

Abstract
Biomaterials displaying precise ratios of different bioactive protein components are critical for applications ranging from vaccines to regenerative medicine, but their design is often hindered by limited choices and cross-reactivity of protein conjugation chemistries. Here, we describe a strategy for inducing multiple different expressed proteins of choice to assemble into nanofibers and gels with exceptional compositional control. The strategy employs novel “βTail” tags, which allow for good protein expression in bacteriological cultures, yet can be induced to co-assemble into nanomaterials when mixed with additional β-sheet fibrillizing peptides. Multiple different βTail fusion proteins could be inserted into peptide nanofibers alone or in combination at predictable, smoothly gradated concentrations, providing a simple yet versatile route to install precise combinations of proteins into nanomaterials. The technology is illustrated by achieving precisely targeted hues using mixtures of fluorescent proteins, by creating nanofibers bearing enzymatic activity, and by adjusting antigenic dominance in vaccines.
Polypeptides that non-covalently assemble into nanofibers and hydrogels are promising materials for a range of biomedical and biotechnological applications, including enzyme catalysis,1 biosensors,2,3 electronics,4 tissue engineering,5,6 drug delivery,7,8 and immunotherapies.9-11 Many of these materials have been based on functional peptide ligands or epitopes fused to fibrillizing peptides, peptide amphiphiles, or other self-assembling molecules, which can be mixed to form fibers and gels that display precise combinations of the desired functionalities.12-14 The ability to integrate and control multiple functional components simultaneously is critical for the use of such materials in biological contexts such as vaccines, tissue engineering, and drug delivery, as each of these applications requires multiple biomolecular cues to be presented in optimized ratios and combinations. However, previous work has focused mainly on short synthetic peptides as the bioactive components of such materials, and an analogous approach for inducing a desired set of expressed globular proteins to assemble into predictably formulated nanofibers, microgels, or gels has been lacking. Materials have been reported that employ folded proteins to drive self-assembly, but they often lack biological function beyond the capacity to self-assemble.15-17 Self-assembled nanofibers displaying a single functional protein,1,4 or two proteins,2,3 have also been reported, as have methods for functionalizing fibrillized peptide nanofibers with proteins post-assembly.10,18 In this contribution, we sought to achieve a general strategy for inducing desired sets of expressed functional proteins to assemble directly into nanofibers or gels, with gradated control over the amount of each protein ultimately being incorporated.
To create multi-component nanofibers out of a set of chosen functional proteins, it is necessary to first prevent their self-assembly or aggregation during expression, maintain them in a soluble state during purification and storage, and then subsequently induce their co-assembly at a desired time and place. To arrive at a predictably formulated multi-component material, proteins with different biophysical properties must integrate into the materials without compositional drift. Owing to the aggregation potential of β-sheet fibrillizing peptides as protein fusion tags,19,20 engineering such a system has been challenging. The strategy we report is based on a fusion tag that allows proteins to be expressed and purified in a soluble form, then later induced to assemble at a desired time point upon mixing them with additional fibrillizing peptide (Figure 1a). The approach can be used to install features into nanofibrillar materials that can only be accomplished with precisely gradated mixtures of proteins, for example finely tuned composite colors using multiple fluorescent proteins.
Figure 1. Engineered fusion proteins with a β-sheet fibrillizing tail integrate into self-assembling peptide nanofibers.
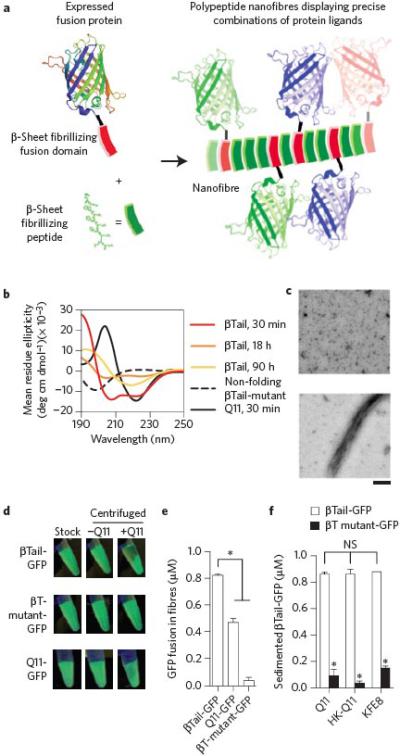
a) Schematic representation of engineered fusion proteins having a β-sheet fibrillizing domain integrating into Q11 nanofibers. b-c) The βTail peptide underwent slow secondary structural transition from an α-helix to a β-sheet, whereas Q11 rapidly assembled into β-sheets, and a mutated βTail adopted a random coil structure (scale bar = 200 nm). A fusion of βTail and Green Fluorescent Protein-UV (βT GFP) efficiently integrated into Q11 nanofibers, whereas a fusion of Q11 and GFP (Q11-GFP) integrated moderately, and a fusion of GFP and a non-folding βTail mutant (βTmutant-GFP) integrated poorly, as demonstrated by (d) digital photographs of 1.5 mL microcentrifuge tubes, and (e) measured by fluorimetry of the supernatant above sedimented Q11 nanofiber solutions. f) βT-GFP also integrated into HK-Q11 and KFE8 nanofibers in a βTail-dependent manner, indicating that co-assembly was not limited to Q11-based nanofibers. N = 3, mean ± s.d. for e and f. * represents p < 0.05, ANOVA with Tukey's post-hoc. GFP adapted from PDB 1EMA in a.
