

Summary
Pancreatic ductal adenocarcinoma (PDAC) is associated with marked fibrosis and stromal myofibroblasts but their functional contribution remains unknown. Transgenic mice with ability to delete αSMA+ myofibroblasts in pancreatic cancer were generated. Depletion starting at either non-invasive precursor (PanIN) or the PDAC stage led to invasive, undifferentiated tumors with enhanced hypoxia, epithelial-to-mesenchymal transition and cancer stem cells, with diminished animal survival. In PDAC patients, lower myofibroblasts in their tumors also correlated with reduced survival. Suppressed immune surveillance with increased CD4+Foxp3+ Tregs was observed in myofibroblasts depleted mouse tumors. While myofibroblasts depleted tumors did not respond to Gemcitabine, anti-CTLA4 immunotherapy reversed disease acceleration and prolonged animal survival. This study underscores the need for caution in targeting carcinoma-associated fibroblasts in PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a near uniformly lethal disease with a dismal median survival of 4-6 months (Hidalgo, 2010; Korc, 2007). Despite years of efforts to design therapeutic approaches for pancreatic cancer, the use of conventional chemotherapy combination regimens with modest benefit remains the only option for the overwhelming majority of patients that present with advanced neoplasms. Revisiting the complex biology of PDAC in an unbiased manner is thus urgently required if we are to develop more effective therapies. The progress in generating genetically engineered mouse models (GEMMs) faithfully mimicking human PDAC provides a unique opportunity to interrogate the functional contribution of the desmoplastic stromal reaction in PDAC, a defining feature of this carcinoma, which accounts for the majority of the tumor tissue volume (Aguirre et al., 2003; Bardeesy et al., 2006a; Bardeesy et al., 2006b; Gidekel Friedlander et al., 2009; Hingorani et al., 2003; Hingorani et al., 2005; Hruban, 2007; Ijichi et al., 2006).
The cellular component of the desmoplastic stroma in PDAC is primarily composed of myofibroblasts, characterized by αSMA expression (Feig et al., 2012; Rasheed et al., 2012). Recent studies have implicated the stroma as a physical barrier to the delivery of cytotoxic chemotherapies to the peri-tumoral milieu (Feig et al., 2012; Provenzano et al., 2012; Whatcott et al., 2012). Nonetheless, while preclinical models have demonstrated the benefit of stromal depletion through blockade of paracrine Hedgehog signaling in accentuating drug delivery (Olive et al., 2009), subsequent clinical trials targeting stromal myofibroblasts in human PDAC resulted in an apparent paradoxical accelerated disease progression, halting clinical trials (Amakye et al., 2013). These studies reinforced the need to critically evaluate the functional contribution of stroma in the initiation and progression of PDAC. In this regard, the functional role of αSMA+ myofibroblasts and type I collagen in PDAC remains unknown.
Results
Myofibroblast depletion leads to increased tumor invasion associated with decreased survival
To interrogate the functional contribution of αSMA+ myofibroblasts in PDAC, we employed a genetic strategy for selective in vivo depletion. Ptf1acre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) mice develop spontaneous PDAC with full penetrance that reliably recapitulates the clinical and histopathological features of the human disease (Ijichi et al., 2006). The mice consistently progress from pancreatic intraepithelial neoplasia (PanIN) at 4.5 weeks of age to invasive cancer at 6 weeks of age, and die at 8 weeks of age without much variation (Ijichi et al., 2006). These mice were crossed with αSMA-tk transgenic mice to selectively target proliferating αSMA+ myofibroblasts upon systemic ganciclovir (GCV) administration (PKT; αSMA-tk mice) (LeBleu et al., 2013). Daily GCV injections were initiated when mice developed PanIN lesions (early) and allowed to progress to PDAC (Figure 1A). Mice also received GCV injections starting at established PDAC stage (late) until they developed significant signs of illness leading to their death or requiring euthanasia (Figure 1A). In both early and late myofibroblast depletion settings, PKT mice presented with significantly more invasive, undifferentiated and necrotic tumors when myofibroblasts were depleted compared to control tumors (Figures 1B-E). Immunohistochemical and immunofluorescence analyses revealed an average of 80% depletion of proliferating myofibroblasts (Figures 1F, S1A-B). Direct visualization of interstitial myofibroblasts using the αSMA-RFP transgenic mice crossed with PKT; αSMA-tk mice also showed approximately 80% depletion of total myofibroblasts (Figure 1G). A significant reduction in αSMA transcript level was also noted in depleted tumors (Figure S1C). Extra-pancreatic organs, such as kidney, lung, small bowel, heart and liver, did not show any depletion in αSMA+ cells (Figure S1D-E). Notably, myofibroblast depletion in PDAC was associated with significant reduction in survival, both in the early and late depletion groups (Figure 1H). Tumor weight was significantly reduced by myofibroblast depletion and was associated with a reduced body weight specifically in PDAC mice with late myofibroblast depletion (Figures S1F-G). Mice with myofibroblast-depleted tumor exhibit an increased frequency of pulmonary emboli, likely contributing to diminished overall survival (Figures S1H).
Figure 1. Myofibroblast depletion augments PDAC and diminishes overall survival.

(A) Tumor progression timeline with experimental treatment time points. Ganciclovir (GCV) administration in PKT;αSMA-tk+ mice allows for myofibroblast depletion in contrast with control PKT;αSMA-tk- mice. (B) Representative micrographs of H&E stained pancreatic samples (scale bar: 100 μm). (C) Pathological scores of early depleted tumors (left. WT, n=5; Control, n=16; Depleted, n=17) and late depleted tumors (right. WT, n=5; Control n=17; Depleted, n=26). Significance was determined by One-way ANOVA with Turkey post-hoc analysis. (D) Necrosis penetrance of early depleted tumors (left. WT, n=5; Control, n=16; Depleted, n=22) and late depleted tumors (right. WT, n=5; Control, n=17; Depleted, n=25). (E) Relative percentage in histological phenotypes in late depleted tumors. Control, n=17; Depleted, n=25. (F) Representative micrographs (scale bar: 50 μm) of Ki-67/αSMA dual-immunofluorescence (left, control; right, depleted) and corresponding quantification (n=4 and 6 for control and depleted, respectively). Arrows point to double positive cells and inserts show high magnification images (scale bar: 25 μm). (G) Representative micrographs (scale bar: 100 μm) of reporter αSMA-RFP tumor samples (left, control; right, depleted) and corresponding quantification (n=3 in each group). (H) Survival analysis of early (left) and late (right) treatment groups. GCV: ganciclovir treatment was initiated. Data is represented as mean +/- SEM. Unless otherwise noted, significance was determine by t-test, * p<0.05, ** p<0.01, ** p< 0.001, ****p < 0.0001. ns: not significant. See also Figure S1.
