

Abstract
Cardio-facio-cutaneous (CFC) syndrome is one of the RASopathies and is caused by alteration of activity through the Ras/mitogen-activated protein kinase (MAPK) pathway due to heterozygous de novo mutations in protein kinases BRAF, MEK1 or MEK2. CFC is a rare multiple congenital anomaly disorder in which individuals have characteristic dysmorphic features, cardiac defects, ectodermal anomalies and developmental delay. We report a 7 ½ month-old boy with a clinical diagnosis of CFC. Bidirectional sequence analysis of MEK2 revealed a novel c.383C→A transition in exon 3 resulting in a nonsynonymous missense substitution, p.P128Q. Other family members, including the proband’s mother and half-sibling, displayed phenotypic features of CFC and were also screened for the MEK2 mutation identified in the proband. SIFT (Sorting Intolerant From Tolerant) analysis determined the novel MEK2 p.P128Q to be deleterious. To corroborate the functional alteration of the novel mutant protein, transient transfection of 293T cells with subsequent Western analysis was used to demonstrate increased kinase activity, as measured by ERK phosphorylation. This first reported case of a vertically transmitted functional CFC MEK mutation further expands our understanding of germline mutations within the Ras/MAPK pathway.
Keywords: acute lymphoblastic leukemia, autosomal dominant, cardio-facio-cutaneous syndrome, BRAF, MEK1, MEK2, RASopathy, Ras/MAPK, signal transduction pathway, multiple generation transmission
INTRODUCTION
Cardio-facio-cutaneous (CFC) syndrome is a multiple congenital anomaly disorder in which individuals have characteristic craniofacial features, cardiac defects, ectodermal anomalies, gastrointestinal dysfunction and neurocognitive delay. CFC is one of the RASopathies, which are a class of human genetic syndromes caused by germline mutations in genes that encode components of the Ras/mitogen-activated protein kinase (MAPK) pathway [Tidyman and Rauen 2009]. Currently, four known genes that encode proteins of the Ras/MAPK pathway have been associated with CFC syndrome: BRAF [Niihori et al. 2006; Rodriguez-Viciana et al. 2006], MEK1 and MEK2 [Rodriguez-Viciana et al, 2006], and KRAS [Niihori et al, 2006]. Other syndromes associated with the pathway are Noonan syndrome, LEOPARD syndrome, gingival fibromatosis 1, capillary malformation-AV malformation syndrome, neurofibromatosis 1 (NF1), Costello syndrome (CS), autoimmune lymphoproliferative syndrome and Legius (NF1-like) syndrome. These RASopathies have many overlapping phenotypic features and are known to be autosomal dominant; stable vertical transmission is known to occur in all of the syndromes except CS and CFC.
In this study, we report a 7 ½ month-old baby boy with a clinical diagnosis of CFC who was found to have a novel p.P128Q MEK2 mutation. Several family members were observed to have phenotypic features consistent with CFC syndrome. Molecular analysis of nine members of this four-generation family revealed the novel MEK2 missense mutation segregating in those family members who had phenotypic features consistent with CFC. This is the first identified vertically transmitted functional CFC MEK mutation reported. This case expands our understanding of germline mutations and their transmission within the Ras/MAPK pathway.
MATERIALS AND METHODS
Family Report
The proband is a Caucasian-Cajun male who was initially evaluated at age 7½ months for short stature and a congenital heart defect. The proband was born to a healthy 27-year-old father and to a 26-year-old mother who reported no consanguinity. The prenatal history was significant for fetal exposures to a half-pack of cigarettes per day. The mother reported that fetal movements were normal, but at 34 weeks, she experienced premature uterine contractions. The proband was delivered by repeat Caesarean section at 36 weeks due to failure to progress. No other complications were noted at delivery, although polyhydramnios was reported. Birth weight was 2910 g. Apgar scores were unknown. The proband was noted to have a cardiac murmur at one month of age and an echocardiogram revealed mild pulmonic stenosis that did not require surgical correction. Also at one month of age, the proband was diagnosed with pyloric stenosis that did require surgical intervention.
When we saw the proband in our clinic, physical examination revealed a dysmorphic 7 ½ month boy with a height of 68 cm (25th centile), weight of 9.5 kg (75th centile) and a head circumference of 45.8 cm (75th centile). His motor milestones were mildly delayed, as indicated by needing minor assistance with sitting. Neurologically, he had good muscle strength and tone, was attentive, alert and able to track objects. His craniofacial features were similar to those of his mother (Table I, Fig. 1A). He had a high forehead with bitemporal narrowing, telecanthus, and a hyperteloric appearance. Ectodermal anomalies were also similar to his mother’s and consisted of sparse, curly hair with sparse eyebrows and eyelashes.
