

Abstract
The role of the endocrine vitamin D pathway in regulating the serum calcium concentration in man is well described. In the presence of a low serum calcium level, the vitamin D metabolic pathway is called upon to produce more of the active vitamin D hormone, 1, 25-dihydroxyvitamin D (1, 25-D), via up-regulation of the CYP27b1-hydroxylase activity in the kidney. The consequence is mobilization of skeletal calcium stores to return the circulating calcium level back to the normal range. On the other hand, when the serum calcium level increases, endocrine forces move to suppress renal 1, 25D production and squelch bone resorption. It is now known that vitamin D metabolites also operate in an autocrine, paracrine and intracrine mode to control vitamin D receptor-directed biological responses at the tissue level. Because the metabolism and action of vitamin D occurs at the tissue level in this situation, use of circulating vitamin D metabolites and biomarkers to ascertain the local action of the vitamin D pathway is often misleading. This mini-review seeks to compare and contrast the operation of the vitamin D pathway at the endocrine and tissue level.
Keywords: metabolism, endocrine, autocrine, hormone, biomarkers
Introduction
Following a summary of vitamin D metabolism and action, and a discussion of the value of the circulating 25-hydroxy vitamin D level as a biomarker of nutritional vitamin D sufficiency, this mini-review will consider four of the generally-accepted biological roles of vitamin D in the human host. At physiological extremes, the endocrine vitamin D system is used to defend the host against the development of low and high blood levels of calcium. In between these two extremes, with calcium levels existing in the normal range, are intracrine, autocrine and/or paracrine actions of vitamin D to sustain skeletal health and promote the innate immune response.
Cutaneous synthesis, metabolism and action of vitamin D
The left panel of Figure 1 depicts the classically-held concept of human vitamin D endocrinology. Vitamin D is synthesized nonenzymatically in the epidermis via photoconversion of 7-dehydrocholesterol to previtamin D, with subsequent thermal isomerization of pre-vitamin D to vitamin D. By yet undetermined means, the preprohormone vitamin D escapes the skin into the general circulation of the host. Circulating vitamin D is subject to a series of metabolic conversion steps. The first metabolic step for vitamin D is catalyzed by one of many cytochrome P450-linked, substrate-dependent, non-saturable 25-hydroxylases, most of which are enriched in the liver of the host (1). 25-Hydroxyvitamin D (25OHD) is a prohormone; it is not biologically active unless serum concentrations of free/bioavailable 25OHD are sufficient to act as an analogue of the vitamin D hormone 1,25-dihydroxyvitamin D (1,25-(OH)2D) at the level of bone (2). 25OHD is the preferred substrate for the mitochondrial CYP27b1-hydroxylase that resides principally in the proximal tubular epithelial cell of the kidney. Unlike the vitamin D 25-hydroxylases in the kidney, the CYP27b1-hydroxylase is not product dependent, saturable and is tightly regulated by two circulating hormones, parathyroid hormone (PTH) and fibroblast growth factor-23 (FGF23) (3); in fact, serum concentrations of substrate 25OHD are 1 000-fold greater than those of product 1,25-(OH)2D. The CYP27b1-hydroxylase can also be expressed in extrarenal tissues in normal physiological circumstances in the human; including the placenta, the parathyroid and perhaps bone and intestine (4). In cases of microbial invasion of the host, cells of the innate immune system may also express the CYP27b1 gene (5). 1,25-(OH)2D is the natural ligand for the vitamin D receptor (VDR). When liganded and dimerized with the RXR (retinoid X receptor), the VDR-RXR acts as the principal transregulator of vitamin D-driven bioresponses in the target cell. Both 25OHD and 1,25-(OH)2D are preferred substrates for the CYP24a1-24-hydroxyalse (6). Like the CYP27b1-hydroxylase, the 24-hydroxylase is a mitochondrial-based mixed function oxidase. Unlike the CYP27b1-hydroxylase, the 24-hydroxylase is ubiquitously distributed among human tissues and serves to convert 25OHD and 1,25-(OH)2D to the non-biologically active metabolites 24,25-(OH)2D and 1,24,25-(OH)3D, respectively. In an endocrine sense, expression of CYP24a1 gene is principally under the control of the 1,25-(OH)2D-liganded VDR-RXR in tissues; 1,25-(OH)2D has the potential to upregulate expression of the CYP24a1 gene many hundred-fold. 24-hydroxylation of any vitamin D metabolite serves as the first enzymatic step in side chain cleavage of the metabolite and its conversion to a water-soluble, excretable catabolite.
Figure 1.