Loss of one allele of Tgfbr2 (versus both in the PKT mice) in the context of KrasG12D activation (PKTHet mice) leads to a similar PDAC phenotype, but with a slower progression rate, with PanIN lesions noted at 4-6 weeks of age, and PDAC at 8-10 weeks of age (Ijichi et al., 2006). GCV treatment was initiated at the onset of PDAC in PKTHet; αSMA-tk mice and continued until they died or were moribund, requiring euthanasia. Depletion of myofibroblasts resulted in undifferentiated tumors (Figures 2A-E) and significantly diminished the survival of PKTHet; αSMA-tk mice (Figure 2F). Similar findings were also observed when myofibroblasts were depleted in Pdx1cre/+;LSL-KrasG12D/+;Trp53R172H/+ (KPC) mice crossed with αSMA-tk (KPC; αSMA-tk) mice. Myofibroblast depletion in this setting also resulted in poorly differentiated tumors and significantly diminished survival (Figures S2A-C). Due to similar phenotypes of the PKT; αSMA-tk mice and KPC; αSMA-tk mice, the remaining experiments were carried out using the PKT mice due to their faster course of disease.
Figure 2. Decreased αSMA correlates with poor prognosis and overall survival in slow-progressing mice with PDAC and patients with pancreatic cancer.

(A, B) Representative micrographs of H&E stained PKTHet tumors at 106 (Control, left) and 103 (Depleted, right) days old (A, scale bar: 100 μm) and pathological scores (B). WT, n=5; Control, n=6; Depleted, n=4. Significance determine by One-way ANOVA with Turkey post-hoc analysis. (C) Relative percentage in histological phenotypes (n=6 and 4, respectively, for control and depleted). (D, E) Representative micrographs of αSMA stained pancreatic samples as in (A) (D, scale bar: 100 μm) and corresponding quantification (E). n=6 and 4, respectively, for control and depleted. V: vessel. (F) Survival analysis in PKTHet mice. GCV: ganciclovir treatment was initiated. (G) Representative micrographs of αSMA stained pancreatic samples resected from patients in each score category :0, 1, 2 and 3 (scale bar: 200 μm). (H) Survival analysis based on histopathological score (left) and αSMA score (right). Low αSMA: scores 0 and 1; high αSMA: scores 2 and 3. Significance determine by Mantel-Cox test. Data is represented as mean +/- SEM. Unless otherwise noted, significance was determine by t-test, * p<0.05, ** p<0.01, ** p< 0.001, ****p < 0.0001. ns: not significant. See also Figure S2 and Table S1.
Based on the striking and somewhat unexpected impact of stromal depletion on survival in PDAC models, we next assessed whether a comparable impact of stromal content was observed in the cognate human setting. Immunohistochemical scoring for interstitial αSMA+ cells from resected PDAC of untreated patients indicates low αSMA is associated with worse survival, supporting the observations in the PKT and KPC mice with myofibroblast depletion (Figure 2G, Table S1). Of note, patients with low αSMA score present significantly more often with poorly differentiated cancers than tumors with well/moderate differentiation (Table S1); and yet despite this correlation, αSMA scoring offered greater significance in differential stratification of patients for survival than histopathological grade (Figure 2H), suggesting that stromal content likely has a direct impact on the natural history of human PDAC.
Myofibroblast depletion reduced type I collagen content and altered ECM organization in PDAC
Global gene expression profiling of tumors from PKT mice without (control) and with myofibroblast depletion (depleted) revealed differentially expressed genes associated with remodeling of the extracellular matrix (ECM), epithelial-to-mesenchymal transition (EMT), angiogenesis, and immune response (Table S2). Comparative analyses of tumor transcriptomes revealed 4,393 differentially expressed genes in early depleted vs. age matched control tumors (1,682 down-regulated and 2,711 up-regulated genes), and 2,344 differentially expressed genes in late depleted vs. age matched control tumors (1,170 down-regulated and 1,174 up-regulated genes) (Figures S3A-D). Tumor collagen content assayed by type I collagen immunostaining (Figure 3A) and Masson Trichrome staining (MTS) (Figure 3B) was significantly decreased in myofibroblast-depleted tumors. Picrosirius Red staining revealed a significant reorganization of collagen fibrils, with decreased stiffness and elastic modulus in myofibroblast-depleted tumors compared to control tumors (Figures 3C, 3D). Interestingly, collagen crosslinking and immunolabeling for the collagen crosslinking enzyme lysyl oxidase (LOX) were unchanged despite reduced stiffness of the extracellular matrix (Figures 3E, 3F), highlighting a complex regulation of type I collagen crosslinking mediated by multiple sources of LOX. These results are consistent with the ability of αSMA+ myofibroblasts to produce type I collagen and induce fibrosis in PDAC. Despite extensive remodeling of type I collagen ECM in myofibroblast-depleted tumors, hyaluronic acid binding protein (HABP), a marker for total hyaluronic acid (HA) content, remained unchanged in the myofibroblast-depleted tumors (Figure S3E). Vimentin labeling however was significantly decreased in myofibroblast-depleted tumors (Figure S3F). Auditing of other mesenchymal cells in the PDAC tumor stroma revealed a decrease in the number of fibroblast-specific protein1 (FSP1)/S100A4+ cells in myofibroblast-depleted tumors (Figure S3G), while the number of fibroblast activation protein (FAP)+ cells remained unchanged (Figure S3H). Co-localization of αSMA with FAP was not detectable in the PDAC tumors (Figure S3H).
Figure 3. PDAC extracellular matrix is significantly remodeled in myofibroblast-depleted tumors.
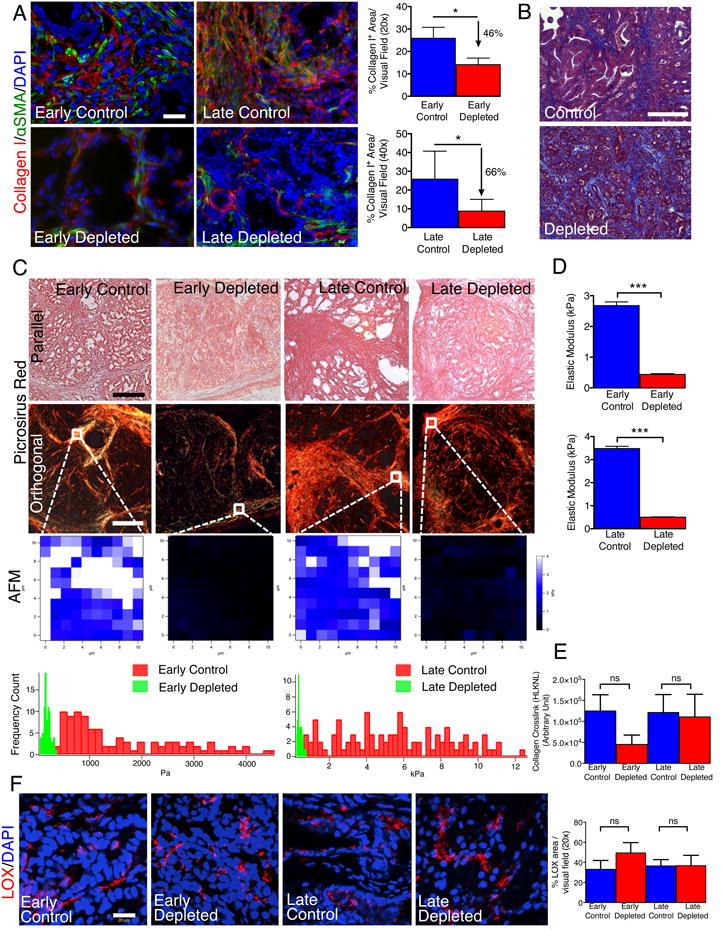
(A) Representative micrographs (scale bar: 50 μm) of Collagen I and αSMA and corresponding quantification (n=4). (B) Representative micrographs (scale bar: 200 μm) of Masson Trichrome staining in late control and late deleted mice. (C) Representative images of Picrosirius Red staining of PKT pancreatic tissues samples viewed under parallel (top row) and polarized light (second row) (scale bar: 75 μm), and representative images of stiffness distribution by atomic force microscopy measurement (AFM, third row) and quantification (bottom row, n=3). (D) Quantification of the elastic modulus of tumors matrix (n=3). (E) Collagen crosslinking quantification (n=4). (F) Representative micrographs for LOX (scale bar: 20 μm) and quantification. Data is represented as mean +/- SEM. Significance was determined by t-test, * p<0.05, ** p<0.01, ** p< 0.001, ****p < 0.0001. ns: not significant. See also Figure S3 and Table S2.