Table 1.
Clinical presentation of family member study participants.
| Pedigree Number | V-2 | IV-8 | IV-9 | V-1 | V-3 | III-6 | II-2 | III-2 | III-3 | III-4 | III-5 | IV-5 | IV-6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Relationship | proband | mother | father | Half brother | Full brother | Mat GM | Mat GGM | Mat GU | Mat GA | Mat GA | Mat GU | First cousin once removed | First cousin once removed |
| Age at evaluation | 7 ½ mo | 27 yrs | 26 yrs | 7 yrs | 8 mo | 42 yrs | 78 yrs | unknown | 58 yrs | unknown | Died 41 yrs | 18 yrs | 14 yrs |
| Sex | M | F | M | M | M | F | F | M | F | F | M | F | M |
| MEK2 mutation | P128Q | P128Q | No mutation | P128Q | Not tested | P128Q | P128Q | Not tested | P128Q | No mutation | Not tested | P128Q | P128Q |
| Neurocognition a | N/A | LD | normal | ADHD/LD | N/A | LD | LD | LD | Tics | - | LD | LD | LD |
| Short stature | - | + | - | - | - | + | + | + | unknown | unknown | unknown | - | - |
| Distinctive facies b | + | + | - | + | + | + | + | + | + | - | + | + | + |
| Cardiac abnormality | Pulmonic Stenosis | Pulmonic Stenosis | - | - | Pulmonic Stenosis | - | - | - | - | - | - | - | - |
| Ectodermal anomalies c | + | + | - | + | + | + | + | + | + | - | + | + | + |
| sparse curly hair | + | + | - | + | + | + | + | + | + | - | + | + | + |
| sparse/lack of eyebrow | + | + | - | + | + | + | + | + | + | - | + | + | + |
| keratosis pilaris | + | - | - | + | + | - | - | unknown | - | - | unknown | + | + |
| hemangiomas | + | + | - | - | - | - | - | unknown | - | - | unknown | - | - |
| CALM | - | + | - | + | + | - | - | unknown | - | - | unknown | - | - |
| nevi | - | 2 | - | - | - | - | - | unknown | - | - | unknown | - | - |
| Pectus excavatum | - | - | - | + | - | - | - | unknown | - | - | unknown | - | - |
| Ocular abnormality d | - | + | - | + | - | + | + | + | + | - | + | + | + |
| Inguinal hernia | - | - | - | + | - | - | - | - | - | - | - | - | - |
| Pyloric stenosis | + | - | - | + | - | - | - | - | - | - | - | - | - |
| Minor penoscrotal inversion | + | N/A | - | + | + | N/A | N/A | unknown | N/A | N/A | unknown | N/A | + |
| Other | Double cervix | Menopause age 40 | infertile | ||||||||||
| Dermatoglyphics e | low TRC | unknown | unknown | low TRC, CPL4F | unknown | t”/t | t”/t | unknown | tt”/tt”’ | unknown | unknown | tt”/t” | t’/t’ |
| Cancer | - | - | - | - | - | - | Breast age 70 | - | - | - | ALL | - | - |
Neurocognition includes learning disability (LD) requiring special education, attention deficient hyperactivity disorder (ADHD), neuromuscular tics or not applicable (N/A) due to young age.
Characteristic facies may include relative macrocephaly, prominent forehead, bitemporal narrowing, shallow orbital ridges, down-slanting palpebral fissures, ptosis, epicanthal folds, telecanthus, hypertelorism, short nose with a depressed nasal bridge and low-set, posteriorly rotated ears.
Ectodermal anomalies may include sparse, curly and/or friable hair, scant eyebrows and eyelashes, keratosis pilaris and/or ulerythema ophryogenes, nevi and/or hemangiomas.
Ophthalmologic abnormalities may include strabismus, myopia and /or hyperopia.
Abbreviations include: Total Ridge Count (TRC), Central Pocket Loop in 4th Finger (CPL4F)
Figure 1.

Clinical images of the family. A) Current clinical images of mutation positive family members. All family members who tested positive for the mutation have similar ectodermal findings as well as have learning disabilities. B) A photograph of the proband’s maternal grandmothers (III-6) family. Photo taken circa late-1960s.
