ice-cold PBS (10 mm sodium phosphate and 130 mm NaCl, pH 7.2). The cells were lysed in PBSD (PBS containing 1.0% dodecyl maltoside and protease inhibitor mixture) for 1 h and spun to remove cell debris. The supernatant was collected, and incubated with 1D4-sepharose immunoaffinity beads for 1 h at 4°C. The beads were washed three times with cold PBSD (without protease inhibitor) and then two times with 10 mm sodium phosphate buffer, pH 6, containing 0.1% dodecyl maltoside. Rhodopsin was eluted from the beads with elution buffer (a competing peptide corresponding to the last 18 aa of rhodopsin in the same buffer). Absorption spectra of purified rhodopsins were acquired on a Varian-Cary 50 spectrophotometer. Samples were heated to 59°C in a temperature controlled cuvette holder from Thermo Scientific, Peltier. Spectra were obtained at 500 nm at 2 min intervals for a total of 32 min. The entire experiment was performed twice and the average results are presented.
Ethical approval.
The procedures used in the animal studies described here have been approved by the University of British Columbia Animal Care Committee
Results
Inactive unliganded T4K and T17M opsins are less toxic than their rhodopsin counterparts
Expression of T4K or T17M rhodopsin in transgenic X. laevis rods causes significant RD in animals exposed to light, but not in those reared in complete darkness (Tam and Moritz, 2009) This suggests that the cytotoxic effects are due to a consequence of activation of either the mutant or endogenous rhodopsin, or possibly a secondary effect of prolonged dark rearing, such as downregulation of rhodopsin expression (Hollyfield and Anderson, 1982). To determine whether RD is directly mediated by activation of the mutant rhodopsin, we created double mutants in which T4K or T17M was combined with K296R. K296 is the site of chromophore attachment and mutation to R296 both prevents binding of 11-cis-retinal and locks opsin into an inactive conformation (Cohen et al., 1992). Primary transgenic X. laevis expressing WT, T17M, K296R, or T17M/K296R opsins (n = 22 per group) were reared in cyclic light for 14 d before quantitative dot-blot analysis of opsin from solubilized retinas and histological analysis of contralateral eyes.
Expression of either human WT rhodopsin or K296R opsin was well tolerated by transgenic X. laevis rods (Fig. 1A). Total rod opsin levels remained relatively constant at all transgene expression levels, indicating that expression of WT or K296R opsins did not cause RD. However, even low-level expression of T17M rhodopsin (∼2% of total rod opsin) was associated with a large decrease in total rod opsin, indicating that this mutant was toxic and caused RD. In comparison, T17M/K296R was much less toxic, requiring >10-fold higher expression (∼20%) to induce loss of total rod opsin. On average, total rod opsin levels in retinas expressing T17M/K296R rhodopsin were more than double the levels of total rod opsin in retinas expressing T17M rhodopsin (p = 1.2 × 10−5, Kruskal–Wallace test followed by multiple comparisons) even though mean expression levels were comparable between the two groups (p = 0.73, Mann–Whitney U test). Altogether, these results indicate substantial rescue from RD when the T17M mutant was constitutively inactivated.
Figure 1.
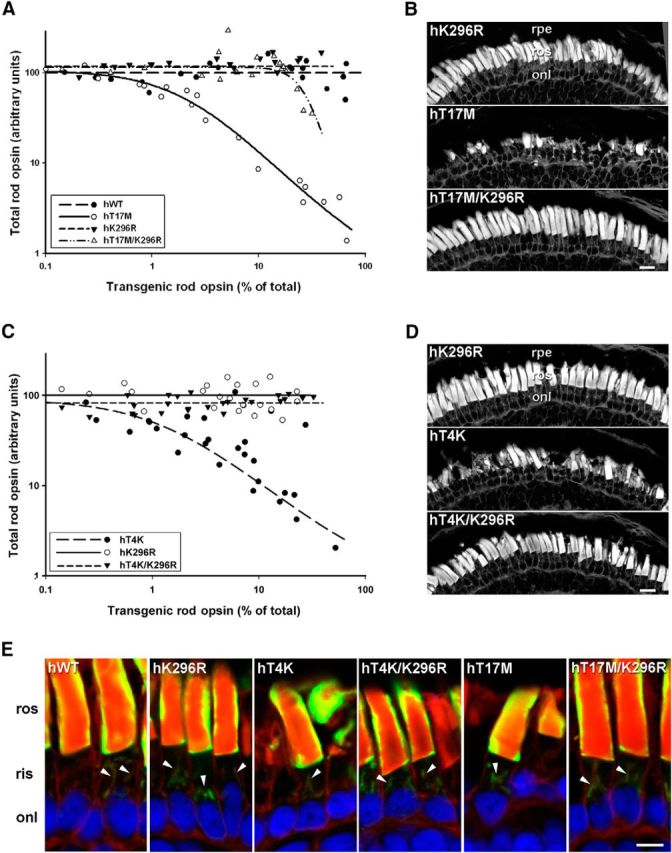
Inactive T4K and T17M opsins exhibited reduced rod toxicity. A, C, Plots of transgenic opsin expression levels versus total rod opsin levels from primary transgenic tadpoles expressing (A) hWT, hT17M, hK296R, or hT17M/K296R rhodopsins (n = 22 per group) or (C) hT4K, hK296R, or hT4K/K296R rhodopsins (n = 29 per group). B, D, Confocal micrographs of cryosections from transgenic retinas stained with WGA. Retinas expressing hK296R, hT17M/K296R, or hT4K/K296R opsin appeared healthy with numerous long closely packed ROS. Retinas expressing hT17M or hT4K rhodopsin exhibited loss or shortening of rod OS. E, Confocal micrographs of retinal cryosections labeled with mAb 2B2 (green) and counterstained with WGA (red) and Hoechst nuclear stain (blue). All mutants (T4K, T17M and K296R, T4K/K296R, and T17M/K296R) localized to the rod OS and Golgi (arrowheads) in a pattern indistinguishable from WT. rpe, Retinal pigment epithelium; ros, rod outer segment; onl, outer nuclear layer. Scale bars: B, D, 20 μm; E, 10 μm.
A similar experiment was conducted comparing T4K, K296R and T4K/K296R rhodopsins (n = 29). Expression of T4K rhodopsin caused RD at low expression levels (<5%). However, constitutively inactive T4K/K296R did not cause RD even at the highest expression levels obtained (>30% of total rod opsin; (Fig. 1C). Mean expression levels were not significantly different (p = 0.23, Mann–Whitney U test) but the average total rod opsin level of retinas expressing T4K/K296R was greater than double that of retinas expressing T4K opsin (p = 7.8 × 10−11, Kruskal–Wallace test followed by multiple comparisons). To confirm that the reduced rod opsin signals observed in dot blots were due to RD, we examined retinal cryosections from contralateral eyes by confocal microscopy. In retinas expressing K296R, T4K/K296R, or T17M/K296R opsins, rod OS were long and densely packed, whereas retinas expressing T4K and T17M rhodopsin clearly exhibited shortening and loss of OS (Fig. 1B,D). Both rhodopsin single (T4K, T17M, and K296R) and double mutants (T4K/K296R and T17M/K296R) localized to the rod OS and Golgi in a pattern indistinguishable from WT (Fig. 1E). Anti-rhodopsin labeling of X. laevis rods typically presents as a “halo” around the OS periphery (due to poor penetration into the OS interior resulting from the extremely high epitope concentration in the tightly packed disk membranes). Collectively, our results demonstrate that inactive, unliganded mutant T4K and T17M opsins are much less toxic than photoactivatable T4K and T17M rhodopsins.