Myofibroblast-depleted PDAC displays suppression of angiogenesis, enhanced tumor hypoxia, EMT program and cancer stem cell-like phenotype
Histopathological analyses of myofibroblast-depleted tumors revealed highly undifferentiated, invasive grade tumors with enhanced necrosis (Figure 1B-E). These features are associated with tumor hypoxia and acquisition of an epithelial-to-mesenchymal transition (EMT) program. Myofibroblast depletion was associated with a decrease in the number of CD31+ vessels, indicative of suppressed vessel density (Figures 4A, S4A-C). Perivascular αSMA+ cells appear unaffected by the αSMA-tk strategy (Figure S4A). Global gene expression profiling performed on normal pancreas fibroblasts (NF) and PDAC-associated fibroblasts (CAF) shows differences in pathways affecting ECM remodeling, angiogenesis and immune response in PDAC (Figure S4D, Table S3). RNA sequencing analyses further validated the increased expression of several pro-angiogenic factors in CAF, supporting their pro-angiogenic role in PDAC (Table S3).
Figure 4. Myofibroblast-depleted tumors display increased invasion associated with intra-tumoral hypoxia.

(A) Representative micrographs (scale bar: 100 μm) of CD31 staining and corresponding quantification (n=4). Dashed lines delineate the stromal compartment where CD31+ vessels are primarily found. (B) Representative micrographs (scale bar: 100 μm) and corresponding quantification (n=4) of CD31 cells and intra-tumoral FITC-Dextran leakage. (C) Representative micrographs (scale bar: 100 μm) of hypoxia indicator, Hypoxyprobe™, and corresponding quantification (n=4). (D) Representative micrographs (scale bar: 50 μm) of αSMA+ cells in tumors with YFP+ linage tagged cancer cells and corresponding quantification (n=4). White arrowheads point to αSMA+YFP+ cells. (E) Relative Twist, Snail and Slug expression in tumors. (F) Representative micrograph (scale bar: 100 μm) of YFP+ tumor spheres (insert shows higher magnification micrograph, scale bar: 25 μm) and corresponding quantification (n=15). (G) Representative scatter plot and quantification of percent CD44+CD133+ cancer cells (Control: n=2, Depleted: n=3). Q1: CD44+CD133-; Q2: CD44+CD133+; Q3: CD44-CD133+; Q4: CD44-CD133-. (H) Tumorigenic ability of YFP+ cells determined by limiting dilution assay. For each cell dose, 6 mice were injected with YFP+ cancer cells from control or myofibroblast depleted tumors and data is presented as the number of mice displaying tumors. Significance was assayed by Χ2 test. Data is represented as mean +/- SEM. Unless otherwise noted, significance was determine by t-test, * p<0.05, ** p<0.01, ** p< 0.001, ****p < 0.0001. ns: not significant. See also Figure S4 and Table S3.
While pericyte numbers, assayed by immunostaining analyses for the pericyte marker NG2 (Figure S4B), and vascular leakage, determined by extravascular FITC-dextran (Figures 4B, S4E), were unchanged, tumor hypoxia assayed by staining for pimonidazole adduct formation was dramatically increased in myofibroblast-depleted tumors compared to control tumors (Figures 4C, S4F). Lineage tracing of cancer cells using the LSL-YFP reporter together with immunolabeling for αSMA allowed for the quantitative analysis of YFP+ cancer cells that have acquired mesenchymal features (YFP+αSMA+ cells). Tumors with 80% depletion of αSMA+ interstitial myofibroblasts showed an increase in EMT program with increased YFP+αSMA+ cells (Figures 4D, S4G) and enhanced expression of the EMT transcriptional regulators, Twist, Snail, and Slug (Figure 4E). The enhanced acquisition of EMT was associated with the loss of mucin staining (Figure S4H), which is in alignment with the decreased histological differentiation within the neoplastic glands (Figure 1B-E). YFP+ cancer cells from myofibroblasts-depleted tumors exhibited an increase in their capacity to form spheres in culture, a feature of cells with EMT program and potential cancer stem cell-like phenotype (Kalluri and Weinberg, 2009; Scheel and Weinberg, 2012) (Figures 4F, S4I). Additionally, the number of CD44+CD133+ cells [suggestive of pancreatic cancer stem cells (Hermann et al., 2007; Simeone, 2008)] was enhanced in myofibroblast-depleted tumors (Figure 4G). FACS sorted YFP+ cells freshly isolated from myofibroblast-depleted tumors showed enhanced tumorigenic potential when implanted in nude mice (Figure 4H). Interestingly, cancer cells with EMT program and expression of αSMA were not eliminated in the PKT; αSMA-tk mice on GCV, but increased in numbers (Figure 4D). This could be explained by the observation that cancer cells with EMT program likely do not proliferate (Tsai et al., 2012; Vega et al., 2004), further confirming their low-cycling-stem-cell status. Further, while overall apoptosis was significantly increased in myofibroblast-depleted tumors (Figures S4J, S4K), it was mostly localized within terminally differentiated compartments, such as islets (Figure S4K).
Reduction of fibrosis does not increase the efficacy of gemcitabine in PDAC
To test whether the decrease in myofibroblasts and type I collagen content along with reduced ECM stiffness leads to increased efficacy of gemcitabine (GEM), we treated control and myofibroblast-depleted mice with GEM. Histopathological score did not improve when PKT mice were treated with gemcitabine alone or in combination with myofibroblast depletion (Figures 5A). TUNEL+ cell number increased with myofibroblast depletion, independent of GEM treatment, and GEM treatment alone did not significantly increase tumor cells apoptosis (Figures 5B). Similarly, the change in FSP1+ cells only correlated with myofibroblasts depletion and was independent of GEM treatment (Figures 5C). Interestingly, CD31+ cell numbers decreased with GEM therapy and decreased further with myofibroblast depletion (Figures 5D). Standard uptake values computed from 18F-FDG PET/CT scanning revealed comparable glycolysis per tumor volume in control, depleted, and GEM therapy in depleted tumors (Figures 5E). Despite a robust decrease in collagen content associated with myofibroblast depletion (fibrosis) (Figure 5F), GEM therapy did not improve overall survival (Figure 5G). Further, GEM therapy of PDAC compared to untreated mice did not result in improved survival, as previously reported using a different PDAC mouse model (Olive et al., 2009) (Figure 5G). GEM therapy in myofibroblast-depleted tumors compared to myofibroblast-depleted tumors also failed to improve overall survival (Figure 5G).