Family history was significant for his mother and older male half-sibling having pulmonic stenosis that did not require surgical correction (Table I). In addition, several family members spanning four generations were also reported to have facial features and ectodermal abnormalities similar to the proband. These same family members also had learning delays requiring special education (Fig. 1; Fig. 2; Table I).
Figure 2.

Pedigree of the family. The proband (indicated by an arrow and black shading) has phenotypic features consistent with a clinical diagnosis of CFC syndrome and has a p.P128Q MEK2 missense mutation. Family members who participated in the study and who have phenotypic features consistent with a clinical diagnosis of CFC syndrome also harbored the same MEK2 mutation (indicated by a blackened symbol). Participating family members that did not have features of CFC tested negative for the mutation (white) and members with gray shading are presumed mutation carriers. I-2, who is shaded in gray, was reported to be born with no arms and had reported features similar to affected individuals in the family. III-3, also shaded in gray, did not want to have his blood drawn but has phenotypic features which are similar to family members who tested positive for the MEK2 mutation. III-5 is deceased but presumed to have been a mutation carrier since he has two affected offspring.
When we evaluated the proband again at 3 years and 4 months, his height, weight and head circumference fell within the normal range and he had no growth delays. However, since the initial evaluation, the mother had become pregnant with the same partner. After a reported normal pregnancy with good fetal movements, good prenatal care and no polyhydramnios, the mother delivered a term male child weighing 3120 g who had a head circumference of 34 cm. At birth, a murmur was noted and the child was subsequently diagnosed with pulmonic stenosis. On physical examination at 8 ½ months, he had craniofacial features that were consistent with those of his mother, the proband and his maternal half-brother (Fig. 1A; Table I).
Bidirectional Sequencing and Analysis
Blood samples from nine family members were obtained after we received approval from the institution review boards from the Louisiana State University Health Sciences Center and Children’s Hospital, and the University of California San Francisco. Genomic DNA was isolated from peripheral blood lymphocytes using the QIAamp DNA Blood Midi kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions.
PCR primers were designed to amplify specific coding exons and intronic flanking regions of BRAF (NM_004333.2), SOS1 (NM_005633.3), KRAS (NM_004985.3), PTPN11 (NM_002834), MEK1 (NM_002755.2) and MEK2 (NM_030662.2). For sequencing, the PCR primers were modified on the 5’ end to include M13 forward (GTAAAACGACGGCCAGT) and reverse (CAGGAAACAGCTATGACC) sequences. PCR and sequencing were performed by GeneDx (Gaithersburg, MD). Because the proband was considered to have either Noonan syndrome due to possible familiar transmission or CFC syndrome based on his phenotypic features, the genes were sequenced in the following order: PTPN11, SOS1, KRAS and BRAF. Subsequently, MEK1 and MEK2 were sequenced.
Bidirectional sequencing was conducted with ABI BigDye v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) according to manufacturer’s guidelines and run on an ABI3730xl capillary sequencing instrument (Applied Biosystems, Foster City, CA). Sequencing data was analyzed using two sequence analysis programs, PolyPhred Software v5.02 (University of Washington, Seattle, WA) and SeqScape® Software (Applied Biosystems, Foster City, CA). Further evaluation of the detected nucleotide mutation consisted of Sorting Intolerant From Tolerant (SIFT; blocks.fhcrc.org/sift/SIFT.html) and screening against known databases: NCBI, Cosmic, UniProtKB/Swiss-Prot and JSNP (www.ncbi.nlm.nih.gov/SNP, www.sanger.ac.uk/genetics/CGP/cosmic/,ca.expasy.org/sprot/, snp.ims.u-tokyo.ac.jp/). Control samples included 50 normal controls (100 alleles) and 60 CFC individuals (120 alleles).
Plasmids
Human MEK2 cDNA (Origene, Rockville, MD) was cloned into a pcDNA3 vector with a Myc-tag at the N-terminus. The MEK2 c.383C→A transition was introduced using Quick-Change Site-Directed Mutagenesis (Stratagene, La Jolla, CA) and verified by direct sequencing.