Vitamin A deprivation prevents light-dependent RD and does not adversely affect T4K opsin biosynthesis
11-cis-retinal acts as a pharmacological chaperone to enhance folding of certain mutant opsins such as P23H in vitro (Noorwez et al., 2004). Furthermore, absence of chromophore results in decreased delivery of P23H opsin to the rod OS and exacerbates RD in transgenic X. laevis (Tam et al., 2010). However, based on the data presented above, we hypothesized that vitamin A deprivation would have the opposite effect (i.e., rescue rod cell death) in retinas expressing T4K opsin, because lack of chromophore also prevents light-induced activation of the mutant protein. We therefore performed an experiment using f1 tadpoles expressing T4K rhodopsin (line F1). For this line, T4K rhodopsin expression was determined by quantitative dot blot to be ∼27% of total rod opsin. Transgenic tapdoles and nontransgenic littermates (present at a 1:1 ratio) were reared in constant darkness for 3 weeks, during which half of the animals received a diet containing vitamin A (vitA+) and the other half received a diet deficient in vitamin A (vitA−). After 3 weeks of dark rearing, a subset of animals were killed for analysis, and the remainder were exposed to cyclic light for 7 d (while continuing on their respective diets) before also being killed for analysis by quantitative dot blot or confocal microscopy. In 3-week-old dark-reared transgenic animals expressing T4K opsin, total rod opsin levels of vitA-retinas were slightly higher than but not significantly different from vitA+ retinas (p = 0.064, Student's t test), and retinal morphology appeared normal in both groups (Fig. 2A,D). In addition, T4K opsin expression was not significantly different between vitA− eyes and vitA+ eyes (p = 0.12, Student's t test). Thus, vitamin A deprivation had no adverse effects on photoreceptor viability or on opsin biosynthesis in general.
Figure 2.

Vitamin A deprivation prevents T4K rhodopsin induced retinal degeneration. F1 tadpoles were dark reared for 3 weeks and then exposed to 12 h cyclic light for 1 week. During this time, tadpoles were either fed a diet with (vitA+) or without (vitA−) vitamin A. A–C, Total rod opsin levels in animals after 3 weeks of dark rearing (line F1, A), or after a subsequent week of 12 h cyclic light exposure (line F1, B; line M3, C) were determined by dot-blot analysis of solubilized retinas. The number of animals analyzed per group is indicated on the graphs. D–F, Confocal micrographs of retinal sections stained with WGA. Both dark-reared vitA+ and vitA− retinas exhibit long and closely apposed rod OS (D). However, after light exposure, the vitA+ group exhibited obvious loss of rod OS, whereas vitA− group remained healthy (line F1, E; line M3, F). rpe, Retinal pigment epithelium; ros, rod outer segment; onl, outer nuclear layer. Scale bar, 20 μm. Error bars indicate SEM.
As expected, exposure to 7 d of cyclic light (following dark rearing) induced severe RD in transgenic animals expressing T4K opsin that were fed the vitA+ diet. Total rod opsin levels of transgenic retinas (n = 5) dropped to 17% of their nontransgenic littermates (n = 5). In stark contrast, retinas from transgenic animals fed the vitA− diet (n = 5) did not exhibit any signs of RD. Total rod opsin levels remained high (128% of nontransgenic littermates, n = 5) and retinal structure appeared normal and healthy (Fig. 2B,E). The results were analyzed by two-way ANOVA and indicated a highly significant interaction between diet and genotype (p = 1.9 × 10−8), such that vitamin A deprivation dramatically prevented RD.
This experiment was repeated with a second line of transgenic animals (line M3), and similar results were obtained. Retinas from transgenic tadpoles fed the vitA+ diet exhibited RD and contained only 48% as much total rod opsin as their nontransgenic littermates. Retinas that were vitamin A-deprived appeared normal and had 113% as much total rod opsin compared with their nontransgenic littermates (Fig. 2C,F). Again, vitamin A deprivation resulted in a highly significant rescue from RD (p = 5.3 × 10−8, two-way ANOVA). It should be noted that the low percentage of nontransgenic offspring in line M3 experiment is due to the presence of three independent transgene integration sites in the founder. Nonetheless, the transgenic groups are likely similar in composition due to large n values and random assignment of tadpoles to the dietary groups. Collectively, these results indicate that vitamin A deprivation prevented light-induced RD in transgenic X. laevis expressing T4K opsin.
T4K and T17M opsins in the active conformation mislocalize to the IS and induce RD in dark-reared animals
If activation of T4K or T17M rhodopsin is cytotoxic, rendering the mutant opsins constitutively active should induce RD under any lighting conditions and abolish rescue by dark rearing. To test this hypothesis, we created double mutant opsins in which T4K or T17M mutations were combined with the K296E mutation. WT rhodopsin is normally constrained in its inactive state by a salt bridge between K296 and Q113. In K296E opsin, the salt bridge is lost and the mutant protein adopts its active conformation even in the absence of light and chromophore (Robinson et al., 1992). In two separate experiments, primary transgenic X. laevis expressing T4K, K296E, or T4K/K296E opsins (n = 19 per group) and T17M, K296E, or T17M/K296E (n = 14 per group) were reared in constant dark for 14 d before quantitative dot-blot and histological analysis.
Because K296E opsins are predicted to be constitutively phosphorylated and bound to arrestin (Li et al., 1995), we did not use mAb 1D4 for quantification due to potential epitope masking. Instead, we used mAb A5-3, which also recognizes only the transgenic human rod opsin and not the endogenous X. laevis rod opsin, but is less sensitive than mAb 1D4. Interestingly, although T4K, T17M, and K296E opsins all produced signals significantly above background and were expressed at levels up to 36, 29, and 27% of total rod opsin respectively, T4K/K296E and T17M/K296E expression levels were below the limit of detection of our dot-blot assay.
The average total rod opsin in T4K/K296E expressing retinas was significantly lower than that of T4K or K296E expressing retinas (p < 0.0012, Kruskal–Wallace test followed by multiple comparisons; Fig. 3A). Similar results were obtained for T17M/K296E-expressing retinas (p < 0.02; (Fig. 3B). Confocal microscopy of retinal cryosections demonstrated that T4K, T17M and K296E retinas from dark-reared animals appeared healthy with tightly packed OS (Fig. 3C,D). In contrast, T4K/K296E and T17M/K296E retinas displayed clear signs of RD, including OS shortening and loss. These results demonstrate that the active conformations of T4K and T17M opsins were deleterious to rods and abolished the protective effects of dark rearing. Note that although the K296E mutant is associated with RP in humans, it did not by itself cause significant RD in these experiments. It is likely that RD would be observed at longer time points, or at higher expression levels (Chen et al., 2006).
Figure 3.

The active conformation of T4K and T17M is unstable and retained in the ER. Transgenic animals were reared in constant dark for 14 d. A, B, Total rod opsin levels obtained from quantitative dot-blot analysis of solubilized retinal extracts from primary transgenic tadpoles expressing T4K, T4K/K296E, or K296E (n = 19; A) or T17M, T17M/K296E, or K296E (n = 14; (B). C, D, Confocal micrographs of transgenic retinas stained with WGA. Both T4K/K296E and T17M/K296E expressing retinas exhibited loss of ROS, whereas T4K, T17M, and K296E expressing retinas contained densely packed rod OS. E, Confocal micrographs of retinal cryosections labeled with mAb 2B2 (green) and counterstained with WGA (red) and Hoechst nuclear stain (blue). K296E opsin localized primarily to ROS and Golgi (arrowheads), whereas both T4K/K296E and T17M/K296E opsins were largely retained in the RIS in an ER-like distribution. Occasional bands of mutant opsin (arrows) were observed in the rod OS of T4K/K296E but not T17M/K296E expressing retinas. rpe, Retinal pigment epithelium; ros, rod outer segment; ris, rod inner segment; onl, outer nuclear layer. Scale bars: C, D, 20 μm; E, 10 μm. Error bars indicate SEM.