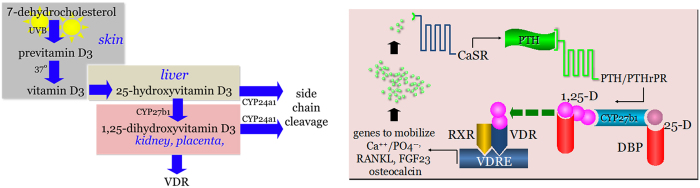
The endocrinology of vitamin D. The left panel depicts the essentials of the endocrine synthesis, metabolism and action of vitamin D. The key enzyme in the synthesis of the active, vitamin D receptor-(VDR) interacting metabolite, 1,25-dihydroxyvitamin D, is the CYP27b1-hydroxylase. In normal human endocrine physiology the CYP27b1-hydroxylase is expressed principally in the kidney but also in the placenta. The catabolic CYP24a1-24-hydroxylase is ubiquitously distributed among human tissues and serves to inactivate 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D. The right panel shows the endocrine responses of the vitamin D pathway to a hypocalcemic challenge and the human host's use of the skeleton to restore blood calcium levels back to normal. Abbreviations include: CaSR, calcium sensing receptor; PTH, parathyroid hormone; PTH/PTHrP, receptor for PTH and parathyroid hormone-related protein; DBP, serum vitamin D binding protein; RXR, retinoid X receptor; VDRE, vitamin D response element; Ca2+, calcium ion; PO43−, phosphate ion; RANKL, receptor activator of nuclear factor kappa-B ligand; FGF23, fibroblast growth factor-23. Figure adapted with the permission of the original authors.
Utility of circulating 25-hydroxyvitamin D
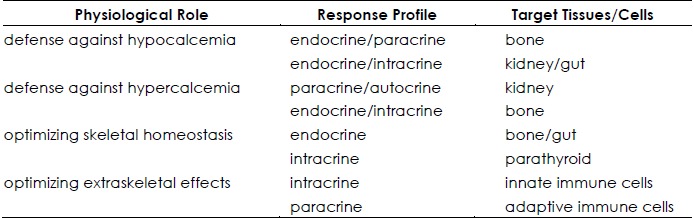
It is generally held that the total circulating 25OHD level is the most informative measure of the vitamin D deficient: sufficient:intoxicated state of the host, taking into account not only vitamin D made by cutaneous photosynthesis, but also that absorbed in the distal small bowel after ingestion of the preprohormone. Also considered true is the fact that the active metabolite of vitamin D,1,25-(OH)2D, is responsible for most, if not all, of the downstream biological responses to vitamin D (left panel, Figure 1). If this is the case, then how does one explain the following facts? One, it is a low serum level of 25OHD, not 1,25-(OH)2D, that is associated with an elevation in the serum Parathyroid Hormone (PTH) level and diminished intestinal calcium absorption efficiency (7). In fact, serum levels of the active hormone 1,25-(OH)2D may be high-normal or even elevated, not diminished, in the presence low 25OHD levels (8). Two, it is also a low serum 25OHD level, and not a low 1,25-(OH)2D, level that is associated with a multitude of adverse outcomes involving skeletal, cardiovascular, metabolic (e.g. diabetes) health as well as overall mortality (9). What then are the possible explanations for these counter-intuitive observations? Perhaps the circulating level of 1,25-(OH)2D is not relevant, because the hormone cannot gain access to the response: signal transduction machinery (VDR) in the target tissue. Alternatively, perhaps the target tissue is not designed to use 1,25-(OH)2D that is derived from the circulation but rather to convert circulating 25OHD to 1,25-(OH)2D inside that cell and use the locally generated active metabolite to drive vitamin D-bioresponses in that cell. Here we will attempt to address these controversial concepts by ascribing the principal actions of the vitamin D signaling system to defend the human host from hypocalcaemia, on the one hand, and hypercalcemia on the other. In between these two extremes, which we rarely observe clinically, is the role of the vitamin D system to maximize skeletal health and immune surveillance in the host (Table 1).
Table 1. Four prototypical physiological roles with response profile and biological targets for vitamin D and its metabolites for each.
Vitamin D in the defense of hypocalcaemia
As shown in the right panel of Figure 1, the activity of the G-coupled protein calcium sensing receptor (CaSR) embedded in the plasma membrane of the PTH-producing parathyroid epithelial cell (so-called chief cell) is increased in the calcium-unliganded state, i.e., when the extracellular ionized calcium concentration drops below the range of normal (10). The activated CaSR signals to increase the release of preformed PTH, and to stimulate expression of the PTH gene (11). The former occurs acutely, releasing PTH into the general circulation. This endocrine PTH signal is received by the G-coupled PTH-PTHrP (parathyroid hormone related protein) receptor in the plasma membrane of the apical (urine-facing) and basolateral (blood-facing) surface of tubular epithelial cells in the kidney (12), as well as in chondrocytes (13), osteoblasts (14) and osteocytes (15) in bone.