Transient Transfections and Western Blot Analysis
Human embryonic kidney (HEK) 293T cells were seeded the day before transfection in six-well dishes. The cells were transfected, in triplicate, with 2 μg total plasmid DNA and 5 μl of Lipofectamine 2000 (Life Technologies, Carlsbad, CA) according to manufacturer’s instructions. In addition, HEK 293T cells were transiently transfected with empty vector, wild-type MEK2, the kinase inactive MEK2 p.K101M negative control, constitutively active MEK2 mutant S222D/S226D, MEK2 p.F57C (a positive CFC control mutant which has known high activity level [Rodriguez-Viciana et al, 2006] and the MEK2 p.P128Q mutant. Cells were serum-starved (0.5% fetal bovine serum), and 24 hours later, lysed in buffer containing Protease and Phosphatase Inhibitor cocktails (Sigma, St. Louis, MO). Expression levels of MEK2-myc, total ERK and phosphorylated-ERK (p-ERK) were analyzed by Western blot. Myc (A-14) and p-ERK (E-4) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and p44/42 MAP Kinase antibody (total ERK, #9102) was purchased from Cell Signaling Technology (Danvers, MA).
RESULTS
MEK2 Mutation
Bidirectional sequence analysis of the proband’s DNA revealed a novel MEK2 c.383C→A transition in exon 3, resulting in a nonsynonymous missense substitution, p.P128Q (Fig. 3). No other mutations resulting in a nonsynonymous amino acid substitution were identified in BRAF, SOS1, KRAS, PTPN11 or MEK1. Sequence analysis of the mother’s and father’s DNA and that of a half-sibling revealed that his mother and his half-sibling carried the same heterozygous MEK2 mutation. Subsequent sequencing of DNA from family members spanning four generations revealed that the MEK2 mutation segregated in family members who had phenotypic features consistent with CFC syndrome (Fig. 1; Fig. 2; Table I). Family members with normal phenotypic features did not have the MEK2 missense mutation (Table I).
Figure 3.
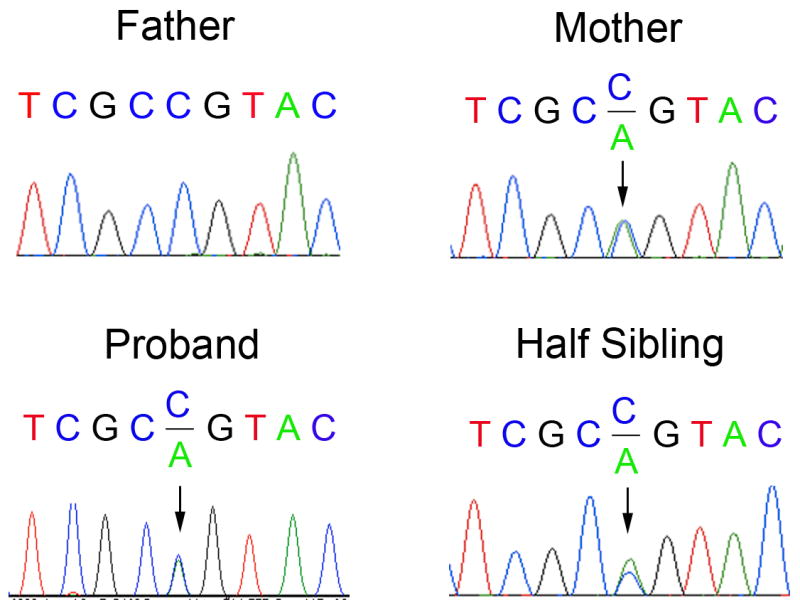
Electropherograms of MEK2 exon 3 sequencing. Electropherograms of the novel MEK2 mutation c.383C>A transition in exon 3 results in a nonsynonymous missense substitution p.P128Q. The proband, his half-sibling brother and mother all have the heterozygous germline mutation. The proband’s father has the wild-type MEK2 sequence.
Functional Characterization of the MEK2 Mutant Protein
SIFT was used to characterize the functional significance of the nonsynonymous amino acid substitution and predicted that the MEK2 p.P128Q would be a deleterious substitution causing an alteration of protein function. To corroborate the functional alteration of the novel MEK2 p.P128Q mutant, HEK 293T cells were transiently transfected with appropriate controls plasmids and MEK2 mutant plasmid. We found that the p.P128Q MEK2 mutant had increased ERK phosphorylation compared to the level induced by empty vector, wild-type MEK2 and the negative kinase inactive control. The level of ERK phosphorylation induced by p.P128Q MEK2 was less than that of the CFC MEK2 p.F57C mutant protein and much less than that of the constitutively active S222D/S226D MEK2 control, both of which are known to have increased activity over wild-type [Rodriguez-Viciana et al, 2006]. This result indicates that the p.P128Q MEK2 CFC mutant protein is a weak hypermorph.