Figure 5. Gemcitabine treatment does not improve survival of mice with myofibroblast-depleted tumors.

(A) Representative micrographs of H&E stained pancreatic samples (scale bar: 100 μm) and pathological scores of indicated experimental groups. Control, n= 16; Depleted, n=17; GEM, n=8 and GEM Depleted, n=8. (B) Representative micrographs (scale bar: 200 μm) of the apoptosis marker TUNEL and corresponding quantification (n=3 in each group). (C) Representative micrographs (scale bar: 100 μm) of FSP1 IHC and corresponding quantification (n=4 in each group). (D) Representative micrographs (scale bar: 100 μm) of CD31 IHC and corresponding quantification (n=3 in each group). (E) Representative 18F-FDG PET/CT scan (top, frontal section; bottom, sagittal section) and associated max standard update value (SUVmax) (n=3 in each group). (F) Hydoxyproline release reflecting collagen content in control and myofibroblast-depleted tumors (n=4 in each group). Significance was determined by t-test. (G). Survival analyses of indicated experimental groups. Data is represented as mean +/- SEM. Unless otherwise noted, significance was determined by One-way ANOVA with Turkey post-hoc analysis, * p<0.05, ** p<0.01, ** p< 0.001, ****p < 0.0001. ns: not significant.
Myofibroblast depletion decreases overall immune infiltration in PDAC, but results in increased frequency of FoxP3+ Treg cells
Gene expression profiling and RNA sequence analyses comparing control tumors with myofibroblast-depleted tumors revealed a significant change in expression of genes associated with tumor immunity, including those associated with acute inflammatory responses, regulation of macrophage activation, regulation of T cell-mediated cytotoxicity, antigen presentation and B cell activation (Figure 6A; Table S2). Tumors with myofibroblast depletion starting at the PanIN stage (early) present with a significant decrease in overall peri-tumoral infiltration of CD45+ cells, CD3+ T cell and CD19+ B cell infiltration, when compared to control tumors (Figure S5A). Such reduction in T cells and B cells was not observed in established PDAC with myofibroblast depletion (late stage depletion) (Figure S5A). NK cell infiltration with myofibroblast depletion was unchanged following myofibroblast depletion (Figure S5A). In both early and late depletion settings, myofibroblast depletion was associated with a significant suppression in the percentage of effector T cells (Teff, CD4+Foxp3-) together with an increase in the percentage of regulator T cells (Treg, CD4+Foxp3+), leading to an overall decrease in Teff/Treg ratio (Figure 6B-D). Myofibroblast depletion resulted in increased Foxp3 and Ctla4 expression assayed by immunohistochemistry (Figure S5B) and real time PCR analysis (Figure 6E) respectively, consistent with the enhanced Treg infiltration. The cytotoxic CD8+/Treg ratio was also decreased in the myofibroblast-depleted tumor (Figure 6F), as well as CD3+/CD11b+ ratio (early depletion, Figure 6G). The percentage of Teff and Treg respectively correlate with disease progression and acceleration of PDAC associated with myofibroblast depletion (Figures 6H, 6I). The percentage of CD11b+Ly6G+ cells and granulocytes (CD11b+Gr1+F4/80-) were also increased in myofibroblast-depleted late tumors (Figures 6J, 6K), while percentage of macrophages (CD11b+Gr1- F4/80+) were suppressed (Figure 6L). The differential tumor immune infiltration in early vs. late depletion is also substantiated by distinct expression patterns in respective tumor immune gene expression signature, with a selectively down-regulated tumor immunity gene signature in late depleted tumor (Figure 6A).
Figure 6. Myofibroblast depletion results in increased frequency of FoxP3+ Treg cells in PDAC.

(A) Heat map of differentially regulated genes pertaining to tumor immunity in early and late treated tumors. (B-D) Percent Teff (B), Treg (C) and Teff/Treg ratio (D) in early and late treated tumors. (E) Relative Ctla4 expression in late treated tumors (n=6). (F-G) CD8+ cytotoxic T cell/Treg ratio (F) and CD3/CD11b ratio (G) in early and late treated tumors. (H, I) Percent Teff (H) and Treg (I) in normal pancreas and in early and late treated tumors. (J-L) Percent CD11b+Ly6G+ (J), CD11b+Gr1+F4/80- (K) and CD11b+Gr1-F4/80+ (L) cells in late treated tumors. Data is represented as mean +/- SEM. Significance was determine by t-test except for multiple groups comparison, in which significance was determined by One-way ANOVA with Turkey post-hoc analysis, * p<0.05, ** p<0.01, ** p< 0.001, ****p < 0.0001. ns: not significant. See also Figure S5.
Anti-CTLA-4 antibody therapy rescues enhanced PDAC progression upon myofibroblast depletion and increases overall survival
Myofibroblast depletion starting at early and late stage tumors resulted in a decreased Teff/Treg ratio associated with increased CTLA-4 expression (Figures 6D, 6E). Therefore, we tested whether the Teff/Treg ratio in PDAC mice with myofibroblasts depletion can be rescued by CTLA-4 checkpoint blockade. Anti-CLTA4 antibody administration significantly improved histopathological scores associated with a reduction in undifferentiated cancer cells (Figure 7A). Despite similar myofibroblast depletion (Figure 7B), suggesting anti-CTLA-4 per se did not alter myofibroblast number, anti-CTLA-4 antibody treatment induced tumor clearance and replacement by normal parenchyma in up to 25% of the organ (Figure 7C). Treatment with anti-CTLA-4 antibodies in the setting of stromal depletion not only resulted in a rescue of the phenotype of myofibroblast-depleted tumors but also attenuated PDAC progression, which was associated with a significant extension in overall survival (an average increase of 60% in life span) (Figure 7D). Such elongation of life span was associated with reduced frequency of pulmonary emboli (Figure 7E). This was noticed in concordance with restored Teff and Treg percentages (Figure 7F) and diminished tumor sphere forming ability within remnant cancer cells in age-matched mice (Figure 7G). Further, the transcriptome of myofibroblast-depleted tumors treated with anti-CTLA-4 clustered with the expression profiles of control tumor rather than myofibroblast-depleted tumors, suggesting a transcriptional reprogramming accompanies the observed phenotypic rescue (clustering using the 2,344 differentially expressed genes in late depleted vs. control) (Figure 7H). Anti-CTLA-4 alone did not alter αSMA+ content, and histopathological analyses revealed a modest increase in percent normal parenchyma (Figures S6A, S6B). Treatment with anti-CTLA-4 alone improved overall survival (median survival of 49.5 days, compared to 47 days in the control group), albeit to a much lesser extent than anti-CTLA-4 treatment in combination with fibrosis depletion (median survival of 65 days) (Figure 7D).