As noted in the section above, in the kidney, PTH is known to up-regulate expression of the CYP27b1 gene that converts to 25OHD to 1,25-(OH)2D. By as yet undetermined means, release of 1,25-(OH)2D back into the general circulation at the basolateral surface of the renal epithelial cell is thought to exert an acute endocrine effect in vivo at the transcriptional level in osteoblasts and osteocytes to up-regulate expression of at least three genes, RANKL (16), FGF23 (3) and undecarboxylated osteocalcin (17). RANKL ligand promotes osteoclastogenesis with subsequent release of calcium (and phosphate) and the matrix-embedded, insulin secretagogue-undecarboxylated osteocalcin into the circulation (18). Interaction of 1,25-(OH)2D with VDR in osteocytes increases the production of FGF23 which, when released locally into the canalicular space of bone (Table 1), suppresses osteoblastic bone-forming activity through interaction with the FGF receptors (19). These concerted actions ensure that the dominant endocrine effect of 1,25-(OH)2D in bone is osteolytic in nature. The central PTH-responding cell in adult bone is the osteoblast, and its principal target is the RANKL gene (20). This way PTH and 1,25-(OH)2D act cooperatively to promote bone resorption and restoration of the serum ionized calcium concentration toward normal. The remodeling skeleton is by far and away the richest and most accessible source of calcium to the circulation. This reservoir can be tapped to yield up to 2 000 mg of calcium on a daily basis, as it sometimes is in patients with humoral hypercalcemia of malignancy.
While bone cell-targeted effects of 1,25-(OH)2D predominate in restoring serum calcium levels toward normal, there are other endocrine effects of the vitamin D hormone that reinforce these actions on the skeleton. Circulating 1,25-(OH)2D appears to have a calcium-sparing effect at the level of the kidney and gut. Acting on the plasma membrane calcium pump, apical calcium channel TRPV5, and calbindin28k targets in the distal tubular epithelial cell, circulating 1,25-(OH)2D acts to enhance distal renal tubular reabsorption of calcium and diminishing urinary excretion of calcium (21). Quantitatively speaking, this represents a maximum savings of 200 mg calcium on a daily basis, far below that which can be mobilized from the skeleton in a similar time span; 1,25-(OH)2D has no apparent impact on the handling of calcium in the more proximal segments of the nephron, where up to 8 000 mg of calcium are reclaimed daily from the urine. Circulating 1,25-(OH)2D also promotes the efficiency of intestinal calcium absorption, principally in the proximal small bowel, by promoting expression of the transcellular chaperone protein genes Calbindin-D9k, the basolateral membrane-based ATPase (PMCA1b) and TRPV6 (22,23). These events are conditional on the ingestion of calcium, with the potential benefit to the calcium economy of the host being relatively small, on the order of 100–150 mg calcium daily (24).
Vitamin D in the modulation of human skeletal health
Since the various vitamin D 25OHD-hydroxylases in the human are substrate dependent and non-limiting in terms of product synthesis, any vitamin D synthesized cutaneously or entering the general circulation via intestinal absorption after oral consumption, and not distributed to storage in fat and muscle (9), is converted to 25OHD. This and the facts that i) 25OHD is bound avidly by the serum vitamin D binding protein (DBP) and ii) the 25OHD-DBP complexes are effectively recovered from the glomerular filtrate (urine) and recycled into the general circulation are responsible for the relatively long half-life of 25OHD in the blood (25,26). While these facts explain the utility of using the total serum 25OHD level as a measurement of the amount of the 25OHD generally available to be converted to 1,25-(OH)2D, they are not an adequate explanation for the direct association of the prohormone, 25-OHD, and not 1,25-(OH)2D, levels with bone health. We propose that there are logical explanations for this physiological conundrum which are dependent on the non-substrate-dependent, extrarenal synthesis of 1,25-(OH)2D in an intracrine fashion (see Table 1); events that are not detectable by routine measurement of 25OHD and its metabolites in the serum.
One explanation is the diminished intracellular conversion of serum 25OHD to 1,25-(OH)2D in the parathyroid cell itself. While signaling through the unliganded CaSR is the major upregulator of secreted PTH, 1,25-(OH)2D acting via the interaction with the VDR in the parathyroid cell is a major inhibitor of PTH synthesis and release (27). Except in extreme, iatrogenic circumstances where 1,25-(OH)2D or related analogues are delivered parenterally at high doses therapeutically (as it is during dialysis of patients with chronic renal failure) (28), the human host does not achieve extracellular (serum) 1,25-(OH)2D levels high enough to have an impact on circulating PTH concentrations. On the other hand, when serum 25OHD levels are raised to the normal range (>20 ng·mL−1 or 50 nmol·L−1), the CYP27b1-hydroxylase in the PTH-producing cell is provided sufficient substrate to generate enough 1,25-(OH)2D intracellularly via the CYP27b1-hydroxylase to engage the VDR, and to activate the 1,25D-VDR-RXR transacting complex to interact with inhibitory vitamin D response elements (VDREs) in and around the PTH gene and to limit PTH production (27). The failure of this intracrine, substrate-driven process of 25OHD conversion to 1,25-(OH)2D via the resident CYP27b1-hydroxylase and subsequent bioaction of 1,25-(OH)2 D, was first described for human macrophages exposed to vitamin D deficient serum (5), but now appears to be applicable to the CYP27b1-VDR-signaling apparatus in a number of different cell types in humans. In this scenario, when the serum 25OHD level is low, as it is in the vitamin D deficient state, the intraparathyroid cell synthesis of 1,25-(OH)2D is reduced, and CaSR-mediated upregulation of PTH gene expression and secretion dominates. Upon boosting the extracellular 25OHD level, as occurs with vitamin D therapy of the host, sufficient free 25OHD enters the parathyroid cell to be converted to 1,25-(OH)2D in an intracrine mode, and to counter CaSR-stimulated PTH gene expression and release. In turn, this event limits PTH stimulation of the renal CYP27b1-hydroxylase and RANKL gene expression in the osteoblast (see right panel, Figure 1). A decrease in local RANKL production diminishes RANKL-driven osteoclastogenesis, chronic bone resorption, and a decrease in skeletal mass and strength (osteoporosis). This set of circumstances would explain the inverse correlation observed between serum PTH and total 25OHD levels in cross-sectional studies of human with osteoporosis (29) as well as the reversal of this association when vitamin D therapy is successful in increasing the 25OHD level to >20–30 ng·mL−1 (50–75 nmol·L−1).