DISCUSSION
CFC syndrome is a RASopathy and is caused by alteration of signaling through the Ras/MAPK pathway [Tidyman and Rauen 2009]. Although CFC syndrome has a distinct phenotype, it shares many overlapping features with other RASopathies especially Noonan syndrome (NS) and Costello syndrome (CS). Craniofacial findings in CFC syndrome may include macrocephaly, broad forehead, bitemporal narrowing, hypoplasia of the supraorbital ridges, down-slanting palpebral fissures with ptosis, short nose with depressed nasal bridge and anteverted nares, low-set, posteriorly rotated ears with prominent helices and a high-arched palate. Ectodermal findings typically consist of sparse, curly hair with sparse eyebrows and eyelashes, keratosis pilaris, hemangioma and nevi. Cardiac anomalies vary with the most prevalent being pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Neurologic abnormalities are universally present to varying degrees and include hypotonia, motor delay, speech delay and/or learning disability. Four genes that encode proteins in the Ras/MAPK pathway have been associated with CFC syndrome: BRAF [Niihori et al, 2006; Rodriguez-Viciana et al, 2006], MEK1 and MEK2 [Rodriguez-Viciana et al, 2006], and KRAS [Niihori et al, 2006]. Although there is some phenotypic variability among family members who harbor the p.P128Q MEK2 mutation, their features are consistent with the diagnosis of CFC syndrome and the MEK2 mutation segregates with those family members who have features of CFC syndrome (Table I).
To date, CFC has been considered sporadic with no mutation-positive cases known to be vertically transmitted. However, many of the RASopathies exhibit autosomal dominant inheritance although de novo mutations certainly do occur. The best studied is NF1 because it is one of the most common dominantly inherited genetic disorder occurring with an incidence of about 1 in 3000 individuals. Approximately half of the affected individuals will have a de novo mutation in the NF1 gene [for review see [Williams et al. 2009]]. This is similar to NS which is caused by mutations in multiple genes encoding components of the Ras/MAPK pathway [Cordeddu et al. 2009; Pandit et al. 2007; Razzaque et al. 2007; Roberts et al. 2007; Schubbert et al. 2006; Tartaglia et al. 2001; Tartaglia et al. 2007]. Although many of the cases of NS are sporadic, vertical transmission of PTPN11, SOS1 and RAF1 are common. This is also true for LEOPARD syndrome with autosomal dominant inheritance of mutations in PTPN11 [Digilio et al. 2002; Legius et al. 2002] or RAF1 [Pandit et al, 2007]. Other RASopathies with autosomal dominant inheritance include capillary malformation– arteriovenous malformation syndrome due to mutations in the gene RASA1 [Eerola et al. 2003; Revencu et al. 2008]; Legius syndrome caused by heterozygous mutations in SPRED1 [Brems et al. 2007] and hereditary gingival fibromatosis type 1 caused by an insertion mutation in the SOS1 gene [Hart et al. 2002].
Although CFC is considered an autosomal dominant syndrome, it was not known how this syndrome impacts reproductive fitness. It is unclear if mutations within the MAPK pathway mediated by Raf (BRAF, RAF1/CRAF, MEK1 and MEK2) are incompatible with reproduction or if lack of reproductive success is due to other phenotypic features of the syndrome such as neurocognitive delay which is present in various degrees [Yoon et al. 2007]. However, this is certainly not the case with RAF1 (CRAF) since autosomal dominant transmission has been documented [Pandit et al, 2007]. The MEK2 mutation identified in this family is a hypermorph and presumed to affect downstream signaling. MEK 1 and MEK 2 are the only known downstream effectors of Raf. MEK1 and MEK2 are threonine/tyrosine kinases with both isoforms having the ability to phosphorylate and activate ERK1 and ERK2 (MAPK). ERK, once activated by MEK, has numerous cytosolic and nuclear substrates [Yoon and Seger 2006]. Previous in vitro functional studies of other CFC MEK mutant proteins have demonstrated increased activity over wild-type MEK in stimulating ERK phosphorylation [Estep et al. 2007; Rodriguez-Viciana et al, 2006].