The very low expression levels of T4K/K296E and T17M/K296E opsins suggested that these double mutants may be misfolded. We therefore examined retinal cryosections of transgenic retinas labeled with mAb 2B2 by confocal microscopy (Fig. 3E). T17M/K296E exhibited almost exclusive IS intracellular localization. Occasionally, very faint bands were observed in OS. T4K/K296E was also retained in the IS, but with more frequent and brighter labeling of OS bands. The lack of the halo effect in the OS is indicative of relatively low transgenic opsin concentrations thus allowing penetration of the antibody into the interior of the OS. These distributions were similar to those previously observed with P23H rhodopsins (Tam and Moritz, 2006, 2007), and suggest that the active conformation of T4K and T17M opsins were unable to pass ER quality control and were misfolded and degraded. In contrast, K296E opsin had a cellular distribution typical of WT rhodopsin (Fig. 3E), indicating that the active conformation per se is not responsible for the altered localization of T4K/K296E and T17M/K296E. Furthermore, the inactive K296R, T4K/K296R, and T17M/K296R opsins were highly expressed and also had normal cellular distributions (Fig. 1E) showing that a lysine at position 296 is not a requirement for opsin biosynthesis.
Photoactivation of T4K and T17M rhodopsins results in loss of structural integrity in the OS both in vitro and in vivo
Engineering the K296E double mutants allowed us to use the rod's ER quality control system to reveal the instability of the active conformations of T4K and T17M opsins. However, rhodopsin must normally be in an inactive conformation during biosynthesis and trafficking since the single T4K and T17M rhodopsins correctly localize to OS and are not characterized by ER accumulation. We therefore examined whether activation of the RP mutants causes protein instability subsequent to the biosynthetic pathway. In rod OS disk membranes, opsin goes through repeated cycles of light activation and regeneration with chromophore before ultimately being phagocytosed by the retinal pigment epithelium. We therefore examined the ability of WT and mutant opsins to regenerate a photopigment as an in vitro measure of protein stability. WT, T4K and T17M opsins were expressed in cell culture, reconstituted with 11-cis-retinal either during (T17M) or after culturing (WT and T4K) but before solubilization and purified. The yield of T17M pigment was much lower than WT and T4K if 11-cis-retinal was not available during opsin biosynthesis, indicating reduced stability of T17M opsin in cultured cells, as previously reported (Sung et al., 1991). The starting concentrations of WT, T4K and T17M rhodopsins used in the assays were 2.0, 0.49, and 0.63 μm respectively. After being bleached with >495 nm light, ∼80% of WT opsin regenerated within 1 h in the presence of excess 11-cis-retinal (Fig. 4A), and this process could be continued for multiple successive bleaches (data not shown). However, there was no detectable regeneration of T4K or T17M rhodopsin under similar conditions (Fig. 4A). These results indicate that whereas WT opsin can cycle between its active and inactive conformations multiple times, both T4K and T17M opsins are rapidly destabilized by activation and are unable to return to a native conformation.
Figure 4.
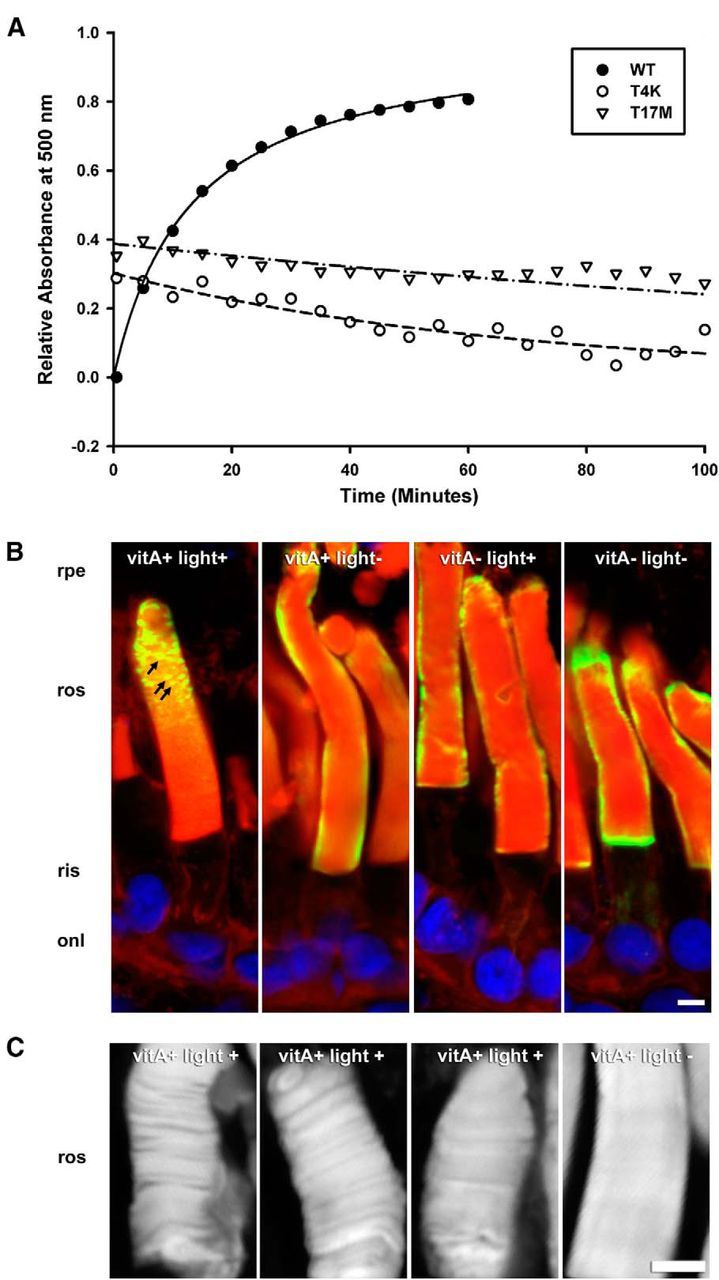
T4K and T17M rhodopsins exhibit instability in vitro and in vivo. A, Regeneration of heterologously expressed hWT, hT4K, and hT17M pigment. A500 measurements were obtained after >495 nm bleach of purified reconstituted pigments. B, C, Confocal micrographs of cryosections from transgenic retinas expressing hT4K opsin labeled with mAb 2B2 (green) and counterstained with WGA (red) and Hoechst nuclear stain (blue; B) or stained with WGA alone (C). Under conditions promoting inactive opsin (vitA− and/or light−), hT4K opsin was uniformly detected at the periphery of the rod OS as is typical for hWT opsin. However, under conditions favoring rhodopsin activation (vitA+ and light+), hT4K opsin labeling frequently appeared punctate (B, arrows). Similarly, WGA labeling of rod OS was smooth and uniform in the absence of light but nonhomogeous indicating disruption of the disk stacks in the presence of light (C). rpe, Retinal pigment epithelium; ros, rod outer segments; onl, outer nuclear layer. Scale bar, 5 μm.