To develop this approach, we first investigated the behavior of Green Fluorescent Protein-UV (GFP) fused with two different putative β-sheet fibrillizing peptides. Previous work showed that proteins appended with β-sheet fibrillizing peptides aggregated into insoluble inclusion bodies during expression.19,20 To determine whether this propensity for aggregation applied to fusions with other fibrillizing peptides as well, we compared the expression of GFP tagged with either the peptide Q11 (QQKFQFQFEQQ), which rapidly assembles into β-sheet nanofibers at physiologic pH (Fig. 1b),21 or the peptide we have termed a “βTail” MALKVELEKLKSELVVLHSELHKLKSEL. The βTail peptide is based on the “i+4” peptide previously reported to slowly transition from a random coil into a β-sheet.22 In our hands, the βTail peptide slowly transitioned from an α-helix to a β-sheet (Fig. 1b-c), with the helical starting conformation being possibly attributable to the additional methionine and alanine residues that we introduced at its N-terminus to enable cloning, as both residues have high helix-forming propensities. We hypothesized that the slow fibrillization kinetics of the βTail tag would minimize aggregation or misfolding during microbial expression. Counter to this expectation, despite differences in fibrillization kinetics, both βTail-GFP and Q11-GFP were recovered from E. coli in similar yields (Supplemental Fig. 1a-d). These results indicated that neither peptide was advantageous over the other with respect to expression.
With respect to subsequent integration into self-assembled peptide nanofibers, however, βTail-GFP was far superior to Q11-GFP. Q11 peptide nanofibers can be efficiently sedimented by centrifugation (Supplemental Fig. 2), so we first monitored the loss of GFP from the supernatant as a measure of tagged GFP integration into Q11 nanofibers (Supplemental Fig. 3). By this measure, greater than 80% of βTail-GFP was partitioned from the supernatant to the nanofibers when incubated with Q11, whereas only modest partitioning was observed for Q11-GFP (Q11:fusion protein ratio 1000:1) (Fig. 1d-e, Supplemental Fig. 4). When the βTail domain was mutated to a non-folding form (βTmutant), or when solutions lacked Q11, partitioning or sedimentation were not observed (Fig. 1d-e, Supplemental Fig. 4). Taken together, these data demonstrated that fusion proteins having a β-sheet fibrillizing domain can integrate into nanofibers when mixed with self-assembling peptides, but that the choice of tag greatly influences the efficiency of incorporation. In contrast, previously reported Q11 peptides bearing short peptide ligands demonstrate near-quantitative co-assembly,23,24 suggesting that bulky protein cargoes influence Q11's assembly properties, possibly by inhibiting fibril growth or trapping the protein in non-fibrillar aggregates of intermediate size and disfavoring the growth of long fibers. Conversely, the slow nucleation and growth of βTail may favor integration of βTail fusion proteins into growing Q11 nanofibers as they self-assemble. Such mechanistic differences are likely worth consideration in future work.
Owing to the reproducible expression and assembly of βTail-GFP, further work focused on exploiting the βTail tag. We investigated whether βTail-GFP could be inserted into nanofibers of other β-sheet fibrillizing peptides beyond Q11, including FKFEFKFE (KFE8),25 RADARADARADARADA (RADA16),26 and QQKFQFQFHQQ (HK-Q11) (Supplemental Fig. 5). βTail-GFP efficiently integrated into nanofibers of HK-Q11 and KFE8, in a βTail-dependent manner (Fig. 1f). However, both βTail-GFP and its mutated counterpart integrated into RADA16 nanofibers with similar efficiency (Supplemental Fig. 6), suggesting that GFP interacted non-specifically with RADA16 nanofibers. Taken together, these results suggested that the βTail fusion tag provides a relatively general approach for integrating protein ligands into β-sheet peptide nanofibers, although the non-specific interaction of the protein with the nanofibers must be considered.
To determine if βTail integration into Q11 nanofibers could be precisely controlled, we next quantified the integration of fluorescent βTail proteins into Q11 nanofibers at different doses using fluorimetry and fluorescent microscopy. The concentration of βTail-GFP integrated into Q11 nanofibers correlated with βTail protein concentration in solution during nanofiber assembly, as measured by loss of fluorescence from the supernatant following centrifugation (Fig. 2a). Similar behavior was observed for two other βTail fusion proteins, βTail-eGFP and βTail-dsRED (Fig. 2a, Supplemental Fig. 1e-h). This was again dependent on the βTail domain and the presence of Q11, since GFP fused to a mutated βTail domain or lacking the βTail domain, as well as βTail-GFP incubated in the absence of Q11, were primarily retained in the supernatant at all tested concentrations (Fig. 2a).
Figure 2. Fluorescent βTail fusion proteins stably integrate into Q11 nanofibers and microgels in smoothly gradated amounts, alone or in combination, without loss of activity.

a) Fluorescent βT fusion proteins integrated into Q11 nanofibers over the range of 0.25-1.5 μM in a βTail-dependent manner, as measured by loss of fluorescence from the supernatant. b) βT-GFP integrated into micron-sized Q11 “microgels” in a βTail- dependent manner. c-d) Fluorescent βTail fusion proteins integrated into Q11 microgels at a predictable dose. e) Different fluorescent βTail proteins co-integrated into Q11 microgels at a precisely tunable dose, as demonstrated by the close correlation between actual gel color and the predicted color, which was determined by using the protein mole ratio in solution during assembly as the RGB pixel ratio. f-g) GFP fluorescence was retained in Q11 microgels when diluted 0-100 fold in 1× PBS, suggesting the materials were in a “kinetically-trapped” state. h-i) Fluorescence intensity of Q11 microgels with βTail-GFP proteins was similar at different time points, demonstrating the stability of these materials. N = 3 for a; N = 5 for g; and N = 10 for d and i. mean ± s.d. Scale bar = 40 μm in b, c, e, f, and h.