The Ras/MAPK pathway is critical in mammalian reproduction. In females, follicle development is dependant upon a properly regulated Ras/MAPK pathway. Sustained and selective activation of Kras G12D in mouse granulosa cells result in abnormal follicle-like structures which result in ovarian failure [Fan et al. 2008]. In addition, ERK1 and ERK2 are essential for the normal function of mouse granulosa cells, as well as the luteinizing response of oocyte resumption of meiosis, normal ovulation and the proper luteinization of the mouse ovary [Fan et al. 2009]. The well-orchestrated control of the Ras/MAPK pathway is just as critical in male reproduction [for review see [Li et al. 2009]]. As some examples, ERK1/2 are present during all stages of mouse spermatogenesis [Lu et al. 1999] and the Ras/MAPK pathway is critical for proper capacitation [de Lamirande and Gagnon 2002] and motility [Almog et al. 2008]. In fact, some activating Ras pathway mutations may confer an advantage to the sperm [Glaser et al. 2003; Glaser and Jabs 2004; Goriely et al. 2009]. Because critical control of the Ras/MAPK pathway is required for mammalian reproduction, it was surprising that a hyperactive MEK2 mutation directly upstream from ERK is compatible with successful reproduction. In addition, this mutation was successfully transmitted from both women and men in this family.
Aberrant upstream Ras signaling resulting in hyperactivated ERK plays a key role in the pathogenesis and progression of approximately 30% of human cancers [Hoshino et al. 1999]. However, neoplasia, either benign or malignant that is observed in CS, NS or NF1, has not been reported in CFC syndrome. Although it is unclear if individuals with CFC are at an increased risk to develop cancer, two individuals with mutations in this pedigree have developed cancer. The proband’s maternal great grandmother (II-2) developed a large B-cell lymphoma at age 70 and the proband’s maternal great uncle died of acute lymphoblastic leukemia (ALL) at age 41 years. This is the third mutation positive CFC individual to have developed ALL [Makita et al. 2007; Van Den Berg 1999].
In conclusion, we present a family of four generations with CFC syndrome caused by a novel p.P128Q MEK2 mutation which has increased functional kinase activity over wild-type in vitro. Evaluation of this family has certainly expanded the phenotype of CFC syndrome to include variable expressivity among family members who harbor the same pathogenetic mutation and it has heightened our awareness that ALL may be of higher risk in individuals with CFC syndrome. Our findings underscore the importance of a thorough genetic evaluation of family members and that activating mutant proteins within the MAPK cascade may be compatible with human reproduction.
Figure 4.
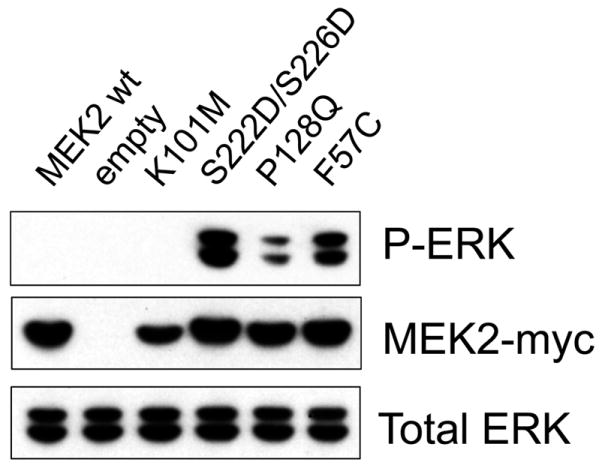
Functional characterization of the MEK2 p.P128Q mutant. Human embryonic kidney 293T cells were transiently transfected with the MEK2 p.P128Q mutant plasmid and appropriate controls. ERK phosphorylation was assayed by Western blotting using phosphospecific antibodies. The p.P128Q MEK2 mutant protein had increased ERK phosphorylation compared to the level induced by empty vector, wild-type MEK2 and the kinase dead control. However, the p.P128Q MEK2 mutant protein was less active less than the CFC MEK2 p.F57C mutant which is known to have increased activity over wild-type. This indicates that p.P128Q MEK2 is a weak hypermorph. Myc-tagged MEK2 served as a marker for transfection efficiency and total ERK served as a loading control.