Figure 7. Anti-CTLA-4 attenuates PDAC and improves survival in mice with myofibroblast-depleted tumors.

(A) Representative micrographs of H&E stained tumors (scale bar: 100 μm) and pathological scores of tumors in indicated experimental groups. Control, n=14; Depleted, n=16; Depleted + anti-CTLA-4, n=6. Significance was determined by One-way ANOVA with Turkey post-hoc analysis. (B) Representative micrographs of αSMA stained pancreatic samples (scale bar: 100 μm) and corresponding quantification. Depleted, n=5; Depleted + anti-CTLA-4, n=7. (C) Relative percent of tissue encompassed by each histology hallmark. Control, n=15; Depleted, n=16; Depleted + anti-CTLA-4, n=6. Significance was determined by One-way ANOVA with Turkey post-hoc analysis. (D) Survival analysis of the indicated experimental groups. (E) Number of pulmonary emboli. Control, n=14; Depleted, n=28; Depleted + anti-CTLA-4, n=8. (F) Percent Teff (left) and Treg (right) cells in tumors of indicated experimental groups. (G) Number of spheres formed from tumors of indicated experimental groups. (H) Heat map of differentially expressed genes in the indicated groups. Data is represented as mean +/- SEM. Unless otherwise noted, significance was determine by t-test, * p<0.05, ** p<0.01, ** p< 0.001, ****p < 0.0001. ns: not significant. See also Figure S6.
Discussion
The desmoplastic reaction is thought to represent a host defense mechanism, similar to wound healing and tissue regeneration, to repair or hopefully impede conversion of neoplastic lesion into invasive carcinoma (Bissell and Radisky, 2001; Dvorak, 1986; Lu et al., 2012). The function of desmoplastic stroma is likely dynamic during cancer progression and its heterogeneous cellular and non-cellular constituents change in relation to the evolving genetic landscape of cancer cells. In this regard, several studies have suggested that αSMA+ myofibroblasts and type I collagen associated with tumor fibrosis are tumor promoting in solid tumors including PDAC (Angeli et al., 2009; Karnoub et al., 2007; Merika et al., 2012; Vong and Kalluri, 2011). We demonstrate that depletion of αSMA+ myofibroblasts resulted in multiple adverse outcomes leading to poor survival. These results suggest that at both early and late stages of pancreatic cancer, fibrosis associated with myofibroblasts and type I collagen constitutes a protective response from the host rather than offering an oncogenic supportive role, as speculated (Armstrong et al., 2004; Omary et al., 2007). This conclusion is supported by the clinical correlation between high αSMA and improved survival, a similar trend was previously reported from analyses also performed on resected tumors from treatment naïve patients (Wang et al., 2013). While other studies have also attempted to specifically address the correlation between relative tumor content in αSMA+ myofibroblasts and PDAC patient outcome, potential therapeutic intervention prior to tumor resection make interpretations unclear (Erkan et al., 2008; Mani et al., 2008).
Myofibroblast depletion leads to extensive remodeling of the tumor extracellular matrix, with a significant decrease in tumor tissue stiffness and total collagen content. Myofibroblasts contribute to the production of type I collagen, albeit, likely not all of it. Contrary to previous assumptions, myofibroblasts do not contribute to the total production of LOX, an enzyme responsible for crosslinking of type I collagen. In this regard, we did not observe a difference in crosslinking despite significant depletion and reorganization of tumor-associated collagen. Therefore, cancer cells or other stromal cells may compensate LOX expression. Similar to LOX, myofibroblasts depletion did not affect HA content of the tumors, suggesting a non-myofibroblast source for this matrix molecule. Targeting HA using PEGPH20, a modified enzyme that degrades HA, yielded promising results when combined with gemcitabine (Jacobetz et al., 2013; Provenzano et al., 2012). In our study, ECM remodeling and the significant reduction in collagen content via myofibroblast depletion did not improve the treatment efficacy of gemcitabine. Therefore, PDAC-associated myofibroblasts/type I collagen do not appear to serve as a physical barrier for exposure of cancer cells to gemcitabine. In this regard, the potential role of HA, independent of type I collagen, in determining interstitial fluid pressure in PDAC tissue needs further mechanistic unraveling and clinical trials with PEGPH20 will offer more insights in the future (Provenzano et al., 2012).
As noted above, significant depletion of myofibroblasts and type I collagen did not alter vessel permeability and perfusion. Gemcitabine efficacy was unaltered in myofibroblasts and type I collagen depleted tumors suggesting that the increased efficacy observed in pre-clinical models treated with gemcitabine and Smoothened inhibitor IPI-926 was likely due to mechanisms independent of myofibroblasts and type I collagen (Olive et al., 2009). It is likely that Smoothened inhibition was not myofibroblasts-specific and could also have impacted other stromal cells such as endothelial cells. In this regard, further analysis of the failed clinical trials could shed further light on this matter (Amakye et al., 2013). It is possible that if higher doses of IPI-926 combined with prolonged treatment period were used in the pre-clinical setting, potential adverse effects may have been encountered (Olive et al., 2009; Amakye et al., 2013). Nevertheless, our direct approach of targeting myofibroblasts and the associated fibrosis offer further insights into complexity of using pharmacological targeting to derive specific answers related to target cells in tumors (Olive et al., 2009).
An interesting corollary that emerges from this study is the dominant contribution of cell division in the accumulation of myofibroblasts in PDAC. The origin of myofibroblasts in PDAC may be diverse (Phillips, 2012; Scarlett et al., 2011), yet using the αSMA-tk strategy to ablate proliferating myofibroblasts, we show 80% depletion of these cells, which account for the majority of myofibroblasts in PDAC tumors. These results offer an opportunity to speculate that drugs such as gemcitabine and Paclitaxel, which target proliferating cancer cells, may also target proliferating myofibroblasts. Such dual targeting may likely compromise the efficacy of gemcitabine over time, due to progressive depletion of protective fibrosis along with proliferating cancer cells, resulting in eventual emergence of resistant cancer cells with EMT program and stem-cell like phenotype.
Intra-tumoral hypoxia was dramatically increased in myofibroblast-depleted tumors. Decreased tumor vasculature, despite intact vessel structural integrity and permeability status, may have directly contributed to the increased tumor hypoxia when myofibroblasts are depleted, consequently promoting enhanced invasiveness and an un-differentiated phenotype of cancer cells (Cheresh and Stupack, 2008; Cooke et al., 2012; Paez-Ribes et al., 2009). Impaired paracrine signaling between myofibroblasts and cancer cells and alteration of ECM microenvironment due to myofibroblast depletion might also have directly contributed to changes in cancer cells lending to the acquisition of EMT program, stem cell-like phenotype and an undifferentiated state. Many studies have indeed suggested that altered matrix microenvironment can influence the acquisition of epigenetics changes in epithelial cells (Bissell et al., 2005; Grassian et al., 2011; Guerra et al., 2011).