A second possibility to explain the direct association of the prohormone 25-OHD, and not 1,25-(OH)2D, levels with bone health, is the diminished local conversion of 25OHD to 1,25-(OH)2D in VDR-expressing bone cells including chondrocytes, osteoblast precursors, mature osteoblasts, osteocytes, and osteoclasts precursors, and mature osteo-clasts (30). The prototypical histomorphometric picture in cancellous (trabecular) bone for vitamin D deficiency is first an increase in uncalcified osteoid, detected by an increase in the osteoid surface:bone surface (OS:BS) ratio (31). In more severe cases, this increase in OS:BS is followed by an increase in osteoid thickness (O.th). Until recently, there have been no studies correlating serum 25OHD levels with findings on bone biopsies. Priemel, et al (32) examined 675 iliac crest bone biopsies and total serum 25OHD concentrations from male and female individuals residing in northern Germany (53° north latitude) at the time of autopsy. As with most large-scale, cross-sectional studies, the serum 25OHD was assessed by immunoassay using Daisorin reagents, a measurement found to be stable for up to ten days postmortem. The mean age of the deceased males and females was 59 and 68 years, respectively. For the population at large, the mean 25OHD level for both males and females was 7.2 ng·mL−1 (18 nmol·L−1), a level considered to be well into the vitamin D deficient range (<20 ng·mL−1 or 50 nmol·L−1) by IOM criteria (33). Surprisingly, only 4% of the population had evidence of an increase in osteoid thickness (O.th.), the hallmark of the mineralization defect observed with osteomalacia. Conversely, the 75% of the population with a 25OHD level <10 ng·mL−1 (<25 nmol·L−1) displayed normal O.th. (≤10%) on biopsy. These results suggest that osteomalacia was not observed in 75% of the study subjects despite 25OHD levels unequivocally in the vitamin D deficient range. This observation suggests that the histomorphometric skeletal lesions observed in most vitamin D deficient adult men and women is not solely one of classical osteomalacia. Thus there are likely other pathophysiological events such as secondary hyperparathyroidism that contribute to the abnormal increases in osteoid surface (OS:BS) seen in 36% of subjects with 25OHD levels <20 ng·mL−1. These observations are reminiscent of patients with the mixed histomorophometric picture of subjects with renal osteo-dystrophy-related mineralization defect (34).
In our view, a more likely explanation for the confounding results is that the serum 25OHD is not the best biomarker for vitamin D metabolite action at the level of bone. There are three explanations for this circumstance. First, as previously mentioned, 25OHD itself is biologically inert except at concentrations high enough to bind to and transactivate the VDR (levels usually >100 ng·mL−1 or 250 nmol·L−1). Second, we do not routinely measure the free/bioavailable fraction of 25-OHD, or that of the active 1,25-(OH)2D metabolite in the blood that is accessible to the interior of target bone cells. Third, all three of the prototypical bone cells, osteoblasts, osteocytes and osteoclasts, have been described to express the CYP27b1-hydroxylase and VDR (35) and, like the human macrophage and parathyroid cell, appear to be capable synthesizing and responding to 1,25-(OH)2D in an intracrine mode (Table 1). In fact, the best biomarker of the intracellular 1,25-(OH)2D fraction available on a routine basis may be the serum PTH level, a hormone whose production is regulated by intracellularly generated and acting 1,25-(OH)2D (36).