Acknowledgments
We thank the outstanding collaboration of this family who participated in this study. We also thank Dr. Michael Lan, Meghan Hurley and the technologists at GeneDx. This work was supported in part by NIH grant HD048502 (K.A.R.) and in part by NIH/NCRR UCSF-CTSI Grant Number UL1 RR024131 (K.A.R.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
References
- Almog T, Lazar S, Reiss N, Etkovitz N, Milch E, Rahamim N, Dobkin-Bekman M, Rotem R, Kalina M, Ramon J, Raziel A, Breitbart H, Seger R, Naor Z. Identification of extracellular signal-regulated kinase 1/2 and p38 MAPK as regulators of human sperm motility and acrosome reaction and as predictors of poor spermatozoan quality. J Biol Chem. 2008;283:14479–89. doi: 10.1074/jbc.M710492200. [DOI] [PubMed] [Google Scholar]
- Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, Kato R, Somers R, Messiaen L, De Schepper S, Fryns JP, Cools J, Marynen P, Thomas G, Yoshimura A, Legius E. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet. 2007;39:1120–6. doi: 10.1038/ng2113. [DOI] [PubMed] [Google Scholar]
- Cordeddu V, Di Schiavi E, Pennacchio LA, Ma’ayan A, Sarkozy A, Fodale V, Cecchetti S, Cardinale A, Martin J, Schackwitz W, Lipzen A, Zampino G, Mazzanti L, Digilio MC, Martinelli S, Flex E, Lepri F, Bartholdi D, Kutsche K, Ferrero GB, Anichini C, Selicorni A, Rossi C, Tenconi R, Zenker M, Merlo D, Dallapiccola B, Iyengar R, Bazzicalupo P, Gelb BD, Tartaglia M. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41:1022–6. doi: 10.1038/ng.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lamirande E, Gagnon C. The extracellular signal-regulated kinase (ERK) pathway is involved in human sperm function and modulated by the superoxide anion. Mol Hum Reprod. 2002;8:124–35. doi: 10.1093/molehr/8.2.124. [DOI] [PubMed] [Google Scholar]
- Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, Marino B, Pizzuti A, Dallapiccola B. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet. 2002;71:389–94. doi: 10.1086/341528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe S, Vanwijck R, Vikkula M. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240–9. doi: 10.1086/379793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estep AL, Palmer C, McCormick F, Rauen KA. Mutation Analysis of BRAF, MEK1 and MEK2 in 15 Ovarian Cancer Cell Lines: Implications for Therapy. PLoS ONE. 2007;2:e1279. doi: 10.1371/journal.pone.0001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan HY, Liu Z, Shimada M, Sterneck E, Johnson PF, Hedrick SM, Richards JS. MAPK3/1 (ERK1/2) in ovarian granulosa cells are essential for female fertility. Science. 2009;324:938–41. doi: 10.1126/science.1171396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan HY, Shimada M, Liu Z, Cahill N, Noma N, Wu Y, Gossen J, Richards JS. Selective expression of KrasG12D in granulosa cells of the mouse ovary causes defects in follicle development and ovulation. Development. 2008;135:2127–37. doi: 10.1242/dev.020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser RL, Broman KW, Schulman RL, Eskenazi B, Wyrobek AJ, Jabs EW. The paternal-age effect in Apert syndrome is due, in part, to the increased frequency of mutations in sperm. Am J Hum Genet. 2003;73:939–47. doi: 10.1086/378419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser RL, Jabs EW. Dear old dad. Sci Aging Knowledge Environ. 2004:re1. doi: 10.1126/sageke.2004.3.re1. [DOI] [PubMed] [Google Scholar]
- Goriely A, Hansen RM, Taylor IB, Olesen IA, Jacobsen GK, McGowan SJ, Pfeifer SP, McVean GA, Meyts ER, Wilkie AO. Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nat Genet. 2009;41:1247–52. doi: 10.1038/ng.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart TC, Zhang Y, Gorry MC, Hart PS, Cooper M, Marazita ML, Marks JM, Cortelli JR, Pallos D. A mutation in the SOS1 gene causes hereditary gingival fibromatosis type 1. Am J Hum Genet. 2002;70:943–54. doi: 10.1086/339689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J, Kohno M. Constitutive activation of the 41-/43-kDa mitogenactivated protein kinase signaling pathway in human tumors. Oncogene. 1999;18:813–22. doi: 10.1038/sj.onc.1202367. [DOI] [PubMed] [Google Scholar]