We also investigated whether there was any indication of protein instability in vivo. We compared mutant T4K opsin localization in rods of animals fed either vitA+ or vitA− diets (line F1). In light-exposed retinas of tadpoles fed the vitA+ diet, T4K opsin labeling in OS was often punctate in nature (Fig. 4B, arrows), suggesting aggregation of the mutant protein. However, in vitA+ fed dark-reared retinas, puncta were not observed, nor was punctate labeling a feature of either light exposed or dark-reared vitamin A-deprived rods. Regardless of nutritional and lighting regimen, T4K opsin exhibited OS localization and did not accumulate in the ER (Fig. 4B). Thus, conditions that promote rhodopsin activation (i.e., light and presence of chromophore) also promoted T4K opsin aggregation. Interestingly, loss of disk structure was also observed under these conditions (Fig. 4C). In control eyes not exposed to light, WGA labeling of OS was uniform indicating tight and regular packing of disk membranes. However, when exposed to light, many retinas exhibited rods in which the disk membranes appeared disorganized with gaps between segments of adjacent disks. It is important to note that the OS T4K opsin aggregates and the loss of OS disk structure are much more prevalent when the animals are dark reared for 3 weeks before light exposure (Fig. 4B) in comparison to those which are exposed to cyclic light from the time of fertilization (Fig. 1E). It is possible that these phenotypes are more severe in the prolonged dark rearing paradigm due to the acute and synchronous light activation of the large pool of mutant rhodopsin.
Thermal stabilization of T4K and T17M rhodopsin partially prevents RD
We hypothesized that if protein instability underlies the toxicity of T4K and T17M rhodopsin, then enhancing the thermal stability of the mutant proteins would protect rods from RD. A genetically engineered bovine rhodopsin (in which amino acids 2 and 282 are mutated to cysteines resulting in formation of a novel C2-C282 disulfide bond between the N-terminus and the third extracellular loop) is more thermally stable than WT rhodopsin, while retaining light-dependent transducin activation capability similar to WT (Xie et al., 2003; Standfuss et al., 2007). We created a similar human rhodopsin cDNA containing N2C and N282C mutations. Mutant and WT opsins were expressed in cell culture, regenerated with 11-cis-retinal and purified in the dark to yield 2.1 and 2.2 μm reconstituted pigment, respectively. The protein preparations were incubated at 59°C and their spectra at 500 nm were obtained at 2 min intervals (Fig. 5A). After 32 min, 91% of WT rhodopsin underwent thermal denaturation, compared with only 15% of N2C/N282C rhodopsin. Thus, like its bovine counterpart, human N2C/N282C rhodopsin is also more thermally stable than WT.
Figure 5.

Increasing the thermal stability of hT4K and hT17M opsins reduces their toxicity. A, Thermal decay of recombinant hWT and hN2C/N282C rhodopsins at 59°C. B, C, Plots of transgenic opsin expression levels versus total rod opsin levels from primary transgenic tadpoles expressing human WT, N2C/N282C, T17M, or N2C/T17M/N282C rhodopsins (B; n = 22 per group) or T4K or N2C/T4K/N282C rhodopsins (C; n = 16 per group). D–F, Confocal micrographs of cryosections from transgenic retinas stained with WGA. Retinas expressing hWT and hN2C/N282C exhibited long densely packed rod OS (D). Retinas expressing hT4K hT17M showed signs of severe degeneration, which were greatly reduced in hN2C/T4K/N282C and hN2C/T17M/N282C retinas, respectively (E, F). G–I, Confocal micrographs of cryosections from transgenic retinas expressing hN2C/N282C (G), hN2C/T17M/N282C (H) or hN2C/T4K/N822C (I) labeled with mAb 2B2 (green) and counterstained with WGA (red) and Hoechst nuclear stain (blue). Stabilized rhodopsins localized primarily to the rod OS. rpe, Retinal pigment epithelium; ros, rod outer segment; ris, rod inner segment; onl, outer nuclear layer. Scale bars: D–F, 20 μm; G–I, 5 μm.
We then placed the mutations T4K or T17M on the N2C/N282C rhodopsin backbone. We generated primary transgenic X. laevis expressing WT, N2C/N282C, T17M, or N2C/T17M/N282C rhodopsins (n = 22 per group) and reared the animals in cyclic light for 14 d before quantitative dot blot and histological analysis. Rods expressed N2C/N282C rhodopsin at a range of levels similar to WT and the transgenic protein was well tolerated, as there was no trend toward loss of total rod opsin at higher transgene expression levels in either group (Fig. 5B). In contrast, T17M rhodopsin caused clear loss of total rod opsin even at expression levels <2% of total rod opsin (Fig. 5B). Expression of N2C/T17M/N282C rhodopsin also caused loss of total rod opsin, but this required approximately an order of magnitude higher expression levels than T17M rhodopsin. The average total rod opsin content of retinas expressing T17M opsin was 43% lower than that of retinas expressing N2C/T17M/N282C (p = 0.013, Kruskal–Wallace test followed by multiple comparisons). In a second experiment, primary transgenic tadpoles expressing T4K or N2C/T4K/N282C (n = 16) were generated and analyzed. Once again, substantially more N2C/T4K/N282C rhodopsin was required to induce RD than T4K rhodopsin (Fig. 5C). The average total rod opsin of retinas expressing hT4K was 58% lower than that of retinas expressing the stabilized form (p = 0.0012, Mann–Whitney U test). Confocal microscopy of retinal cryosections confirmed the lack of RD in retinas expressing WT or N2C/N282C rhodopsins (Fig. 5D) as well as the differences in severity of RD between retinas expressing the stabilized versus nonstabilized T4K and T17M rhodopsins (Fig. 5E,F). Introduction of the two cysteine residues into the stabilized rhodopsins had no noticeable effects on biosynthesis or trafficking because both the range of expression levels and cellular localization of the stabilized rhodopsins (Fig. 5G–I) were similar to WT.
BiP is not upregulated in light-exposed T4K rhodopsin-expressing rod photoreceptors
Cell death associated with the expression of mutant rhodopsins, such as P23H and T17M has been proposed to involve the ER stress response (Lin et al., 2007; Kunte et al., 2012). We did not observe ER retention of the glycosylation-deficient rhodopsins suggesting that protein misfolding is not a major contributing factor to pathogenesis. Nonetheless, to determine whether the light-induced cell death mechanism that we observed is distinct from processes involving ER stress, we looked for upregulation of BiP, also known as GRP78 (a marker for ER stress), in light-exposed rod photoreceptors expressing T4K rhodopsin. Animals derived from a transgenic line (line F1) were used rather than primary transgenics to ensure a uniform phenotype. Transgenic animals exposed to 48 or 72 h of 12 h cyclic light were compared with WT animals reared under the same lighting regimen and to dark-reared transgenic animals. After 48 and 72 h of 12 h cyclic light, retinal degeneration had begun as judged by altered nuclear morphology and shortened or missing rod OS. Retinal cryosections were labeled with an anti-BiP antibody and analyzed by quantitative confocal microscopy (Fig. 6). Results were analyzed by two-way ANOVA (time vs rearing/genotype), which indicated a significant difference between the three rearing/genotype conditions (p = 0.019) that was not time point-dependent (p = 0.645) A post hoc Dunnett's test indicated BiP levels in rods of light-exposed T4K animals were indistinguishable from WT (Dunnett's test, p = 0.991). These results suggest that ER stress does not play a significant role in T4K rhodopsin induced RD. However, there was a small but significant decrease in BiP expression in rods of T4K animals reared in darkness relative to WT (p = 0.023). BiP downregulation in dark-reared T4K animals was likely due to lighting conditions rather than the transgene.
