

Abstract
Hydrogel scaffolds are commonly used as 3D carriers for cells because their properties can be tailored to match natural extra-cellular matrix. Hydrogels may be used in tissue engineering and regenerative medicine to deliver therapeutic cells to injured or diseased tissue through controlled degradation. Hydrolysis and enzymolysis are the two most common mechanisms employed for hydrogel degradation, but neither allows sequential or staged release of cells. In contrast, photodegradation allows external real-time spatial and temporal control over hydrogel degradation, and allows for staged and sequential release of cells. We synthesized and characterized a series of macromers incorporating photodegradbale ortho-nitrobenzyl (o-NB) groups in the macromer backbone. We formed hydrogels from these macromers via redox polymerization and quantified the apparent rate constants of degradation (kapp) of each via photorheology at 370 nm, 10 mW/cm2. Decreasing the number of aryl ethers on the o-NB group increases kapp, and changing the functionality from primary to seconday at the benzylic site dramatically increases kapp. Human mesenchymal stem cells (hMSCs) survive encapsulation in the hydrogels (90% viability post-encapsulation). By exploiting the differences in reactivity of two different o-NB linkers, we quantitatively demonstrate the biased release of one stem cell population (green-fluroescent protein expressing hMSCs) over another (red-fluorescent protein expressing hMSCs).
Keywords: cell delivery, ortho-nitrobenzyl, photodegradable hydrogels, regenerative medicine
INTRODUCTION
Hydrogels are commonly used as 3D carriers for cells, due to their high water content, tunable mechanical properties, and the rapid diffusivity of nutrients through the network.1 Erodable hydrogels are important in tissue engineering for creating complex cell niches, and in therapeutics, where degradation allows the predictable release of encapsulated cells and tethered therapeutics. Hydrolysis and enzymolysis are the two most common mechanisms employed for hydrogel degradation. Both mechanisms are effective for sustained hydrogel degradation, however the rate of degradation cannot be adjusted or arrested after the hydrogel is fabricated, nor is degradation spatially controlled. In contrast to enzymolyis and hydrolysis, photodegradation allows external real-time spatial and temporal control over hydrogel properties. Kloxin et. al. incorporated 2-methoxy-5-nitro-4-(1-hydroxyethyl)phenoxybutanoate (an ortho-nitrobenzyl (o-NB) linker) into the backbone of a poly(ethylene glycol) (PEG) macromer.2 Physical properties of hydrogels formed from this macromer are temporally and spatially controllable via light exposure (single and two-photon),3,4 and photodegradation is compatible with live cells.2,5 While photodegradation allows dynamic control of hydrogel properties, there are practical limitations to the single o-NB linker reported. The 2-methoxy-5-nitro-4-(1-bromoethyl)phenoxybutanoate-based macromers degrade over several minutes or longer when exposed to 365 nm light with reasonable (10 mW/cm2) intensity. This wavelength and dose of light has been shown to be compatible with live-cell encapsulation.6 However, long-wave UV may still interact with and alter structures or chemicals within the cell7 and therefore it is best to minimize cell exposure to it. Attenuation is also significant due to the high molar absorptivity of this o-NB linker. Finally, with only a single photolyzable linker, there is no way to independently execute two separate events, such as the sequential release of different therapeutics or cells. Therefore, new or additional o-NB groups with lower molar absorptivities combined with more efficient degradation may improve cell and tissue compatibility, and multiple photodegradable groups may also allow multi-stage processes.
o-NB groups are widely used as photocages in the presence of live cells,8–12 and also as degradable linkages in polymer materials13. Several different o-NB groups have been reported14–16, and small modifications to the structure of the o-NB linker have a significant impact on the photolysis rate.14 The benzylic position (photodegradation site) of o-NB groups can be primary or secondary (Chart 1a, R1= −H or R1= −CH3). Additionally, the o-NB group may contain no other aromatic substituents (R2=R3=−H), one or two aryl ethers, or other substituents. Each of these structural changes can affect the degradation rate. For example, the half-life of an o-NB linker with a primary benzylic group is approximately 25 times that of one with a secondary benzylic group (X= −NHR, buffer, pH=7).13 When X=RSO3-, the apparent rate constant of photodegradation (kapp) is nearly 40 times higher for a secondary benzylic group compared to a primary group (solvent: CDCl3).17 Increasing the number of aryl ethers from one (R1=−CO2H, X= −SR, R2=−OR, R3=−H) to two (R2=−OR, R3=−OR) decreases the quantum yield of the o-NB group at 365 nm by 30%.15
Chart 1.
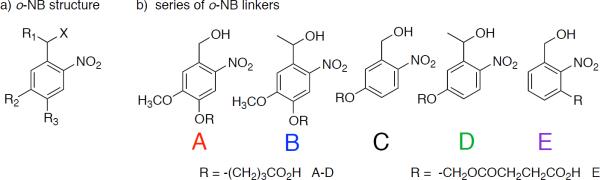
a) Structural variables in o-NB linkers b) series of o-NB linkers designed to have varying degradation rates.