When rocked gently on a rocker table, Q11 nanofibers in aqueous suspensions associated into micron-sized, spherical bundles rich in β-sheet structure (Supplemental Fig. 7), which we refer to as “Q11 microgels”. These associations of nanofibers are not to be confused with polymer microgels, which are chemically cross-linked polymer particles rather than the physically entangled nanofibers discussed here. βTail-GFP efficiently integrated into Q11 microgels in a βTail-dependent manner, as demonstrated by co-localization of GFP fluorescence with Q11 microgels assembled in the presence of βTail-GFP, but not βTmutant-GFP (Fig. 2b). The fluorescence intensity of Q11 microgels correlated with the concentration of βTail-GFP, βTail-dsRED or βTail-eGFP that was added during nanofiber assembly (Fig. 2c-d). Notably, the three proteins could also be mixed without sacrificing this predictability, which we illustrate using composite color as a readout. For example, in digital images, a neutral grey color is achieved by combining equivalent amounts of red, green, and blue pixels. Analogously, when equimolar βTail-GFP, βTail-dsRED, and βTail-eGFP are assembled into microgels, grey Q11 microgels are produced in fluorescence overlay images (Figure 2e). Varying the βTail-GFP, βTail-eGFP, and βTail-dsRED molar ratio in solution during Q11 assembly enabled fine-tuning of microgel color, as demonstrated by microgels with colors ranging from pink to orange to turquoise that closely matched the predicted color (Fig. 2e). In contrast, replacing βTail-GFP with the non-folding βTmutant-GFP abolished this precise color matching (Fig. 2e). Together, these results demonstrated that multiple different βTail fusion proteins could be integrated into peptide nanofibers at a predictable dose, alone or in combination, via simple mixing of different molecular components at the onset of assembly, thereby providing a general route to biomaterials displaying precise ratios of expressed proteins.
To determine if βTail fusion proteins were non-covalently associated with Q11 nanofibers in a reversible state or stably integrated into the fibers, we characterized the loss of βTail-GFP from Q11 microgels upon dilution using fluorescent microscopy. Fluorescence of Q11 microgels containing βTail-GFP was unchanged when diluted from 0-100 fold (Fig. 2f-g). Fluorescence of Q11 microgels containing βTail-GFP was also unchanged when incubated for 0-120 h (Fig. 2h-i), and βTail-GFP was efficiently sedimented with Q11 nanofibers assembled for 8-48 h (supplemental Fig. 8). Taken together, these observations indicated that βTail-GFP was stably integrated into Q11 nanofibers, likely in a kinetically trapped state, rather than being bound to nanofibers in a reversible manner.
We used immunogold labeling and transmission electron microscopy (TEM) to verify the co-localization of expressed βTail fusion proteins and synthetic βTail peptides within Q11 nanofibers. Q11 nanofibers assembled in the presence of βTail-GFP were specifically labeled with a primary/secondary antibody system directed against GFP (Fig. 3a), and nanofibers assembled in the presence of biotinylated-βTail specifically bound streptavidin-gold (Fig. 3b). Similar to βTail fusion proteins, tryptophan-terminated βTail (W-βTail) could also be precisely dosed into Q11 nanofibers (Fig. 3c), and this was again dependent on both the βTail sequence and the presence of Q11. Taken together, these results supported earlier observations that βTail-GFP was sedimented with Q11 nanofibers due to specific association of the βTail domain with β-sheet peptide nanofibers.
Figure 3. TEM, CD, and fluorescence measurements confirmed the rapid co-assembly of βTail polypeptides into Q11 nanofibers.

a) Immunogold staining identified gold beads co-localized with nanofibers assembled in the presence of βTail- GFP and then stained with an anti-GFP 1° and a gold-labeled 2° antibody, whereas few gold beads were observed near Q11 nanofibers assembled in the presence of a βTail peptide and then stained similarly (scale bar = 100 nm). b) Gold beads conjugated to streptavidin bound to Q11 nanofibers assembled in the presence of biotinylated βTail, whereas few gold beads were co-localized with Q11 nanofibers (scale bar = 200 nm). c) Tryptophan-terminated βTail (W-βTail, W-βT) integrated into Q11 nanofibers over the range of 25-100 μM in a βTail-dependent manner, as measured by loss of fluorescence from the supernatant. d) W-βTail and βTail-GFP integrated into Q11 nanofibers within minutes, as measured by loss of fluorescence from the supernatant. βTail secondary structure changed from α-helical to β-sheet following overnight co-assembly with Q11 (e), individual peptides shown left and center, after overnight co-assembly shown right, where red=spectrum of mixed peptides and black=this mixed sample with the Q11 spectrum subtracted, revealing structure of βTail. In contrast, βTmutant secondary structure remained predominantly random coil (f, same coloring scheme as e). N = 3, mean ± s.d. for c, d.