Vitamin D in defense of microbial invasion
The landmark studies of Liu et al (5) led us consider vitamin D sufficiency as a crucial element in the maintenance of the normal human innate immune response to microbial pathogens. Microbial pathogens shed membrane elements (pathogen-associated membrane patterns or PAMPs) that, in turn, stimulate pattern recognition receptor (PRR) activation on the surface on innate immune cells (most notably the macrophage), some with downstream initiation of expression of a vitamin D signaling cascade. Take the example of exposure of the human host to Mycobacterium tuberculosis (M. tb.) (left panel, Figure 2). M. tb. PAMPs interact with the PRR toll-like receptor 2/1 (TLR 2/1) (37,38). This cell surface interaction event results in signal transduction through the MyD88-directed NF κ B pathway, with induction of expression of two vitamin D-related genes, the CYP27b1 and the VDR. The former gene product is capable of synthesizing 1,25-(OH)2D intracellularly, and the latter capable of binding 1,25-(OH)2D and transactivating VDR-directed antimicrobial genes. When the extracellular concentration of 25OHD falls below the equivalent of approximately 20 ng·mL−1 or 50 nmol·L−1, the intracellular production of 1,25-(OH)2D via the substrate-driven, macrophage CYP27b1-hydroxylase becomes limiting, and is unable to generate enough active vitamin D metabolite to effectively ligand enough VDR in that cell to promote expression of the antimicrobial genes (39). The end result is failure of the macrophage to mount effective autophagy-related vesicular killing of ingested M. tb. (40). This failure can be rescued in a concentration-dependent fashion by the addition of 25OHD to the extracellular medium (5), or by exchanging vitamin D deficient human serum with vitamin D sufficient serum (>30 ng·mL-1 or 70 nmol·L−1) from the same host after treatment of the host with vitamin D in vivo (39).
Figure 2.
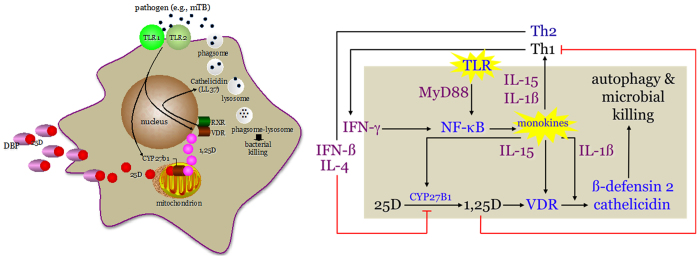
The intra-, auto- and paracrinology of the vitamin D system in the local inflammatory microenvironment. The left panel shows the intracellular metabolism of extracellular substrate 25-dihydroxyvitamin D (25D) and intracrine action of the CYP27b1-hydroxylase product 1,25-dihydroxyvitamin D metabolite (1,25D) in the Toll-like receptor-(TLR2-1 dimer) activated human macrophage. TLR2-1 stimulation initiates expression of the vitamin D receptor (VDR) and CYP27b1-hydroxylase genes. Intracellularly-synthesized 1,25Ddrives expression of the anti-microbial gene cathelicidin (LL37) to kill ingested Mycobacterium tuberculosis (M. tb). The right panel shows the signaling pathways in the intracrine, autocrine and paracrine modes that mediate the concerted TLR-driven, interferon-gamma-(IFN-γ) amplified innate immune response in the macrophage and the IFN-β- and 1,25D-directed feedback inhibited adaptive immune response of activated, VDR-expressing Th lymphocytes (Th). Other abbreviations include: DBP, serum vitamin D binding protein; and myD88, myeloid differentiation primary response gene (88). Figure adapted with the permission of the original authors.
As noted in Table 1, the antimicrobial capacity of the macrophage is subject to intracrine, autocrine and paracrine feedforward and feedback immune regulatory circuits. This regulatory network is depicted schematically in the right panel of Figure 2. Stimulation of the TLR2/1 signaling pathway leads not only to expression of the CYP27b1 and VDR, but also to cytokines IL-15 and IL-1β. In an autocrine mode, these two monokines act to promote 1,25-(OH)2D-VDR-directed generation of the antimicrobials β-defensin2 and cathelicidin (41), and in a paracrine fashion to mobilize and activate cells of the adaptive immune response. Activation of the Th1 subset of lymphocytes promotes the local production of IFN-γ, the most potent known stimulator of the macrophage CYP27b1-hydroxylase (42,43). On the other hand, the proliferation Th2 cells under the influence of locally-released inflammatory cytokines, leads to counter production of the lymphokines IFN-β and IL-4 which inhibit 1-hydroxyalse activity, principally by promoting expression of an appropriately-spliced CYP24a1-24-hydroxylase gene (43). Finally, when intracellular, IFN-γ-driven production of 1,25-(OH)2D is robust enough to allow escape of the hormone into the local, pericellular inflammatory microenvironment, 1,25-(OH)2D is concentrated in sufficient quantities to interact with the VDR, which is expressed in activated, but not quiescent, lymphocytes. The major bioresponse in the affected lymphocyte populations is inhibition of proliferation with the rank order of 1,25-(OH)2D-directed inhibition being Th1>Th2. As such, the predominant paracrine actions of 1,25-(OH)2D is to squelch the adaptive immune response (44). It is proposed that this negative feedback control of IFN-β and IL-4 on 1,25-(OH)2D production, teamed with the aforementioned antiproliferative actions of the hormones on cells of adaptive immune response, acts to prevent an overzealous adaptive immune response to intracellular pathogens like M. tb., while at the same time promoting the intracellular destruction of the pathogen. In this scenario, vitamin D sufficiency of the host is paramount in providing the optimal IFN-γ-mediated feedforward antimicrobial response to M. tb. and IFN-β-mediated feedback control on 1,25-(OH)2D synthesis by the macrophage, a circumstance that not infrequently occurs in certain granuloma-forming diseases including tuberculosis (45).