Figure 6.

BiP is not upregulated in light-exposed T4K-expressing rod photoreceptors relative to WT. F1 T4K-rhodopsin expressing tadpoles or their WT siblings were dark-reared for 2 weeks and then exposed to 48 or 72 h of 12 h cyclic light (i.e., 2 or 3 light cycles). Cryosections were labeled with anti-BiP, and imaged by confocal microscopy. The integrated intensity of the BiP signal within rod inner segment regions selected using WGA and DAPI counterstains as guides (left) was determined and averaged over n = 4 animals per condition. The autofluorescence associated with outer segments was excluded from the analysis. Two-way ANOVA indicated a significant difference in BiP levels between the three rearing/genotype conditions (p = 0.019). This effect was not time point-dependent (p = 0.645), and therefore results of the two time points are combined in the plot shown (n = 8 per group). Post hoc Dunnett's test indicated the difference is due to lower BiP levels in the T4K dark-reared group relative to the WT cyclic light group (p = 0.023), with no significant difference between the T4K and WT cyclic light groups (p = 0.991). Error bars indicate SEM.
Amino acid substitutions tolerated at T4 are not tolerated at T17
We have previously demonstrated that although glycosylation at N2 is not essential for rhodopsin biosynthesis or trafficking, altering the secondary structure of the extreme N-terminus can result in reduced rod cell viability. Rhodopsins containing amino substitutions N2S and T4V did not cause RD, whereas T4K and T4N did (Tam and Moritz, 2009). In contrast, both of the mutations previously examined affecting rhodopsin's second glycosylation site (N15S and T17M) were toxic to rod cells. Thus, the existing evidence suggests that the carbohydrate moiety itself is required at N15. Threonine, valine, and isoleucine are Cβ branched amino acids that prefer to reside in β-strands, whereas asparagine, methionine, and lysine prefer α helices. Thus, we further probed the preference for β strand conformation at the extreme N-terminus by examining T4I rhodopsin. We also examined T17V rhodopsin to determine whether a similar tolerance for valine exists at rhodopsin's second glycosylation site. Primary transgenic X. laevis expressing WT, T4I, T17M, or T17V rhodopsin (n = 20 per group) were reared in cyclic light for 14 d before quantitative dot blot and histological analysis. Expression of both WT and T4I rhodopsin was well tolerated by rods; total opsin levels remained high at all transgenic opsin expression levels (Fig. 7A). In contrast, increasing expression of T17V rhodopsin correlated with loss of total rhodopsin. This result is similar to that obtained with T17M opsin. On average, total opsin levels in eyes expressing T17V and T17M rhodopsin were reduced 65 and 62% respectively (p = 7.0 × 10−9 and p = 2.8 × 10−9, Kruskal–Wallace test followed by multiple comparisons) compared with eyes expressing WT opsin. Confocal images of retinal cryosections from contralateral eyes confirmed the dot blot results. While T4I retinas appeared similar to WT retinas, T17V retinas had significant RD, including loss and shortening of rod OS (Fig. 7B). Both T4I and T17V rhodopsins trafficked primarily to the OS (Fig. 7C). Altogether, these results confirm that amino acid substitutions that favor β strand structure around N2 are well tolerated but that the structural requirements surrounding N15 are different and may require the presence of the carbohydrate moiety itself.
Figure 7.

Conservative amino acid substitutions are tolerated at T4 but not at T17. Transgenic animals expressing hWT, hT4I, hT17M, or hT17V rhodopsins (n = 20 per group) were generated and raised in cyclic light. A, Plot of transgenic opsin expression levels versus total rod opsin levels derived from solubilized transgenic eye extracts. B, C, Confocal micrographs of cryosections from transgenic retinas stained with WGA (B) or labeled with mAb 2B2 (green) and counterstained with WGA (red) and Hoescht nuclear dye (blue; C). Expression of hT17V rhodopsin resulted in loss of rod OS, whereas expression of hT4I did not affect rod density or morphology (B). Both T4I and T17V rhodopsins localized predominantly to the rod OS (C). rpe, Retinal pigment epithelium; ros, rod outer segment; ris, rod inner segment; onl, outer nuclear layer. Scale bars: B, 20 μm; C, 5 μm.
Discussion
Rhodopsin mutants have been grouped into several distinct classes based on their biochemical properties in cultured cells, and each class likely initiates a distinct set of pathogenic mechanisms leading to rod death. P23H rhodopsin belongs to class II, characterized by protein misfolding, ER retention and inefficient pigment generation (Sung et al., 1991). P23H rhodopsin exhibits similar properties in transgenic animals (Frederick et al., 2001; Tam and Moritz, 2006, 2007; Price et al., 2011) and is thought to cause cell death via ER stress pathways. RD caused by P23H rhodopsin is exacerbated by light exposure due to loss of the stabilization of mutant opsin by chromophore binding during biosynthesis (Tam et al., 2010). In contrast, we previously demonstrated that T4K and T17M rhodopsins are not defective in biosynthesis and trafficking (Tam and Moritz, 2009), but also cause light-exacerbated RD. In this paper, we examine the mechanisms underlying this light-induced RD. We demonstrate that mutants were less toxic when thermally stabilized or constitutively inactive (via preventing light exposure, withholding chromophore or genetic ablation of chromophore binding). Moreover, the mutants exhibited instability in vitro and in vivo. In all cases, except for constitutively active variants, T4K and T17M opsins predominantly localized to OSs. Our results demonstrate a novel mechanism in which mutant rhodopsins successfully traffic through the biosynthetic machinery, but become destabilized by light activation in the OS, leading to pathogenesis
Our results provide a mechanistic link between the human sector RP phenotype and light exposure. In these patients, only the superior retina retains significant function (Fishman et al., 1992; Li et al., 1994; van den Born et al., 1994). In a postmortem human retina heterozygous for T17M opsin, rods exhibited normal OS length in the superior retina, but were absent in the inferior retina (Li et al., 1994). These observations prompted the hypothesis that the asymmetric degeneration reflects the greater light exposure received from overhead sources such as the sun. Moreover, acute bright light induces RD in the T4R dog and the T17M mouse (Cideciyan et al., 2005; White et al., 2007). Here we have directly linked T4K and T17M induced RD with light activation of the mutant rhodopsins, since rod death was significantly blocked by abolishing their photoactivation. In contrast, inactivating P23H rhodopsin had minimal effect on RD and eliminated the rescuing effect of dark rearing (Tam and Moritz, 2007), further indicating that P23H and T4K/T17M pathogenesis are mechanistically different.
In transgenic X. laevis, light-activated T4K and T17M rhodopsins exhibited altered stability, and caused abnormalities in OS structure. Conformational changes occurring during activation of rhodopsin do not involve its N-terminal domain (Scheerer et al., 2008; Choe et al., 2011). However, anchoring the N-terminus to the third extracellular loop reduced the toxicity of T4K and T17M rhodopsins, suggesting these mutations promote movement of the N-terminus and instability. Destabilization may lead to aggregates of T4K rhodopsin in the OS, similar to those observed in transgenic frogs expressing P23H rhodopsin or P23H rhodopsin-GFP (Tam and Moritz, 2007; Haeri and Knox, 2012), which are associated with aberrant disk structure (Haeri and Knox, 2012). Protein aggregation or unfolding may cause reduced rhodopsin packing density and rupturing or remodeling of OS membranes. Possible toxic downstream effects include impeded clearance of N-retinylidene-PE from disk interiors, exposure of cell surface phosphatidylserine triggering of phagocytosis (Ruggiero et al., 2012) and accelerated OS shortening, or release of cytochrome C from disk interiors leading to apoptosis (Panfoli et al., 2012). More studies are required to establish how T4K or T17M rhodopsin instability in the OS translates into cellular toxicity.