βTail-GFP and W-βTail rapidly integrated into Q11 nanofibers, approaching a maximum within 1 min or 30 min, respectively, when incubated at a Q11:βTail ratio of 1000:1 or 10:1 (Fig. 3d). These kinetics were much faster than those of βTail self-assembly (Fig. 1b-c), suggesting a mechanism wherein Q11 β-sheet nanofibers provided a template for βTail fibrillization that eliminated the need for slow βTail nucleation to precede self-assembly. Using circular dichroism, we also found that the βTail peptide adopted an α-helical 2° structure in the absence of Q11, and transitioned to a predominantly β-sheet 2° structure when incubated with a 10-fold molar excess of Q11 overnight (Fig. 3e) or for 30 minutes (Supplemental Fig. 9), the latter demonstrating that the βTail 2° structural transition was consistent with the kinetics of βTail integration into Q11 nanofibers observed in Fig. 3d. In contrast, a non-folding βTail peptide mutant adopted a random coil conformation alone, or when incubated with a 10-fold molar excess of Q11 overnight (Fig. 3f). These observations suggested that Q11 and βTail rapidly co-assembled together into mixed β-sheet nanofibers, and were consistent with a previous report demonstrating significant changes to CD spectra following co-assembly of two different peptides into mixed β-sheets.27 We inferred that Q11 nanofibers bearing βTail-fusion proteins were also mixed (i.e. heterogeneous) β-sheets, since the 1000:1 molar ratio of Q11 to βTail in these materials was significantly greater than the 2.5:1 Q11 to βTail molar ratio required for structural transition after overnight assembly (Supplemental Fig. 10). Importantly, these observations suggesting co-fibrillization of Q11 and βTail into heterogeneous β-sheets further supported earlier observations that βTail fusion proteins were stably integrated into Q11 nanofibers (Fig. 2f-g), likely in a kinetically trapped state, rather than reversibly bound to the nanofiber surface.
We next expressed βTail-tagged cutinase (βTail-cutinase) (Supplemental Fig. 1i-j), a fungal esterase, and characterized its assembly into Q11 nanofibers to establish the potential of βTail tags to integrate protein ligands with different activities into β-sheet peptide nanofibers. The amount of βTail-cutinase integrated into Q11 nanofibers correlated with βTail-cutinase concentration in solution during Q11 assembly, and this was again dependent on the presence of Q11 (Fig. 4a). Q11 nanofibers assembled in the presence of βTail-cutinase demonstrated enzymatic activity, as measured by their ability to hydrolyze p-nitrophenyl butyrate to p-nitrophenol,28 and cutinase activity was predictably dosed into Q11 nanofibers by varying the βTail-cutinase concentration in solution during assembly (Fig. 4b-c). Notably, βTail-cutinase and βTail-GFP co-integrated into Q11 nanofibers at predictable doses and without loss of activity, again by varying protein molar ratio in solution during Q11 assembly (Fig. 4d). Importantly, these results suggested the broad potential versatility provided by this approach, since two proteins having vastly different amino acid composition, tertiary structure, and bioactivity could co-integrate into peptide nanofibers via the βTail domain, leading to biomaterials with independently controllable activities related to each protein.
Figure 4. The βTail platform is capable of integrating an enzyme into nanofibers in precise amounts, without loss of enzyme activity.

a) A fusion of βTail and the fungal enzyme cutinase (βTail-cutinase, βT-cut) integrated into Q11 nanofibers over the range of 0.25-1.5 μM in a βTail-dependent manner, as measured by loss of protein from the supernatant following centrifugation. b) Q11 nanofibers assembled in the presence of βTail-cutinase demonstrated cutinase activity, as measured by hydrolysis of p-nitrophenyl butyrate (colorless) to p-nitrophenol (yellow). c) Nanofiber cutinase activity was precisely varied by changing the concentration of βTail-cutinase present during Q11 assembly, as determined from the initial velocity of pNB hydrolysis plots in (b). d) βTail- GFP (βT-GFP) and βTail-cutinase (βT-cut) could be mixed into Q11 nanofibers in predictable ratios without loss of activity, as demonstrated by the direct correlation between nanofiber fluorescence or cutinase activity and βTail-GFP or βTail-cutinase concentration, respectively. N = 3, mean ± s.d.
Peptide nanofibers displaying peptide epitopes have received significant attention recently as compositionally defined vaccines.9,11 The usefulness of self-assembled peptides as vaccines arises from their ability to generate strong antibody responses without inflammation and without additional immune adjuvants.29 In part their activity is due to their particulate nature, which facilitates uptake and activation by antigen presenting cells, and in part it arises from the materials’ repetitive antigen display, which facilitates B cell activation, though the detailed mechanism of action of the materials continues to be investigated. Here we sought to determine if antibody responses could be raised against protein antigens similarly to peptide antigens, and whether the modular construction of the βTail system could be used to adjust antigen dose to optimize antibody responses. C57BL/6 mice immunized with Q11 nanofibers bearing μg doses of βTail-GFP or βTail-cutinase elicited higher GFP-reactive antibody titers (Fig. 5a) or cutinase-reactive antibody titers (Fig. 5b), respectively, than mice immunized with an identical dose of either protein without Q11 (i.e. soluble antigen). Antibody responses against βTmut-GFP mixed with (but not co-assembled into) Q11 nanofibers were significantly diminished compared to βT-GFP co-assembled into Q11 nanofibers, and statistically similar to those against unfibrillized βT-GFP in PBS (Fig. 5a), indicating that incorporation of the protein into the nanofibers was necessary for enhanced immunogenicity. Mice immunized with nanofibers bearing βTail proteins underwent isotype switching predominanted by IgG1, whereas a weaker IgG1 polarization was elicited by non-fibrillized βTail proteins with or without Q11 nanofibers (Fig. 5c-d, Supplemental Fig. 11). Significant steps were taken to reduce endotoxin content of these materials to below 1 EU/mL, which is the maximum allowable dose for pre-clinical vaccines.30 However, to further rule out the role of endotoxin in the observed immune responses, we immunized C57BL/6 mice lacking Toll-like receptor-4 with βTail-GFP/Q11 nanofibers. These mice also raised significant GFP-reactive antibody titers (Supplemental Fig. 12), demonstrating that endotoxin contaminants are not mediators of the observed responses. Taken together, these results demonstrated that polypeptide nanofibers bearing a protein antigen can elicit adaptive immune responses in the absence of additional immunostimulatory factors, which was consistent with a recent report by our group demonstrating that Q11 nanofibers bearing covalently conjugated GFP can act as self-adjuvanting vaccines.10
Figure 5. Nanofibers displaying βTail fusion proteins raised antibody responses in mice that could be adjusted by changing the protein content.