Vitamin D in defense of hypercalcemia
The endocrine effects of 1,25-(OH)2D to counter the pathophysiological state of hypercalcemia in the human host are perhaps best exemplified by diseases where the extrarenal overproduction of 1,25-(OH)2D occurs (46). In these disorders, the CYP27b1-hydroxylase gene is over-expressed in inflammatory cells, usually a macrophage. The macrophage is resistant to the circulating hormones that normally regulate 1,25-(OH)2D production via the CYP27b1 gene product in the proximal renal distal tubular epithelial cell (42). In contrast to renal tubular epithelial cells, the CYP27b1-hydroxylase in the macrophage is not subject to stimulatory control by circulating PTH or inhibition by circulating FGF23, because innate immune cells express neither the PTH-PTHrPR nor the FGFRs (30). On the other hand, macrophage CYP27b1 enzymatic activity is largely substrate 25(OH)D-dependent (42); much of the substrate dependence of the macrophage CYP27b1 is the consequence of alternative splicing of the CYP24a1 gene in this cell into a translated isoform missing the N-terminal mitochondrial targeting sequence (47). By comparison, when 1,25-(OH)2D is produced for export and endocrine function by the CYP27b1-hydroxylase resident in the proximal tubular epithelial cell of the kidney, the CYP27b1 gene is under stringent endocrine inhibition by elevated circulating PTH and low FGF23 levels (Table 1). Moreover, 1,25-(OH)2D is the major stimulator of the degradatory CYP24A1 pathway (left panel, Figure 1). With the possible exception of cells of the human innate immune response (6), nearly every cell in the host that expresses the VDR also expresses an active 24-hydroxy-lase degradation pathway.
From a physiological perspective, persistent increases in the serum calcium concentration increases the excitatory threshold, inhibiting nerve and neuromuscular firing; as such, hypercalcemia is manifested clinically by obtundation and dysfunction of electro-myocardial signaling in the host, both of which are life-threatening if not reversed. Endocrine suppression of the 1,25-(OH)2D synthetic pathway and upregulation of the CYP24a1-24-hydroxylase catabolic pathway are two of the principal mechanisms employed by the host to counter hypercalcemia (left panel, Figure 1), actually moving to reverse the host response to the hypocalcemic state (right panel, Figure 1). When the serum ionized calcium concentration increases above normal, signaling through the CaSR in the parathyroid cell is suppressed, PTH synthesis and release is inhibited, and PTH-driven CYP27b1-hydroxylase activity in the proximal renal tubular epithelial cell diminishes. This decrease in circulating PTH and 1,25-(OH)2D cooperate to reduce RANKL and FGF23 gene expression (48) and to coordinate a reciprocal downturn in osteoclastogenesis and an upturn osteoblastic bone forming activity, respectively, resulting in a net decrease in bone resorption to counter the hypercalcemic state. As mentioned previously, there is a complimentary negative feedback loop within the cell to diminish local concentrations of the biologically active hormone 1,25-(OH)2D and its substrate 25OHD (left panel, Figure 1). The CYP24a1-24-hydroxylase converts 1,25-(OH)2D and 25OHD to non-biologically active metabolites, “short circuiting” the calcemic actions of the 1,25-(OH)2D metabolite.
Summary
There is no doubt that the endocrine vitamin D synthetic/metabolic pathway is used by the human host to maintain the serum calcium level in a prescribed physiological range (Table 1). The endocrine pathway to synthesis of the active vitamin D metabolite 1,25-(OH)2D is activated in the presence of hypocalcemia and suppressed in the hypercalcemic state. What is far less clear is how vitamin D pathway operates in the normocalcemic state. We propose that it is a function of the intracrine production and intracrine/paracrine action of 1,25-(OH)2D at extrarenal sites, including the intestine, parathyroid, innate immune cells and bone, sites that are out of the reach or our serum-based assays. Otherwise, how do we explain so-called “vitamin D deficient” individuals with “low” circulating levels of 25OHD, but with relatively elevated 1,25-(OH)2D, that have impaired intestinal absorption and compromised bone health (e.g., osteoporosis), events that are thought to be dependent on 1,25-(OH)2D?