The most dramatic impact of myofibroblast depletion was on the composition of the immune infiltrate in the tumor microenvironment. The interplay between cancer associated fibroblasts and immune cells has long been recognized as a major contributor of cancer development (Coussens and Werb, 2002; Erez et al., 2010; Kalluri and Zeisberg, 2006). Here, we demonstrate that the immune response (innate and adaptive) associated with PDAC tumors was significantly impaired when fibrosis was reduced starting at the PanIN stage or at the PDAC stage. Myofibroblast-depleted tumors are associated with a decreased Teff/Treg ratio and a significant elevation in Ctla4 expression. Myofibroblast depletion coupled with inhibition of checkpoint blockade using anti-CTLA-4 antibodies significantly ameliorated tumor burden in mice with established PDAC and improved overall survival. Our data suggest that while myofibroblasts depletion results in suppression of immune surveillance, an opportunist immune profile dominated by suppressor T cells emerges in the tumor, offering a viable checkpoint blockade target using anti-CTLA-4 antibodies. While this antibody shows marginal efficacy in control PDAC mice with normal levels of fibrosis, depletion of fibrosis unravels more robust efficacy, likely due to significant changes in the microenvironment. Small proof-of-concept trials with Ipilimumab have revealed some anecdotal responses (Royal et al., 2010) and our study suggests that stratifying patients based on their fibrosis score might offer better responses. This is supported by the fact that Ipilimumab showed dramatic efficacy in metastatic melanoma (Robert et al., 2011), a cancer with low levels of overall stroma. In summary, our study together with other ongoing efforts to elucidate pancreatic tumor immunity and immunotherapy (Bayne et al., 2012; Clark et al., 2007; Hiraoka et al., 2006; Roberts et al., 2013; Vonderheide et al., 2013) will likely offer new insights into potential efficacy of combination therapies involving immunotherapy in patients with pancreas cancer.
Materials and Methods
Animal studies
The disease progression and genotyping for the Ptf1acre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) mice was previously described (Ijichi et al., 2006). PKT mice were crossed to R26-stop-EYFP (purchased from the Jackson Laboratory, Bar Harbor, ME), αSMA-tk (LeBleu et al., 2013), and αSMA-RFP (LeBleu et al., 2013) mice. Mice received daily intraperitoneal (i.p.) injections with 50 mg/kg of body weight of ganciclovir (GCV, InvivoGen) at 4-4.5 weeks of age (30.3 ± 1.7 days) for 14 days or less (early depletion) or at 6 weeks of age (40.7 ± 1.7 days) for 10 days or less (late depletion). One hour prior to sacrifice, mice were injected i.p. with 60 mg/kg of body weight pimonidazole (Hypoxyprobe™, Hypoxyprobe Inc.). Pdx1cre/+;LSL-KrasG12D/+;Trp53R172H/+ (KPC) mice were previously described (Hingorani et al., 2005) and crossed to αSMA-tk mice. KPC mice were treated with ganciclovir starting at 7 weeks of age (49 ± 2.5 days). Gemcitabine (50 μg/kg of body weight, LC laboratories, Woburn, MA) was given i.p. at day 1 and day 7, either alone or in combination with GCV. An initial 200 μg anti-CTLA-4 (BioXCell, clone 9H10, BE0131) antibody or 200 μg Hamster IgG (BioXCell, BE0091) in 200 μl PBS were administered i.p., followed by two injection of 100 μg of anti-CTLA-4 or 100 μg of Hamster IgG in 100 μl PBS every other day. All mice were housed under standard housing conditions at the Beth Israel Deaconess Medical Center (BIDMC) and MD Anderson Cancer Center animal (MDACC) facilities and all animal procedures were reviewed and approved by the BIDMC and the MDACC Institutional Animal Care and Use Committee. Acquisition and analysis of 18F-FDG PET/CT in mice is detailed in the Supplemental Experimental Procedures.
Histology and histopathology
Formalin-fixed tumors were submitted to the BIDMC and the MDACC Histology Core Facilities and paraffin-embedded sections were cut for Hematoxylin & Eosin (H&E) staining and Masson's Trichrome staining (MTS). For histopathological scoring, H&E stained slides were scored for the penetrance of each histological hallmark on a scale of 0-3. The predominant tumor phenotype gave the pathological score for the whole tumor (1=well differentiated, 2=moderately differentiated, 3=poorly differentiated). Necrosis was also scored on a scale of 0-3. Lung emboli were evaluated and H&E counted in one section per mouse evaluated. Picrosirius Red staining for collagen was achieved using 0.1% Picrosirius Red (Direct Red 80; Sigma) and counterstained with Weigert's Hematoxylin. Mucin staining was achieved using mucicarmine, metalin yellow and counter stained with Weigert's Hematoxylin.
Immunohistochemistry
Harvested tumors were formalin fixed prior to paraffin embedding. 4-5 μm sections were deparaffinized, rehydrated, and boiled one hour in 10 mM citrate buffer at pH 6.0. Staining for αSMA was processed using the M.O.M. Kit (Vector Labs) according to the manufacturers' recommendations. For all other stains the tissue sections were blocked with 1% bovine serum albumin in Tris buffered saline for 30 minutes prior to incubation with the primary antibody. Sections were then incubated with biotin-conjugated anti-rabbit/rat/goat IgG and ABC reagent (Vector Laboratories, West Grove, PA) for 30-45 min at room temperature. DAB was used as a detection system (Vector Laboratories) according to the manufacturer's instructions. The following primary antibodies were used: mouse anti-αSMA (Sigma) 1:200, rabbit anti-FSP1 (a gift from Dr. Eric Nielson, Northwestern University Feinberg School of Medicine, Chicago, IL) 1:50, biotinylated anti-HABP (Amsbio) 1:200, rabbit anti-CD31 (Abcam) 1:50, and rabbit anti-vimentin (Cell Signaling) 1:100. For all stainings DAB positivity was analyzed in 5-8 visual fields at an original magnification of 10×, 20× or 40×. Control and treated mice within an experimental set (at least 3 mice/group) were analyzed. All stainings were quantified by NIH ImageJ analysis software with the same threshold for each stain; results expressed as percent staining per visual field.
Immunofluorescence
Harvested tumors were embedded in O.C.T. medium (TissueTek, Torrance, CA). 4-5 μm frozen sections were fixed in ice-cold acetone for 20 minutes, blocked with 1% bovine serum albumin in phosphate buffered saline, and immunostained using standard protocols. Primary antibodies used are: goat anti-Collagen 1 (Southern Biotech) 1:200, mouse anti-αSMA-FITC/mouse anti-αSMA-Cy3 (Sigma) 1:200, rat anti-mouse Ki67 (abcam) 1:200, rabbit anti-FAP (abcam) 1:200; rat anti-foxp3 (eBioscience) 1:150; rabbit anti-NG2 (Millipore) 1:200; rat anti-CD31 (BD Pharmingen) 1:200, mouse 4.3.11.3 (hypoxyprobe) 1:50, and rabbit anti-LOX (Imgenex) 1:200. Stainings for pimonidazole adduct (hypoxyprobe) was processed using the M.O.M. Kit (Vector Labs) according to the manufacturers' recommendations. Apoptosis was assessed using in Situ Cell Death Detection Kit, TMR red (Roche). DAPI was used to stain cell nuclei.