Although T4K and T17M rhodopsins behaved similarly in most respects, T17M-induced degeneration was less amenable to rescue. Both thermal stabilization (N2C/N282C) and inactivation (K296R) were more effective at preventing T4K-induced RD, whereas constitutive activation (K296E) made T17M opsin more prone to ER retention. Moreover, substitutions eliminating the N2 glycosylation site (N2S, T4I, and T4V) were tolerated, whereas substitutions eliminating the N15 site (N15S, T17M, and T17V) were not (Tam and Moritz, 2009; this paper). Altogether, these results likely reflect the requirement for glycosylation at N15 and that loss of this carbohydrate leads to more severe perturbation of rhodopsin than the T4K substitution. Notably, loss of dehydrodolichyl diphosphate synthase activity, which is required for rhodopsin glycosylation, causes RD in zebrafish and humans (Zelinger et al., 2011; Züchner et al., 2011).
We propose that two mechanisms underlie T17M rhodopsin toxicity: light induced instability, and a second pathway not abrogated by constitutive inactivation. T17M rhodopsin misfolding and ER stress have been implicated in the disease pathology in transgenic mice since genes involved in the unfolded protein response (including BiP, CHOP, and Xbp-1) are upregulated (Kunte et al., 2012). However, in this and previous studies (Tam and Moritz, 2009), T17M rhodopsin expressed at high levels and trafficked to the OS of X. laevis rods indicating protein misfolding was minimal. Moreover, in a human retina expressing this mutant, the rhodopsin labeling density in OS resembles that of controls indicating T17M rhodopsin successfully trafficks to the OS (Li et al., 1994). In transgenic mice, T17M rhodopsin localization is unclear due to lack of antibodies that can distinguish mutant from WT rhodopsin. Classification of T17M as a misfolding mutant originates from cell-culture studies where mutant rhodopsin is retained in the ER (Sung et al., 1991). However, plasma membrane trafficking increases dramatically when cells are cultured in the dark with 11-cis-retinal (Li et al., 1998). In T17M rhodopsin mice, 2.5 min of bright light induces rod apoptosis (White et al., 2007). It is unlikely that such a brief stimulus could cause cell death solely via rhodopsin misfolding in the ER, especially since the same effect was not seen in P23H rhodopsin mice (White et al., 2007). Thus, in vitro results likely reflect the intrinsic instability of T17M opsin compared with WT opsin, but some property of biosynthesis in rods (rod-specific chaperones, chromophore, or WT opsin) promotes folding in vivo. We propose that a small proportion of T17M opsin may misfold, induce ER stress, and contribute to toxicity, but the majority of light-induced toxicity is mediated by OS T17M rhodopsin.
RD in dogs expressing T4R rhodopsin resembles RD induced by T4K rhodopsin in transgenic frogs in its light sensitivity but differs in that lack of chromophore aggravates rather than rescues degeneration (Zhu et al., 2004; Cideciyan et al., 2005). Like P23H opsin, T4R opsin may require chromophore for stability during biosynthesis (Tam et al., 2010), such that RD is mechanistically different in the absence of 11-cis-retinal. Localization of T4R opsin in the T4R/RPE65−/− dog was not reported, so it is unknown whether unliganded T4R opsin is retained in the ER. Although considered a conservative substitution, switching Lys for Arg or vice versa can cause protein misfolding (Kim et al., 2008), and alterations in protein–protein interactions (Morrow et al., 2000; Sun et al., 2013). We have previously shown that the fate of P23H rhodopsin is influenced by species related amino acid differences in rhodopsin sequence (Tam and Moritz, 2007). Thus mutations affecting T4 may have different outcomes in canine versus human rhodopsin.
Inhibiting rod death by blocking mutant rhodopsin activation presents the intriguing therapeutic strategy of sacrificing rod vision to save cone vision. A feasible approach for humans may be to develop compounds that specifically prevent the activation of rod but not cone opsins, rendering rods nonfunctional, but sparing them from cell death and thus preserving cones. Retinoids that differentially affect rods and cones have been identified: 9-demethyl retinal inhibits formation of the active metaII intermediate of rod opsin (Vogel et al., 2000), but not red or blue cone opsins (Das et al., 2004). Others (11-cis-locked retinals) photoisomerize without inducing a conformational change (Jang et al., 2001). Even partial reductions in rhodopsin activation may be beneficial in extending the lifetime of rods and cones.
Based on this study, vitamin A supplementation is contraindicated for RP patients with the T4K genotype. Vitamin A has been recommended for patients with typical RP, based on a study in which vitamin A palmitate delayed vision loss (Berson et al., 1993). However, patients with sector RP (potentially including those with T4K and T17M mutations) were excluded from the study. Rhodopsin is a complex molecule with multiple functional domains; different mutations cause different downstream functional and structural defects such as protein instability in the ER (Tam and Moritz, 2007; Price et al., 2011) and in the OS (this paper), trafficking defects (Sung et al., 1994; Tam et al., 2006), and aberrant signal transduction (Sieving et al., 2001; Jin et al., 2003). Thus, heterogeneity in disease mechanisms should be considered in the design and application of RP therapies. Treatments that are not broadly effective may be highly effective or detrimental for specific genotypes. For example, vitamin A deprivation has opposite effects on RD in T4K and P23H rhodopsin transgenic X. laevis (i.e., rescue vs exacerbation). Our study further suggests that correlating patient genotype with treatment outcomes may be required in clinical trials for RP (such as the current trials for valproic acid).
Footnotes
This work was funded by grants from the Canadian Institutes of Health Research (MOP-64400) and the Foundation Fighting Blindness of Canada (O.L.M.), the National Institutes of Health (NIH R01EY019515; to M.K.) and an unrestricted grant from Research to Prevent Blindness to MUSC (M.K.). We thank Dr R. S. Molday for providing mAb 2B2, Dr W. C. Smith for providing mAbs B630N and A5-3, Jenny Wong for technical assistance, and Colette Chiu for animal care.
The authors declare no competing financial interests.