C57BL/6 mice immunized with Q11 nanofibers bearing βTail-GFP or βTail-cutinase elicited higher titers of anti-GFP (a) or anti-cutinase (b) antibodies, respectively, when compared to mice immunized with βTail-GFP in PBS (a), non-fibrillizing βTmut-GFP plus Q11 (a) or βTail-cutinase in PBS (b). Mice immunized with a single fibrillized antigen also underwent robust isotype switching towards IgG1 (c, d). e) Mice immunized with Q11 nanofibers bearing both βTail-GFP and βTail-cutinase elicited higher titers of cutinase and GFP-reactive antibodies when compared to mice immunized with an identical dose of soluble βTail-GFP and βTail-cutinase in PBS, though boosting with βTail-GFP was necessary to overcome antigenic dominance of the cutinase domain. f) Mice immunized with two co-fibrillized antigens underwent isotype switching towards IgG1, whereas IgG1 polarization was diminished for soluble antigens. N=5 (a, b, e) and n=4 (c, d, and e), mean ± s.d. * p<0.05, ** p<0.01, Student's t-test (b and e), ANOVA with Tukey's post- hoc (a, c, d, and f).
The adjuvanticity of nanofibers bearing a single βTail-fusion protein and the ability to precisely integrate multiple different βTail-fusion proteins into Q11 nanofibers suggested that these materials would provide a tailorable multi-antigen vaccine platform. Multi-antigen vaccines are of clinical interest because they can provide simultaneous immunity against multiple pathogens, or multiple different antigens from a single pathogen, while also minimizing the number of immunizations that are required. C57BL/6 mice immunized with Q11 nanofibers bearing both βTail-GFP and βTail-cutinase raised high cutinase-reactive antibody titers when compared to mice receiving an equivalent dose of soluble βTail antigens, whereas low titers of GFP-reactive antibodies were observed in both fibril-adjuvanted and soluble antigen groups (Fig. 5e, left half). Together, these observations indicated preferential immunological responses, or “antigenic dominance”, favoring cutinase when co-administered with GFP. We relied on the modular nature of βTail-Q11 nanofibers to determine if repeated exposure to antigen, or a “booster vaccine”, with modified antigen formulation could be use to adjust this antigenic dominance. Mice receiving a booster of nanofibers bearing βTail-GFP elicited higher GFP-reactive antibody titers within two weeks, when compared to mice receiving a booster of soluble βTail-GFP (Supplemental Fig. 13). These elevated GFP-reactive antibody titers persisted for at least 6 weeks in the fibrillized antigen group, without significantly diminishing cutinase-reactive antibody titers (Fig. 5e, right half). Mice immunized with nanofibers bearing both βTail-GFP and βTail-cutinase raised predominantly IgG1 reactive antibodies against each antigen by week 20, whereas mice immunized with soluble βTail-GFP and βTail-cutinase exhibited weaker IgG1 polarization (Fig. 5f), similar to mice immunized with nanofibers bearing only βTail-GFP or βTail-cutinase (Fig. 5c-d). These results demonstrated that peptide nanofibers bearing multiple βTail fusion proteins could be efficiently tailored as multi-antigen immunogens, in part because the dose of each antigen could be precisely adjusted in primary and booster formulations. An ability to control antigenic dominance in this way is advantageous in the general context of biomaterials, for example to avoid eliciting immune responses against tags such as fluorescent proteins, or for developing efficacious multi-antigen or multi-pathogen vaccines, which can be challenging with live or attenuated whole-pathogen vaccines or adjuvants loaded via non-specific antigen adsorption, as is the case with aluminum salts (alum).
Although supramolecular assemblies of multiple proteins have been reported previously,15-17 we sought a strategy to produce β-sheet nanofibers displaying smoothly gradated combinations of multiple different proteins that retain their native folding and biological activity. In light of the general nature and versatility afforded by this approach, we envision that it could be widely used to create multi-antigen vaccines for disease prophylaxis and immunotherapy, as well as new biomaterials for diverse medical and technological applications, including drug delivery, synthetic extracellular matrices for tissue engineering and 3-D culture, and biosensors.
Materials and Methods
Detailed materials and methods are available in the Supplemental Information.