Flow cytometry
Weighed tumors were minced and allowed to digest in a 2 ml mixture of collagenase (400 U type II collagenase, Worthington) and 0.2 mg/ml DNase I in RPMI media at 37°C for 1 hr. The mixture was gently vortexed every 10-20 minutes. The tissue lysate was filtered through a 40 μm mesh prior to immunostaining. The resulting single cell suspension was stained with fixable viability dye eFluor 780, anti-CD45.2 Pacific Blue, anti-CD3 PE-Cy7, anti-CD3 Alexa Fluor 700, anti-Foxp3 Alexa Fluor 700, anti-CD11c eFluor 615, anti-NK1.1 PE (all from eBioscience), anti-Granzyme B APC and anti-CD4 Qdot 605 (from Life Technologies), anti-CD8 Brilliant Violet 650, anti-CD11b Brilliant Violet 570, anti-CD19 Brilliant Violet 650, anti-F4/80 FITC (all from BioLegend), anti-Ly6C APC, anti-Ly6G PE-Cy7, and anti-Ki67 PE (BD Biosciences). The percent positive cells were analyzed by FlowJo and gated on CD45 positivity. To analyze the number of CD133+CD44+ cells, the single cell suspension was incubated in the dark, on ice with Aqua LIVE/DEAD Fixable Dead Cell Stain (Molecular Probes) 1:1000 for 30 min, followed by staining with anti-CD44 APC (eBioscience, 1: 400) and anti-CD133 PE (eBioscience, 1: 200). Unstained, LIVE/DEAD only, and single stain served as control. Doublets were gated out using forward scatter width/height and sideward scatter width/height event characteristics.
Tumor spheres and limiting dilution tumor formation assay
Tumors were digested as described above and cell suspension filtered through a 100 μm mesh. 2 million cells were plated in low adherence dish with 1% FBS/DMEM+PSA. 3 to 4 weeks later, the formed spheres were counted at 200× magnification. For the limiting dilution assay, flow cytometry purified YFP+ cancer cells (from PKT mice crossed to R26-stop-EYFP reporter mice) were pelleted and resuspended in PBS prior to subcutaneous injection in Nude mice. Six Nude mice received injection of YFP+ cells (100, 1000 or 10,000 cells) from tumors of PKT mice without myofibroblast depletion (Control, 2 donor mice were used) or with myofibroblast depletion (Depleted, 2 donor mice were used).
Gene expression profiling
Total RNA was isolated from tumors of PKT control mice (n=3 at early stage, n=3 at late stage) and myofibroblast-depleted mice (n=4 at early stage, n=3 at late stage, n=2 at late stage and treated with anti-CTLA4) using RNeasy Plus Mini Kit (Qiagen) and submitted to the Microarray Core Facility at MD Anderson Cancer Center. Gene expression analysis was performed using Mouse Ref6 Gene Expression BeadChip (Illumina), and the Limma package from R Bioconductor (Carey et al., 2005) was used to analyze differentially expressed genes of myofibroblast-depleted mice versus control mice (p ≤ 0.05, fold change ≥ 1.5). Gene Ontology/pathway analyses of differentially expressed genes are performed using the web-accessible program DAVID (Database for Annotation, Visualization and Integrated Discovery) (Huang da et al., 2009). Pathways that enrich differentially expressed genes are selected (p<0.05). For RNA sequencing analyses, sequencing of the whole transcriptome was performed using the SOLiD system. Lifescope Genomic Analysis Software (v 2.5.1) was utilized to map the raw data to the mouse genome (build mm10) and to quantify the counts per gene. The count data was used to perform differential expression analysis using the R package, DEseq. Quantitative real-time PCR analyses are detailed in the Supplemental Experimental Procedures.
Vascular leakage
Vascular leakage was assayed and quantified as previously described (Cooke et al., 2012). Quantification was performed using a grading system where 0-5, representing 0%-100% extravascular FITC-dextran. Results were plotted as scores.
Clinical Studies
Resected tumors were obtained from 53 patients (Table S1) with invasive pancreatic adenocarcinoma who underwent surgical resection at Johns Hopkins Hospital after approval by the Johns Hopkins Hospital Institutional Review Board. The cases were obtained under an IRB exempt protocol. Clinical information was obtained from the electronic medical records. Tissue sections from paraffin embedded specimen were stained for αSMA (Sigma) as described above and staining intensity was scored blinded. Each tissue section was surveyed entirely for αSMA stain intensity in interstitial fibroblasts. αSMA+ vessels of all sizes were easily distinguishable from interstitial fibroblasts. Overall, there were few αSMA+ vessels detected in the tumors, and these were not included in the score. A 0-3 scale was used, with 0: no detected staining, 1: weak staining, 2: moderate staining, 3: strong staining. Patients were then divided into two groups: a ‘low αSMA’ group, as defined by scores of 0 and 1, and a ‘high αSMA’ group, as defined by scores of 2 and 3. Kaplan-Meier plots were drawn for each group and statistical differences evaluated using the Log-rank Mantel-Cox test.
Statistics
Statistical analyses of pathological scores, flow cytometry, and IHC quantifications were performed by using the student's t-test, one-way ANOVA or Fisher's exact test with GraphPad Prism. Limiting dilution assay was evaluated by SPSS with Χ2 test. For survival analyses, Kaplan-Meier plots were drawn and statistical differences evaluated using the Log-rank Mantel-Cox test. p< 0.05 was considered statistically significant.
Accession numbers
The microarray data are deposited at Gene Expression Omnibus under accession numbers GSE52812 and GSE55871.
GSE52812: Gene expression changes comparing pancreas tumors from Ptf1acre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) mice with pancreas tumors from PKT mice with myofibroblast depletion (PKT; αSMA-tk mice).
GSE55871: Gene expression changes comparing myofibroblasts from pancreas tumors of Ptf1acre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) mice with myofibroblasts from pancreas tumors of PKT mice with myofibroblast depletion (PKT; αSMA-tk mice).
Supplementary Material
Highlights.
Depletion of myofibroblasts in mouse pancreas cancer led to invasive tumors.
Decrease in fibrosis and myofibroblasts led to enhanced EMT and cancer stem cells.
Low myofibroblasts in tumors associates with poor survival in mice and patients.
Immunotherapy combined with loss of myofibroblasts prolongs survival.
Significance.
Pancreas cancer is associated with large amounts of stroma composed of collagen and myofibroblasts but the functional contribution of collagen and myofibroblasts in pancreas cancer remains unknown. Specific depletion of myofibroblasts using compound genetic mouse models of PDAC leads to aggressive tumors with diminished animal survival. Lower myofibroblasts in human PDAC also correlates with reduced patient survival. Detailed studies show that myofibroblasts loss decreases the ability of immune system to control cancer due to presence of regulatory T cells. Myofibroblasts depletion did not improve Gemcitabine efficacy but immunotherapy to revive immune attack prolonged survival of mice. This study demonstrates a protective role of myofibroblasts and suggests that targeting carcinoma-associated fibroblasts in pancreas cancer should be approached with caution.