References
- Adamus G, Zam ZS, Arendt A, Palczewski K, McDowell JH, Hargrave PA. Anti-rhodopsin monoclonal antibodies of defined specificity: characterization and application. Vis Res. 1991;31:17–31. doi: 10.1016/0042-6989(91)90069-H. [DOI] [PubMed] [Google Scholar]
- Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, Willett W. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111:761–762. doi: 10.1001/archopht.1993.01090060049022. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Meng EC. Structure. Rhodopsin sees the light. Science. 2000;289:733–734. doi: 10.1126/science.289.5480.733. [DOI] [PubMed] [Google Scholar]
- Chen J, Shi G, Concepcion FA, Xie G, Oprian D, Chen J. Stable rhodopsin/arrestin complex leads to retinal degeneration in a transgenic mouse model of autosomal dominant retinitis pigmentosa. J Neurosci. 2006;26:11929–11937. doi: 10.1523/JNEUROSCI.3212-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang WC, Hiramatsu N, Messah C, Kroeger H, Lin JH. Selective activation of ATF6 and PERK endoplasmic reticulum stress signaling pathways prevent mutant rhodopsin accumulation. Invest Ophthalmol Vis Sci. 2012;53:7159–7166. doi: 10.1167/iovs.12-10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe HW, Kim YJ, Park JH, Morizumi T, Pai EF, Krauss N, Hofmann KP, Scheerer P, Ernst OP. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Jacobson SG, Aleman TS, Gu D, Pearce-Kelling SE, Sumaroka A, Acland GM, Aguirre GD. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc Natl Acad Sci U S A. 2005;102:5233–5238. doi: 10.1073/pnas.0408892102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen GB, Oprian DD, Robinson PR. Mechanism of activation and inactivation of opsin: role of Glu113 and Lys296. Biochemistry. 1992;31:12592–12601. doi: 10.1021/bi00165a008. [DOI] [PubMed] [Google Scholar]
- Conover WJ. Practical nonparametric statistics. Ed 3. New York: Wiley; 1999. [Google Scholar]
- Das J, Crouch RK, Ma JX, Oprian DD, Kono M. Role of the 9-methyl group of retinal in cone visual pigments. Biochemistry. 2004;43:5532–5538. doi: 10.1021/bi036097u. [DOI] [PubMed] [Google Scholar]
- Fishman GA, Stone EM, Sheffield VC, Gilbert LD, Kimura AE. Ocular findings associated with rhodopsin gene codon 17 and codon 182 transition mutations in dominant retinitis pigmentosa. Arch Ophthalmol. 1992;110:54–62. doi: 10.1001/archopht.1992.01080130056026. [DOI] [PubMed] [Google Scholar]
- Franke RR, Sakmar TP, Oprian DD, Khorana HG. A single amino acid substitution in rhodopsin (lysine 248→leucine) prevents activation of transducin. J Biol Chem. 1988;263:2119–2122. [PubMed] [Google Scholar]
- Frederick JM, Krasnoperova NV, Hoffmann K, Church-Kopish J, Rüther K, Howes K, Lem J, Baehr W. Mutant rhodopsin transgene expression on a Null Background. Invest Ophthalmol Vis Sci. 2001;42:826–833. [PubMed] [Google Scholar]
- Haeri M, Knox BE. Rhodopsin mutant P23H destabilizes rod photoreceptor disk membranes. PLoS One. 2012;7:e30101. doi: 10.1371/journal.pone.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrave PA. The amino-terminal tryptic peptide of bovine rhodopsin: a glycopeptide containing two sites of oligosaccharide attachment. Biochim Biophys Acta. 1977;492:83–94. doi: 10.1016/0005-2795(77)90216-1. [DOI] [PubMed] [Google Scholar]
- Heckenlively JR, Rodriguez JA, Daiger SP. Autosomal dominant sectoral retinitis pigmentosa: two families with transversion mutation in codon 23 of rhodopsin. Arch Ophthalmol. 1991;109:84–91. doi: 10.1001/archopht.1991.01080010086038. [DOI] [PubMed] [Google Scholar]
- Hollyfield JG, Anderson RE. Retinal protein synthesis in relationship to environmental lighting. Invest Ophthalmol Vis Sci. 1982;23:631–639. [PubMed] [Google Scholar]
- Jang GF, Kuksa V, Filipek S, Bartl F, Ritter E, Gelb MH, Hofmann KP, Palczewski K. Mechanism of rhodopsin activation as examined with ring-constrained retinal analogs and the crystal structure of the ground state protein. J Biol Chem. 2001;276:26148–26153. doi: 10.1074/jbc.M102212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Cornwall MC, Oprian DD. Opsin activation as a cause of congenital night blindness. Nat Neurosci. 2003;6:731–735. doi: 10.1038/nn1070. [DOI] [PubMed] [Google Scholar]
- Kim PS, Lee J, Jongsamak P, Menon S, Li B, Hossain SA, Bae JH, Panijpan B, Arvan P. Defective protein folding and intracellular retention of thyroglobulin-R19K mutant as a cause of human congenital goiter. Mol Endocrinol. 2008;22:477–484. doi: 10.1210/me.2007-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll KL, Amaya E. Transgenic Xenopus embryos from sperm nuclear transplantations reveal FGF signaling requirements during gastrulation. Development. 1996;122:3173–3183. doi: 10.1242/dev.122.10.3173. [DOI] [PubMed] [Google Scholar]
- Kunte MM, Choudhury S, Manheim JF, Shinde VM, Miura M, Chiodo VA, Hauswirth WW, Gorbatyuk OS, Gorbatyuk MS. ER stress is involved in T17M rhodopsin-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2012;53:3792–3800. doi: 10.1167/iovs.11-9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Franson WK, Gordon JW, Berson EL, Dryja TP. Constitutive activation of phototransduction by K296E opsin is not a cause of photoreceptor degeneration. Proc Natl Acad Sci U S A. 1995;92:3551–3555. doi: 10.1073/pnas.92.8.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Sandberg MA, Pawlyk BS, Rosner B, Hayes KC, Dryja TP, Berson EL. Effect of vitamin A supplementation on rhodopsin mutants threonine-17-> methionine and proline-347-> serine in transgenic mice and in cell cultures. Proc Natl Acad Sci U S A. 1998;95:11933–11938. doi: 10.1073/pnas.95.20.11933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZY, Jacobson SG, Milam AH. Autosomal dominant retinitis pigmentosa caused by the threonine-17-methionine rhodopsin mutation: retinal histopathology and immunocytochemistry. Exp Eye Res. 1994;58:397–408. doi: 10.1006/exer.1994.1032. [DOI] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie D, Arendt A, Hargrave P, McDowell JH, Molday RS. Localization of binding sites for carboxyl terminal specific anti-rhodopsin monoclonal antibodies using synthetic peptides. Biochemistry. 1984;23:6544–6549. doi: 10.1021/bi00321a041. [DOI] [PubMed] [Google Scholar]
- McKee TD, Lewis MR, Kono M. Engineering a “steric doorstop” in rhodopsin: converting an inverse agonist to an agonist. Biochemistry. 2007;46:12248–12252. doi: 10.1021/bi701003b. [DOI] [PubMed] [Google Scholar]
- Moritz OL, Tam BM, Knox BE, Papermaster DS. Fluorescent photoreceptors of transgenic Xenopus laevis imaged in vivo by two microscopy techniques. Invest Ophthalmol Vis Sci. 1999;40:3276–3280. [PubMed] [Google Scholar]
- Moritz OL, Biddle KE, Tam BM. Selection of transgenic Xenopus laevis using antibiotic resistance. Transgenic Res. 2002;11:315–319. doi: 10.1023/A:1015612022976. [DOI] [PubMed] [Google Scholar]
- Morrow JA, Arnold KS, Dong J, Balestra ME, Innerarity TL, Weisgraber KH. Effect of arginine 172 on the binding of apolipoprotein E to the low density lipoprotein receptor. J Biol Chem. 2000;275:2576–2580. doi: 10.1074/jbc.275.4.2576. [DOI] [PubMed] [Google Scholar]
- Nelson RM, Long GL. A general method of site-specific mutagenesis using a modification of the Thermus aquaticus polymerase chain reaction. Anal Biochem. 1989;180:147–151. doi: 10.1016/0003-2697(89)90103-6. [DOI] [PubMed] [Google Scholar]