Acknowledgments
Authors' roles: JSA produced the first and updated drafts of the manuscript. All other authors reviewed and revised the final draft. We thank Martin Hewison for reviewing the manuscript. This work was supported by 1) NIH/NCRR/NCATS UCLA CTSI Grant Number UL1TR000124, and 2) NIH/NIAID Grant Number R01AI73539. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
References
- Zhu J, DeLuca HF. Vitamin D 25-hydroxylase—Four decades of searching, are we there yet? Arch Biochem Biophys. 2012;523:30–36. [DOI] [PubMed] [Google Scholar]
- Endres DB. Investigation of hypercalcemia. Clin Biochem. 2012;45:954–963. [DOI] [PubMed] [Google Scholar]
- Quarles LD. Role of FGF23 in vitamin D and phosphate metabolism: implications in chronic kidney disease. Exp Cell Res. 2012;318:1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JS, Hewison M. Extrarenal expression of the 25-hydroxyvitamin D-1-hydroxylase. Arch Biochem Biophys. 2012;523:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zügel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–1773. [DOI] [PubMed] [Google Scholar]
- Jones G, Prosser DE, Kaufmann M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): its important role in the degradation of vitamin D. Arch Biochem Biophys. 2012;523:9–18. [DOI] [PubMed] [Google Scholar]
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281. [DOI] [PubMed] [Google Scholar]
- Chapuy MC, Preziosi P, Maamer M, Arnaud S, Galan P, Hercberg S, Meunier PJ. Prevalence of vitamin D insufficiency in an adult normal population. Osteoporos Int. 1997;7:439–443. [DOI] [PubMed] [Google Scholar]
- Adams JS, Hewison M. Update in vitamin D. J Clin Endocrinol Metab. 2010;95:471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EM. The calcium-sensing receptor: physiology, pathophysiology and CaR-based therapeutics. Subcell Biochem. 2007;45:139–167. [DOI] [PubMed] [Google Scholar]
- Kumar R, Thompson JR. The regulation of parathyroid hormone secretion and synthesis. J Am Soc Nephrol. 2011;22:216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajwa A, Forster MN, Maiti A, Woolbright BL, Beckman MJ. Specific regulation of CYP27B1 and VDR in proximal versus distal renal cells. Arch Biochem Biophys. 2008;477:33–42. [DOI] [PubMed] [Google Scholar]
- Wysolmerski JJ. Parathyroid hormone-related protein: an update. J Clin Endocrinol Metab. 2012;97:2947–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takyar FM, Tonna S, Ho PW, Crimeen-Irwin B, Baker EK, Martin TJ, Sims NA. EphrinB2/EphB4 inhibition in the osteoblast lineage modifies the anabolic response to parathyroid hormone. J Bone Miner Res. 2012. [Epub ahead of print]. [DOI] [PubMed]
- Qing H, Ardeshirpour L, Pajevic PD, Dusevich V, Jahn K, Kato S, Wysolmerski J, Bonewald LF. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J Bone Miner Res. 2012;27:1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Takahashi F, Takahashi N. Bone effects of vitamin D — Discrepancies between in vivo and in vitro studies. Arch Biochem Biophys. 2012;523:22–29. [DOI] [PubMed] [Google Scholar]
- Fukumoto S, Martin TJ. Bone as an endocrine organ. Trends Endocrinol Metab. 2009;20:230–236. [DOI] [PubMed] [Google Scholar]
- Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, Chen D, Faugere MC, Aja S, Hussain MA, Bruning JC, Clemens TL. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 2010;142:309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesseling-Perry K, Juppner H. The osteocyte in CKD: New concepts regarding the role of FGF23 in mineral metabolism and systemic complications. Bone. 2012. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- Silva BC, Costa AG, Cusano NE, Kousteni S, Bilezikian JP. Catabolic and anabolic actions of parathyroid hormone on the skeleton. J Endocrinol Invest. 2011;34:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Tebben PJ, Thompson JR. Vitamin D and the kidney. Arch Biochem Biophys. 2012;523:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleet JC, Schoch RD. Molecular Mechanisms for regulation of intestinal calcium and phosphate absorption by vitamin D. Feldman D, ed. Vitamin D. 3rd ed. London: Elsevier 2011. [Google Scholar]
- Christakos S. Recent advances in our understanding of 1,25-dihydroxyvitamin D(3) regulation of intestinal calcium absorption. Arch Biochem Biophys. 2012;523:73–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordin BE. Evolution of the calcium paradigm: the relation between vitamin D, serum calcium and calcium absorption. Nutrients. 2010;2:997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen TK. The role of plasma-binding proteins in the cellular uptake of lipophilic vitamins and steroids. Horm Metab Res. 2006;38:279–290. [DOI] [PubMed] [Google Scholar]