Peptide synthesis
The β-sheet fibrillizing peptides Q11 (QQKFQFQFEQQ),21 HK-Q11 (QQKFQFQFHQQ) (Supplemental figure 5), KFE8 (FKFEFKFE),25 and RADA16 (RADARADARADARADA),26 as well as βTail peptides, βTail (MALKVELEKLKSELVVLHSELHKLKSEL, adapted from Pagel et al.22), W-βTail (WGSGSMALKVELEKLKSELVVLHSELHKLKSEL), and βTail mutant (MAGKPEGEKPKSEGGPGHSEGHKPKSEG), were synthesized using a standard Fmoc solid-phase peptide synthesis protocols. Peptide molecular weight was verified using MALDI-TOF-MS with α-cyano-4-hydroxycinnamic acid as the matrix.
Cloning, expression, and purification of β Tail proteins
See Supplemental information for detailed cloning, expression, and purification methods. Plasmids containing βTail-GFPuv were constructed starting with a pET-21d vector containing the recombinant genetic fusion cutinase-(Gly-Ser linker)-GFPuv-His6 (described previously in Hudalla et al.10). The cutinase domain was excized and replaced with the βTail or βTmutant sequence using synthetic oligonulceotides. βTail-cutinase was prepared by amplifying the cutinase gene out of the cutinase-(Gly-Ser linker)-GFP vector and cloning it into the βTail-(Gly-Ser linker)-GFP vector to replace the GFP domain. Vectors encoding βTail-eGFP, βTail-dsRED, and Q11-GFP were synthesized and subcloned into pET-21D by Genscript (New Jersey, USA). The sequence of each recombinant fusion was confirmed by sequencing performed at the University of Chicago Sequencing Facility (nucleotide and amino acid sequences provided in Supplemental Information). Fusion proteins were expressed in Origami B (DE3) E. coli, and purified using metal-affinity chromatography on HisPur cobalt resin (Thermo Scientific, IL). Endotoxin content was reduced using Triton X-114 cloud-point precipitation, according to previously reported methods.10
Nanofiber preparation
Lyophilized Q11 was dissolved in deionized water at a final concentration of 10 mM by vortexing and sonicating for 5 min. Aqueous Q11 solutions were diluted 10-fold with 1× PBS containing GFP (Vector Labs cat# MB-0752), βTmutant-GFP, or one or more βTail fusion proteins at a total protein concentration between 0.25-1.5 μM. Similarly, aqueous Q11 solutions were diluted 10-fold in 1× PBS containing synthetic βTail, tryptophan-terminated βTail (W-βTail), tryptophan-terminated mutant βTail (W-βTmut) or biotinylated βTail peptides at a concentration between 10-100 μM. These solutions were then incubated under static conditions for nanofiber assembly, or on a rocker table (speed setting 4, LabNet Rocker 35, New Jersey) to induce microgel formation.
Characterizing protein integration into nanofibers
Q11 nanofibers assembled overnight in the presence of βTail fusion proteins were sedimented by centrifugation at 12000×g for 5 min. The supernatant was removed and analyzed for βTail-GFP, βTail-eGFP, βTail-dsRED, βTmutant-GFP, Q11-GFP, GFP, or W-βTail content by measuring fluorescence emission with a SpectraMax M5 (excitation 395 nm/emission 503 nm for GFP, 474 nm/503 nm for eGFP; 556 nm/586 nm for dsRED; 280 nm/325 nm for W-βTail. Plastic plates were used for GFP and eGFP fluorescence measurements; glass-bottom 96-well plates were used for dsRED and W-βTail fluorescence measurements). Emission intensity was converted to protein concentration using appropriate protein or peptide standards. To measure nanofiber fluorescence, pelleted nanofibers were resuspended in fresh 1× PBS, and fluorescence was measured using the above excitation and emission wavelengths. The μBCA assay kit (Pierce) was used according to the manufacturer's instructions to determine βTail-cutinase concentration in the supernatant. The fluorescence of resuspended fibers plus the supernatant was equal to that of a GFP standard with the same concentration (Supplemental Fig. 3), and so supernatant fluorescence was used to assess protein concentration in the fibers. Specifically, protein concentration in the nanofibers was reported as the difference between the protein concentration in solution during nanofiber assembly and the protein concentration in the supernatant after centrifugation. Microgel fluorescence was analyzed using a Zeiss Axioscope inverted epifluorescent microscope, as described in detail in the Supplemental Information. Negligible cross-talk was observed between fluorescent channels (Supplemental Fig. 14). Due to differences in the quantum efficiency of each protein, exposure time was adjusted until the grayscale image intensity of microgels containing 0.33 μM βTail-GFP, βTail-eGFP, or βTail-dsRED alone was similar (e.g. 0.75 sec for βTail-GFP, 1.5 sec for βTail-eGFP, and 2.5 sec for βTail-dsRED). These exposure times were then used to collect grayscale images of microgels formed from solutions containing βTail-GFP, βTail-eGFP, and βTail-dsRED at different molar ratios with each fluorophore filter cube. Grayscale microgel images were pseudocolored red, green, or blue according to the filter cube set used, and then merged using ImageJ software (NIH). Predicted colors were calculated by using the protein mole ratio in solution during assembly as the RGB pixel ratio.
Cutinase activity
Cutinase activity was measured using p-nitrophenyl butyrate,28 as described in detail in the Supplemental Information. The initial velocity, vo, of pnitrophenyl butyrate hydrolysis to p-nitrophenol was calculated from the linear portion of a plot of A405 nm versus time.