- Noorwez SM, Malhotra R, McDowell JH, Smith KA, Krebs MP, Kaushal S. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J Biol Chem. 2004;279:16278–16284. doi: 10.1074/jbc.M312101200. [DOI] [PubMed] [Google Scholar]
- Oprian DD. Expression of opsin genes in COS cells. Methods Neurosci. 1993;15:301–306. doi: 10.1016/B978-0-12-185279-5.50027-3. [DOI] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: a G-protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Panfoli I, Calzia D, Ravera S, Morelli AM, Traverso CE. Extra-mitochondrial aerobic metabolism in retinal rod outer segments: new perspectives in retinopathies. Med Hypotheses. 2012;78:423–427. doi: 10.1016/j.mehy.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Price BA, Sandoval IM, Chan F, Simons DL, Wu SM, Wensel TG, Wilson JH. Mislocalization and degradation of human P23H-rhodopsin-GFP in a knockin mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2011;52:9728–9736. doi: 10.1167/iovs.11-8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridge KD, Palczewski K. Visual rhodopsin sees the light: structure and mechanism of G-protein signaling. J Biol Chem. 2007;282:9297–9301. doi: 10.1074/jbc.R600032200. [DOI] [PubMed] [Google Scholar]
- Robinson PR, Cohen GB, Zhukovsky EA, Oprian DD. Constitutively active mutants of rhodopsin. Neuron. 1992;9:719–725. doi: 10.1016/0896-6273(92)90034-B. [DOI] [PubMed] [Google Scholar]
- Roof DJ, Adamian M, Hayes A. Rhodopsin accumulation at abnormal sites in retinas of mice with a human P23H rhodopsin transgene. Invest Ophthalmol Vis Sci. 1994;35:4049–4062. [PubMed] [Google Scholar]
- Ruggiero L, Connor MP, Chen J, Langen R, Finnemann SC. Diurnal, localized exposure of phosphatidylserine by rod outer segment tips in wild-type but not Itgb5−/− or Mfge8−/− mouse retina. Proc Natl Acad Sci U S A. 2012;109:8145–8148. doi: 10.1073/pnas.1121101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakami S, Maeda T, Bereta G, Okano K, Golczak M, Sumaroka A, Roman AJ, Cideciyan AV, Jacobson SG, Palczewski K. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem. 2011;286:10551–10567. doi: 10.1074/jbc.M110.209759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieving PA, Fowler ML, Bush RA, Machida S, Calvert PD, Green DG, Makino CL, McHenry CL. Constitutive “light” adaptation in rods from G90D rhodopsin: a mechanism for human congenital nightblindness without rod cell loss. J Neurosci. 2001;21:5449–5460. doi: 10.1523/JNEUROSCI.21-15-05449.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standfuss J, Xie G, Edwards PC, Burghammer M, Oprian DD, Schertler GF. Crystal structure of a thermally stable rhodopsin mutant. J Mol Biol. 2007;372:1179–1188. doi: 10.1016/j.jmb.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LJ, Makris GS, Dickinson P, Mulhall LE, Forrest S, Cotton RG, Loughnan MS. A new codon 15 rhodopsin gene mutation in augtosomal dominant retinitis pigmentosa is associated with sectorial disease. Arch Ophthalmol. 1993;111:1512–1517. doi: 10.1001/archopht.1993.01090110078029. [DOI] [PubMed] [Google Scholar]
- Sullivan LS, Bowne SJ, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Lewis RA, Garcia CA, Ruiz RS, Blanton SH, Northrup H, Gire AI, Seaman R, Duzkale H, Spellicy CJ, Zhu J, Shankar SP, Daiger SP. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci. 2006;47:3052–3064. doi: 10.1167/iovs.05-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun AQ, Luo Y, Backos DS, Xu S, Balasubramaniyan N, Reigan P, Suchy FJ. Identification of functionally relevant lysine residues that modulate human farnesoid X receptor activation. Mol Pharmacol. 2013;83:1078–1086. doi: 10.1124/mol.113.084772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Schneider BG, Agarwal N, Papermaster DS, Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Makino C, Baylor D, Nathans J. A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci. 1994;14:5818–5833. doi: 10.1523/JNEUROSCI.14-10-05818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Moritz OL. Characterization of rhodopsin P23H-induced retinal degeneration in a Xenopus laevis model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006;47:3234–3241. doi: 10.1167/iovs.06-0213. [DOI] [PubMed] [Google Scholar]
- Tam BM, Moritz OL. Dark rearing rescues P23H rhodopsin-induced retinal degeneration in a transgenic Xenopus laevis model of retinitis pigmentosa: a chromophore-dependent mechanism characterized by production of N-terminally truncated mutant rhodopsin. J Neurosci. 2007;27:9043–9053. doi: 10.1523/JNEUROSCI.2245-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Moritz OL. The role of rhodopsin glycosylation in protein folding, trafficking, and light-sensitive retinal degeneration. J Neurosci. 2009;29:15145–15154. doi: 10.1523/JNEUROSCI.4259-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Xie G, Oprian DD, Moritz OL. Mislocalized rhodopsin does not require activation to cause retinal degeneration and neurite outgrowth in Xenopus laevis. J Neurosci. 2006;26:203–209. doi: 10.1523/JNEUROSCI.3849-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Qazalbash A, Lee HC, Moritz OL. The dependence of retinal degeneration caused by the rhodopsin P23H mutation on light exposure and vitamin a deprivation. Invest Ophthalmol Vis Sci. 2010;51:1327–1334. doi: 10.1167/iovs.09-4123. [DOI] [PubMed] [Google Scholar]
- van den Born LI, van Schooneveld MJ, de Jong LA, Riemslag FC, de Jong PT, Gal A, Bleeker-Wagemakers EM. Thr4Lys rhodopsin mutation is associated with autosomal dominant retinitis pigmentosa of the cone-rod type in a small Dutch family. Ophthalmic Genet. 1994;15:51–60. doi: 10.3109/13816819409098864. [DOI] [PubMed] [Google Scholar]
- Vogel R, Fan GB, Sheves M, Siebert F. The molecular origin of the inhibition of transducin activation in rhodopsin lacking the 9-methyl group of the retinal chromophore: a UV-Vis and FTIR spectroscopic study. Biochemistry. 2000;39:8895–8908. doi: 10.1021/bi000852b. [DOI] [PubMed] [Google Scholar]
- White DA, Fritz JJ, Hauswirth WW, Kaushal S, Lewin AS. Increased sensitivity to light-induced damage in a mouse model of autosomal dominant retinal disease. Invest Ophthalmol Vis Sci. 2007;48:1942–1951. doi: 10.1167/iovs.06-1131. [DOI] [PubMed] [Google Scholar]
- Xie G, Gross AK, Oprian DD. An opsin mutant with increased thermal stability. Biochemistry. 2003;42:1995–2001. doi: 10.1021/bi020611z. [DOI] [PubMed] [Google Scholar]
- Zelinger L, Banin E, Obolensky A, Mizrahi-Meissonnier L, Beryozkin A, Bandah-Rozenfeld D, Frenkel S, Ben-Yosef T, Merin S, Schwartz SB, Cideciyan AV, Jacobson SG, Sharon D. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am J Hum Genet. 2011;88:207–215. doi: 10.1016/j.ajhg.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Jang GF, Jastrzebska B, Filipek S, Pearce-Kelling SE, Aguirre GD, Stenkamp RE, Acland GM, Palczewski K. A naturally occurring mutation of the opsin gene (T4R) in dogs affects glycosylation and stability of the G-protein-coupled receptor. J Biol Chem. 2004;279:53828–53839. doi: 10.1074/jbc.M408472200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züchner S, Dallman J, Wen R, Beecham G, Naj A, Farooq A, Kohli MA, Whitehead PL, Hulme W, Konidari I, Edwards YJ, Cai G, Peter I, Seo D, Buxbaum JD, Haines JL, Blanton S, Young J, Alfonso E, Vance JM, et al. Whole exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am J Hum Genet. 2011;88:201–206. doi: 10.1016/j.ajhg.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]