Acknowledgments
We wish to thank Travis Hardcastle, Lauren Bizarro, Judith Kaye, Laura Gibson and Sara Lovisa for genotyping and animal husbandry support, Donna Reynolds for immunohistochemistry stainings, and Travis Hardcastle, Dr. Ganiraju C. Manyam, Dr. Jing Wang and the Bioinformatics and Computational Biology Department at MDACC for bioinformatic support. This study was primarily supported by the NIH grant UO1 CA151925. B.C.O. received funding from the OncoSuisse MD/PhD Scholarship 323630-128865/1 and the Swiss National Science Foundation Fellowship PBBEP3_144809. C.K. is funded by a Research Fellowship of the Deutsche Forschungsgemeinschaf (DFG). A.M. is supported by the United States National Institutes of Health (NIH) grant CA113669. R.K. is supported by the Cancer Prevention and Research Institute of Texas and the Metastasis Research Center at MD Anderson Cancer Center, NIH grants DK55001, DK81976, CA125550, CA155370 and CA163191.
Footnotes
Conflict of interest disclosure: Dr. James P. Allison is an Inventor of IP owned by the University of California, Berkeley, and licensed to Bristol Meyers-Squibb.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes & development. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nature medicine. 2013;19:1410–1422. doi: 10.1038/nm.3389. [DOI] [PubMed] [Google Scholar]
- Angeli F, Koumakis G, Chen MC, Kumar S, Delinassios JG. Role of stromal fibroblasts in cancer: promoting or impeding? Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2009;30:109–120. doi: 10.1159/000218708. [DOI] [PubMed] [Google Scholar]
- Armstrong T, Packham G, Murphy LB, Bateman AC, Conti JA, Fine DR, Johnson CD, Benyon RC, Iredale JP. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:7427–7437. doi: 10.1158/1078-0432.CCR-03-0825. [DOI] [PubMed] [Google Scholar]
- Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proceedings of the National Academy of Sciences of the United States of America. 2006a;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes & development. 2006b;20:3130–3146. doi: 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer cell. 2012;21:822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, Kenny PA, Radisky DC. Microenvironmental regulators of tissue structure and function also regulate tumor induction and progression: the role of extracellular matrix and its degrading enzymes. Cold Spring Harbor symposia on quantitative biology. 2005;70:343–356. doi: 10.1101/sqb.2005.70.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, Radisky D. Putting tumours in context. Nature reviews Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey VJ, Dudoit R, Irizarry R, Huber W, Gentleman R. Bioinformatics and Computational Biology Solutions using R and Bioconductor 2005 [Google Scholar]
- Cheresh DA, Stupack DG. Regulation of angiogenesis: apoptotic cues from the ECM. Oncogene. 2008;27:6285–6298. doi: 10.1038/onc.2008.304. [DOI] [PubMed] [Google Scholar]
- Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer research. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- Cooke VG, LeBleu VS, Keskin D, Khan Z, O'Connell JT, Teng Y, Duncan MB, Xie L, Maeda G, Vong S, et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer cell. 2012;21:66–81. doi: 10.1016/j.ccr.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. The New England journal of medicine. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer cell. 2010;17:135–147. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- Erkan M, Michalski CW, Rieder S, Reiser-Erkan C, Abiatari I, Kolb A, Giese NA, Esposito I, Friess H, Kleeff J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2008;6:1155–1161. doi: 10.1016/j.cgh.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, Vasile E, DePinho RA, Jacks T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer cell. 2009;16:379–389. doi: 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassian AR, Coloff JL, Brugge JS. Extracellular matrix regulation of metabolism and implications for tumorigenesis. Cold Spring Harbor symposia on quantitative biology. 2011;76:313–324. doi: 10.1101/sqb.2011.76.010967. [DOI] [PubMed] [Google Scholar]
- Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, Canamero M, Rodriguez-Justo M, Serrano M, Barbacid M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer cell. 2011;19:728–739. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell stem cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Hidalgo M. Pancreatic cancer. The New England journal of medicine. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:5423–5434. doi: 10.1158/1078-0432.CCR-06-0369. [DOI] [PubMed] [Google Scholar]
- Hruban RH, P MB, Klimstra DS. Tumors of the pancreas. Washigton, DC: American Registry of Pathogloy; 2007. [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, Wright CV, Moses HL. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes & development. 2006;20:3147–3160. doi: 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–120. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of clinical investigation. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature reviews Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- Korc M. Pancreatic cancer-associated stroma production. American journal of surgery. 2007;194:S84–86. doi: 10.1016/j.amjsurg.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nature medicine. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. The Journal of cell biology. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merika EE, Syrigos KN, Saif MW. Desmoplasia in pancreatic cancer. Can we fight it? Gastroenterology research and practice. 2012;2012:781765. doi: 10.1155/2012/781765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. The Journal of clinical investigation. 2007;117:50–59. doi: 10.1172/JCI30082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips P. Pancreatic stellate cells and fibrosis. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum (India): 2012. [PubMed] [Google Scholar]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasheed ZA, Matsui W, Maitra A. Pathology of pancreatic stroma in PDAC. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum (India): 2012. [PubMed] [Google Scholar]
- Robert C, Thomas L, Bondarenko I, O'Day S, J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. The New England journal of medicine. 2011;364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, Espeli M, Kraman M, McKenna B, Wells RJ, et al. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. The Journal of experimental medicine. 2013;210:1137–1151. doi: 10.1084/jem.20122344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33:828–833. doi: 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlett CJ, Colvin EK, Pinese M, Chang DK, Morey AL, Musgrove EA, Pajic M, Apte M, Henshall SM, Sutherland RL, et al. Recruitment and activation of pancreatic stellate cells from the bone marrow in pancreatic cancer: a model of tumor-host interaction. PloS one. 2011;6:e26088. doi: 10.1371/journal.pone.0026088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Seminars in cancer biology. 2012;22:396–403. doi: 10.1016/j.semcancer.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone DM. Pancreatic cancer stem cells: implications for the treatment of pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:5646–5648. doi: 10.1158/1078-0432.CCR-08-0584. [DOI] [PubMed] [Google Scholar]
- Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer cell. 2012;22:725–736. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes & development. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide RH, Bajor DL, Winograd R, Evans RA, Bayne LJ, Beatty GL. CD40 immunotherapy for pancreatic cancer. Cancer immunology, immunotherapy : CII. 2013;62:949–954. doi: 10.1007/s00262-013-1427-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vong S, Kalluri R. The role of stromal myofibroblast and extracellular matrix in tumor angiogenesis. Genes & cancer. 2011;2:1139–1145. doi: 10.1177/1947601911423940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WQ, Liu L, Xu HX, Luo GP, Chen T, Wu CT, Xu YF, Xu J, Liu C, Zhang B, et al. Intratumoral alpha-SMA enhances the prognostic potency of CD34 associated with maintenance of microvessel integrity in hepatocellular carcinoma and pancreatic cancer. PloS one. 2013;8:e71189. doi: 10.1371/journal.pone.0071189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whatcott CJ, Posner RG, Von Hoff DD, Han H. Desmoplasia and chemoresistance in pancreatic cancer. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum (India): 2012. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.