Transmission Electron Microscopy
TEM was used to visualize βTail-GFP or biotinylated-βTail integration into Q11 nanofibers using established methods, with minimal modifications. Briefly, nanofibers were adsorbed onto 200 mesh lacey carbon grids, blocked with 2% acetylated bovine serum albumin (BSA)/0.1% cold water fish skin gelatin, and placed onto a series of droplets containing (1) monoclonal mouse anti-GFP antibody (Santa Cruz Biotechnology cat. #sc-9996, 1:4 in PBS), (2) goat anti-mouse IgG-15 nm gold particles (EMS cat. # 25133), and streptavidin-5 nm gold particles (Invitrogen cat. # A32360, all particles diluted 1:4 in PBS), and (3) 1% uranyl acetate in water. Triplicate PBS washes were performed between staining steps. Grids were analyzed with an FEI Tecnai F30 TEM. Detailed methods are provided in the Supplemental Information.
Circular dichroism
Circular dichroism was performed using an Aviv 202 circular dichroism spectrometer in the University of Chicago Biophysics Core. Solutions containing 10 μM or 25 μM βTail or βTmutant, 250 μM Q11, 25 μM βTail plus 250 μM Q11, or 25 μM βTmutant plus 250 μM Q11 in 1× phosphate buffer plus 137 mM potassium fluoride were analyzed after 30 min, 18 h, and 90 h (for βTail assembly kinetics, Fig 1) or 30 min and overnight assembly (for Q11 + βTail and the corresponding controls, Fig. 3) under static conditions at room temperature. Each sample was analyzed 3 times, and the averaged spectrum was reported.
Immunizations
Immune responses were measured in C57BL/6 mice (detailed methods provided in the Supplemental Information). Endotoxin content of all immunizations was determined with the Limulus Amoebocyte Lysate assay kit (Lonza) immediately before immunization, and all immunizations contained less than 1 EU/mL endotoxin. Female wild-type C57BL/6 mice (8-10 weeks old, Taconic Farms, IN) or female Toll-like receptor 4 knockout mice (20-24 weeks old, Jackson Laboratories) were given two 50 μL subcutaneous injections near the shoulder blades at each primary and booster immunization, similar to previously reported methods.10 The GFP dosing regimen was: 2.98 μg βTail-GFP with or without 1 mM Q11 at day 0, and 2.64 μg βTail-GFP with or without 1 mM Q11 at day 31; the cutinase dosing regimen was: 3.0 μg βTail-cutinase with or without 1 mM Q11 at days 0, 28, and 63. For nanofibers bearing both βTail-GFP and βTail-cutinase, the dosing regimen was 2.6 μg βTail-GFP and 3.0 μg βTail-cutinase at days 0, 28; 3.7 μg βTail-GFP and 3.0 μg βTail-cutinase at day 63, and 3.7 μg βTail-GFP at day 91. Blood was collected weekly via the submandibular vein. Institutional guidelines for the care and use of laboratory animals were strictly followed under a protocol approved by the University of Chicago's Institutional Animal Care and Use Committee.
ELISA
ELISA was conducted as previously reported,10 with minimal modifications (See Supplemental Information for detailed ELISA methods). For mice immunized with βTail-GFP, serum collected at week 7 was analyzed; for mice immunized with βTail-cutinase, serum collected at week 11 was analyzed. For isotyping ELISAs, goat anti-mouse IgG1 (Sigma cat M5532), IgG2a/c (M5657), IgG2b (M5782), IgG3 (M5907), or IgM (M6157) were utilized.
Supplementary Material
Acknowledgements
This research was supported by the National Institutes of Health (NIBIB, 1R01EB009701; NCI, U54 CA151880; NIAID, 1F32AI096769 and 5R21AI09444), the Chicago Biomedical Consortium with support from the Searle Funds at the Chicago Community Trust, and the National Science Foundation (CHE-0802286). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Biomedical Imaging and BioEngineering, the National Institute of Allergy and Infectious Disease, the National Cancer Institute, or the National Institutes of Health.
Footnotes
Contributions. J.H.C. directed the work, designed experiments, analyzed the data, and wrote the paper. G.A.H. designed experiments, conducted experiments, analyzed the data, and wrote the paper. T.S., J.Z.G., Y.F.T, and H.H. performed experiments and analyzed results. A.S.C. designed experiments and analyzed data.
Competing financial interests. JHC and GAH are named as inventors on a patent application filed by the University of Chicago that covers the technology described in this paper.