- Legius E, Schrander-Stumpel C, Schollen E, Pulles-Heintzberger C, Gewillig M, Fryns JP. PTPN11 mutations in LEOPARD syndrome. J Med Genet. 2002;39:571–4. doi: 10.1136/jmg.39.8.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MW, Mruk DD, Cheng CY. Mitogen-activated protein kinases in male reproductive function. Trends Mol Med. 2009;15:159–68. doi: 10.1016/j.molmed.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Sun QY, Breitbart H, Chen DY. Expression and phosphorylation of mitogen-activated protein kinases during spermatogenesis and epididymal sperm maturation in mice. Arch Androl. 1999;43:55–66. doi: 10.1080/014850199262733. [DOI] [PubMed] [Google Scholar]
- Makita Y, Narumi Y, Yoshida M, Niihori T, Kure S, Fujieda K, Matsubara Y, Aoki Y. Leukemia in Cardio-facio-cutaneous (CFC) syndrome: a patient with a germline mutation in BRAF proto-oncogene. J Pediatr Hematol Oncol. 2007;29:287–90. doi: 10.1097/MPH.0b013e3180547136. [DOI] [PubMed] [Google Scholar]
- Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, Okamoto N, Hennekam RC, Gillessen-Kaesbach G, Wieczorek D, Kavamura MI, Kurosawa K, Ohashi H, Wilson L, Heron D, Bonneau D, Corona G, Kaname T, Naritomi K, Baumann C, Matsumoto N, Kato K, Kure S, Matsubara Y. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38:294–6. doi: 10.1038/ng1749. [DOI] [PubMed] [Google Scholar]
- Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, Pogna EA, Schackwitz W, Ustaszewska A, Landstrom A, Bos JM, Ommen SR, Esposito G, Lepri F, Faul C, Mundel P, Lopez Siguero JP, Tenconi R, Selicorni A, Rossi C, Mazzanti L, Torrente I, Marino B, Digilio MC, Zampino G, Ackerman MJ, Dallapiccola B, Tartaglia M, Gelb BD. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39:1007–12. doi: 10.1038/ng2073. [DOI] [PubMed] [Google Scholar]
- Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, Kamisago M, Momma K, Katayama H, Nakagawa M, Fujiwara Y, Matsushima M, Mizuno K, Tokuyama M, Hirota H, Muneuchi J, Higashinakagawa T, Matsuoka R. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. 2007;39:1013–7. doi: 10.1038/ng2078. [DOI] [PubMed] [Google Scholar]
- Revencu N, Boon LM, Mulliken JB, Enjolras O, Cordisco MR, Burrows PE, Clapuyt P, Hammer F, Dubois J, Baselga E, Brancati F, Carder R, Quintal JM, Dallapiccola B, Fischer G, Frieden IJ, Garzon M, Harper J, Johnson-Patel J, Labreze C, Martorell L, Paltiel HJ, Pohl A, Prendiville J, Quere I, Siegel DH, Valente EM, Van Hagen A, Van Hest L, Vaux KK, Vicente A, Weibel L, Chitayat D, Vikkula M. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat. 2008;29:959–65. doi: 10.1002/humu.20746. [DOI] [PubMed] [Google Scholar]
- Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, Li L, Yassin Y, Tamburino AM, Neel BG, Kucherlapati RS. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39:70–4. doi: 10.1038/ng1926. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, McCormick F, Rauen KA. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–90. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner LE, Nguyen H, West B, Zhang KY, Sistermans E, Rauch A, Niemeyer CM, Shannon K, Kratz CP. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331–6. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–8. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, Pandit B, Oishi K, Martinelli S, Schackwitz W, Ustaszewska A, Martin J, Bristow J, Carta C, Lepri F, Neri C, Vasta I, Gibson K, Curry CJ, Siguero JP, Digilio MC, Zampino G, Dallapiccola B, Bar-Sagi D, Gelb BD. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39:75–9. doi: 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230–6. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Berg H, Hennekam RCM. Acute lymphoblastic leukaemia in a patient with cardiofaciocutaneous syndrome. Journal of Medical Genetics. 1999;36:799–800. doi: 10.1136/jmg.36.10.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123:124–33. doi: 10.1542/peds.2007-3204. [DOI] [PubMed] [Google Scholar]
- Yoon G, Rosenberg J, Blaser S, Rauen KA. Neurological complications of cardio-facio-cutaneous syndrome. Dev Med Child Neurol. 2007;49:894–9. doi: 10.1111/j.1469-8749.2007.00894.x. [DOI] [PubMed] [Google Scholar]
- Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]