- Chun RF. New perspectives on the vitamin D binding protein. Cell Biochem Funct. 2012;30:445–456. [DOI] [PubMed] [Google Scholar]
- Bienaime F, Prie D, Friedlander G, Souberbielle JC. Vitamin D metabolism and activity in the parathyroid gland. Mol Cell Endocrinol. 2011;347:30–41. [DOI] [PubMed] [Google Scholar]
- Salusky IB. Are new vitamin D analogues in renal bone disease superior to calcitriol? Pediatr Nephrol. 2005;20:393–398. [DOI] [PubMed] [Google Scholar]
- Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, Murad MH, Weaver CM. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:1911–1930. [DOI] [PubMed] [Google Scholar]
- Anderson PH, Turner AG, Morris HA. Vitamin D actions to regulate calcium and skeletal homeostasis. Clin Biochem. 2012;45:880–886. [DOI] [PubMed] [Google Scholar]
- Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. [DOI] [PubMed] [Google Scholar]
- Priemel M, von Domarus C, Klatte TO, Kessler S, Schlie J, Meier S, Proksch N, Pastor F, Netter C, Streichert T, Püschel K, Amling M. Bone mineralization defects and vitamin D deficiency: histomorphometric analysis of iliac crest bone biopsies and circulating 25-hydroxyvitamin D in 675 patients. J Bone Miner Res. 2010;25:305–312. [DOI] [PubMed] [Google Scholar]
- Rosen CJ, Gallagher JC. The 2011 IOM report on vitamin D and calcium requirements for north america: clinical implications for providers treating patients with low bone mineral density. J Clin Densitom. 2011;14:79–84. [DOI] [PubMed] [Google Scholar]
- Ott SM. Bone histomorphometry in renal osteodystrophy. Semin Nephrol. 2009;29:122–132. [DOI] [PubMed] [Google Scholar]
- Anderson PH, Atkins GJ, Turner AG, Kogawa M, Findlay DM, Morris HA. Vitamin D metabolism within bone cells: effects on bone structure and strength. Mol Cell Endocrinol. 2011;347:42–47. [DOI] [PubMed] [Google Scholar]
- Ritter CS, Armbrecht HJ, Slatopolsky E, Brown AJ. 25-Hydroxy-vitamin D(3) suppresses PTH synthesis and secretion by bovine parathyroid cells. Kidney Int. 2006;70:654–659. [DOI] [PubMed] [Google Scholar]
- Liu PT, Krutzik SR, Modlin RL. Therapeutic implications of the TLR and VDR partnership. Trends Mol Med. 2007;13:117–124. [DOI] [PubMed] [Google Scholar]
- Liu PT, Modlin RL. Human macrophage host defense against Mycobacterium tuberculosis. Curr Opin Immunol. 2008;20:371–376. [DOI] [PubMed] [Google Scholar]
- Adams JS, Ren S, Liu PT, Chun RF, Lagishetty V, Gombart AF, Borregaard N, Modlin RL, Hewison M. Vitamin d-directed rheostatic regulation of monocyte antibacterial responses. J Immunol. 2009;182:4289–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, Lee HM, Krutzik SR, Schenk M, Sieling PA, Teles R, Montoya D, Iyer SS, Bruns H, Lewinsohn DM, Hollis BW, Hewison M, Adams JS, Steinmeyer A, Zügel U, Cheng G, Jo EK, Bloom BR, Modlin RL. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med. 2011;3:104ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen CJ, Adams JS, Bikle DD, Black DM, Demay MB, Manson JE, Murad MH, Kovacs CS. The nonskeletal effects of vitamin D: an Endocrine Society scientific statement. Endocr Rev. 2012;33(3):456–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JS, Gacad MA. Characterization of 1 alpha-hydroxylation of vitamin D3 sterols by cultured alveolar macrophages from patients with sarcoidosis. J Exp Med. 1985;161:755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edfeldt K, Liu PT, Chun R, Fabri M, Schenk M, Wheelwright M, Keegan C, Krutzik SR, Adams JS, Hewison M, Modlin RL. T-cell cytokines differentially control human monocyte antimicrobial responses by regulating vitamin D metabolism. Proc Natl Acad Sci U S A. 2010;107:22593–22598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JS, Hewison M. Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. Nat Clin Pract Endocrinol Metab. 2008;4:80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JS. Vitamin D metabolite-mediated hypercalcemia. Endocrinol Metab Clin North Am. 1989;18:765–778. [PubMed] [Google Scholar]
- Singer FR, Adams JS. Abnormal calcium homeostasis in sarcoidosis. N Engl J Med. 1986;315:755. [DOI] [PubMed] [Google Scholar]
- Wu S, Ren S, Nguyen L, Adams JS, Hewison M. Splice variants of the CYP27b1 gene and the regulation of 1,25-dihydroxyvitamin D3 production. Endocrinology. 2007;148(7):3410–3418. [DOI] [PubMed] [Google Scholar]
- Ito N, Findlay DM, Anderson PH, Bonewald LF, Atkins GJ. Extracellular phosphate modulates the effect of 1α,25-dihydroxy vitamin D(3) (1,25D) on osteocyte like cells. J Steroid Biochem Mol Biol. 2012. [Epub ahead of print]. [DOI] [PubMed]