References
- 1.Baxa U, Speransky V, Steven AC, Wickner RB. Mechanism of inactivation on prion conversion of the Saccharomyces cerevisiae Ure2 protein. Proc Natl Acad Sci U S A. 2002;99:5253–5260. doi: 10.1073/pnas.082097899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leng Y, et al. Integration of a Fluorescent Molecular Biosensor into Self-Assembled Protein Nanowires: A Large Sensitivity Enhancement. Angewandte Chemie International Edition. 2010;49:7243–7246. doi: 10.1002/anie.201002452. [DOI] [PubMed] [Google Scholar]
- 3.Men D, et al. Seeding-induced self-assembling protein nanowires dramatically increase the sensitivity of immunoassays. Nano Lett. 2009;9:2246–2250. doi: 10.1021/nl9003464. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin AJ, et al. Cytochrome display on amyloid fibrils. J Am Chem Soc. 2006;128:2162–2163. doi: 10.1021/ja0565673. [DOI] [PubMed] [Google Scholar]
- 5.Horii A, Wang X, Gelain F, Zhang S. Biological designer self-assembling peptide nanofiber scaffolds significantly enhance osteoblast proliferation, differentiation and 3-D migration. PLoS One. 2007;2:e190. doi: 10.1371/journal.pone.0000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, et al. Peptide nanofibers preconditioned with stem cell secretome are renoprotective. J Am Soc Nephrol. 2011;22:704–717. doi: 10.1681/ASN.2010040403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matson JB, Stupp SI. Drug release from hydrazone-containing peptide amphiphiles. Chem Commun (Camb) 2011;47:7962–7964. doi: 10.1039/c1cc12570b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veiga AS, et al. Arginine-rich self-assembling peptides as potent antibacterial gels. Biomaterials. 2012;33:8907–8916. doi: 10.1016/j.biomaterials.2012.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Black M, et al. Self-assembled peptide amphiphile micelles containing a cytotoxic T-cell epitope promote a protective immune response in vivo. Adv Mater. 2012;24:3845–3849. doi: 10.1002/adma.201200209. [DOI] [PubMed] [Google Scholar]
- 10.Hudalla GA, et al. A Self-Adjuvanting Supramolecular Vaccine Carrying a Folded Protein Antigen. Adv Healthc Mater. 2013 doi: 10.1002/adhm.201200435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rudra JS, Tian YF, Jung JP, Collier JH. A self-assembling peptide acting as an immune adjuvant. Proc Natl Acad Sci U S A. 2010;107:622–627. doi: 10.1073/pnas.0912124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collier JH, Rudra JS, Gasiorowski JZ, Jung JP. Multi-component extracellular matrices based on peptide self-assembly. Chem Soc Rev. 2010;39:3413–3424. doi: 10.1039/b914337h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woolfson DN, Mahmoud ZN. More than just bare scaffolds: towards multi-component and decorated fibrous biomaterials. Chem Soc Rev. 2010;39:3464–3479. doi: 10.1039/c0cs00032a. [DOI] [PubMed] [Google Scholar]
- 14.Cui H, Webber MJ, Stupp SI. Self-assembly of peptide amphiphiles: from molecules to nanostructures to biomaterials. Biopolymers. 2010;94:1–18. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brodin JD, et al. Metal-directed, chemically tunable assembly of one-, twoand three-dimensional crystalline protein arrays. Nat Chem. 2012;4:375–382. doi: 10.1038/nchem.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King NP, et al. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science. 2012;336:1171–1174. doi: 10.1126/science.1219364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinclair JC, Davies KM, Venien-Bryan C, Noble ME. Generation of protein lattices by fusing proteins with matching rotational symmetry. Nat Nanotechnol. 2011;6:558–562. doi: 10.1038/nnano.2011.122. [DOI] [PubMed] [Google Scholar]
- 18.Sangiambut S, et al. A Robust Route to Enzymatically Functional, Hierarchically Self-Assembled Peptide Frameworks. Adv Mater. 2013 doi: 10.1002/adma.201204127. [DOI] [PubMed] [Google Scholar]
- 19.Kim W, et al. A high-throughput screen for compounds that inhibit aggregation of the Alzheimer's peptide. ACS Chem Biol. 2006;1:461–469. doi: 10.1021/cb600135w. [DOI] [PubMed] [Google Scholar]
- 20.Wu W, Xing L, Zhou B, Lin Z. Active protein aggregates induced by terminally attached self-assembling peptide ELK16 in Escherichia coli. Microb Cell Fact. 2011;10:9. doi: 10.1186/1475-2859-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collier JH, Messersmith PB. Enzymatic modification of self-assembled peptide structures with tissue transglutaminase. Bioconjug Chem. 2003;14:748–755. doi: 10.1021/bc034017t. [DOI] [PubMed] [Google Scholar]
- 22.Pagel K, et al. How metal ions affect amyloid formation: Cu2+- and Zn2+-sensitive peptides. Chembiochem. 2008;9:531–536. doi: 10.1002/cbic.200700656. [DOI] [PubMed] [Google Scholar]
- 23.Jung JP, Moyano JV, Collier JH. Multifactorial optimization of endothelial cell growth using modular synthetic extracellular matrices. Integr Biol (Camb) 2011;3:185–196. doi: 10.1039/c0ib00112k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jung JP, et al. Co-assembling peptides as defined matrices for endothelial cells. Biomaterials. 2009;30:2400–2410. doi: 10.1016/j.biomaterials.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marini DM, Hwang W, Lauffenburger DA, Zhang S, Kamm RD. Left-Handed Helical Ribbon Intermediates in the Self-Assembly of a β-Sheet Peptide. Nano Letters. 2002;2:295–299. [Google Scholar]
- 26.Zhang S, et al. Self-complementary oligopeptide matrices support mammalian cell attachment. Biomaterials. 1995;16:1385–1393. doi: 10.1016/0142-9612(95)96874-y. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi Y, Ueno A, Mihara H. Heterogeneous assembly of complementary peptide pairs into amyloid fibrils with alpha-beta structural transition. Chembiochem. 2001;2:75–79. doi: 10.1002/1439-7633(20010105)2:1<75::AID-CBIC75>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 28.Kolattukudy PE, Purdy RE, Maiti IB. In: Methods in Enzymology. Lowenstein John M, editor. Vol. 71. Academic Press; 1981. pp. 652–664. [Google Scholar]
- 29.Chen J, et al. The use of self-adjuvanting nanofiber vaccines to elicit high-affinity B cell responses to peptide antigens without inflammation. Biomaterials. 2013;34:8776–8785. doi: 10.1016/j.biomaterials.2013.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malyala P, Singh M. Endotoxin limits in formulations for preclinical research. J Pharm Sci. 2008;97:2041–2044. doi: 10.1002/jps.21152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.