Although general trends are clear, it is difficult to determine which structural changes will have the greatest effect on the rate of degradation – primary versus secondary benzylic group, or the number of aryl ethers. In order to better understand the effect of structural changes on the degradation of o-NB groups, we developed a series of photodegradable o-NB based linkers with varying functionality at the benzylic position (primary or secondary) and varying number of aryl ether groups (Chart 1b). Each o-NB group contains two reactive handles, a benzylic alcohol and a carboxylic acid, to allow for selective conjugation of each site to either a polymerizable group or therapeutic agent (benzylic position), or to a polymer chain or macromer (carboxylic acid).
RESULTS AND DISCUSSION
We synthesized 4-(4-(hydroxymethyl)-2-methoxy-5-nitrophenoxy) butanoic acid (A) and 4-(4-(1-hydroxyethyl)-2-methoxy-5-nitrophenoxy) butanoic acid (B) according to a modified literature protocol (Scheme 1).2,18 First, we esterify the phenol of either acetovanillone (R= −CH3) or vanillin (R= −H) with ethyl-4-bromobutyrate under basic conditions. We use 70% nitric acid to nitrate the ring ortho to the aldehyde/ketone. We then hydrolyze the ester using aqueous trifluoracetic acid and reduce the aldehyde/ketone using sodium borohydride. We reversed the order of hydrolysis and reduction compared to the reported procedure, which increases the overall yield from these two steps from ~36% to ~80%.19
Scheme 1.
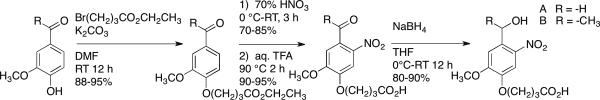
Synthesis of 4-(4-(hydroxymethyl)-2-methoxy-5-nitrophenoxy)butanoic acid and 4-(4-(1-hydroxyethyl)-2-methoxy-5-nitrophenoxy)butanoic acid.
We initially attempted to synthesize 4-(3-(hydroxymethyl)-4-nitrophenoxy) butanoic acid (C) and 4-(3-(1-hydroxyethyl)-4-nitrophenoxy) butanoic acid (D) using the same synthetic route as above, however, the single aryl ether does not sufficiently activate the ring towards electrophilic substitution with 70% nitric acid alone.20 Adding sulfuric acid to the reaction mixture catalyzes the substitution, but results in a mixture of nitrated products. To properly guide nitration to the ortho- position, we react the phenol with oxalyl chloride to form a dimer (Scheme 2) and then nitrate in a mixture of nitric and sulfuric acid.21 This reaction is temperature sensitive; for example, lowering the reaction temperature from −10 °C to −20 °C results in a significant increase in yield (~20% vs. 85%) and eliminates side-product formation. Once we obtain the nitrated product, the rest of the reaction scheme follows the above-described procedure.
Scheme 2.

Synthesis of 4-(3-(hydroxymethyl)-4-nitrophenoxy) butanoic acid and 4-(3-(1-hydroxyethyl)-4-nitrophenoxy) butanoic acid.
To produce an o-NB linker with no aryl ether moieties, we synthesized both 1,3-dihydroxymethyl-2-nitrobenzene (E) and 3-hydroxymethyl-2-nitrobenzoic acid. The synthesis of 1,3-dihydroxymethyl-2-nitrobenzene (E) has already been reported in the literature.22,23 To synthesize 3-hydroxymethyl-2-nitrobenzoic acid, we first oxidize 2-nitro-m-xylene using potassium permanganate to obtain 2-nitro-1,3-benzene dicarboxylic acid. We then partially esterify the dicarboxylic acid in refluxing ethanol overnight and isolate 3-(ethoxycarbonyl)-2-nitrobenzoic acid by flash column chromatography. Although the yield of the partial esterification reaction is low (10 %), we are able to recover a large portion of the starting material (~70%). Following purification, we selectively reduce the carboxylic acid using 1M borane in tetrahydrofuran to produce a benzylic alcohol, and hydrolyze the ester to obtain the heterobifunctional linker, 3-hydroxymethyl-2-nitrobenzoic acid.
We measured the absorbance spectra of each linker and its degradation product via UV/Vis spectroscopy (Supporting information Figure S1). The molar absorptivities (ε) of each linker and its degradation product at 365 nm, 405 nm and 436 nm (peak output wavelengths of a mercury vapor lamp) are summarized in Table 1. Increasing the number of aryl ethers on an o-NB linker results in a red-shift in the absorption spectrum, as does going from a primary benzyl alcohol to a secondary (although this effect is much less pronounced). The molar absorptivity of each molecule decreases with increasing wavelength. The molar absorptivity of the degraded products of each o-NB linker is generally higher than the undegraded molecules, except for the degradation products of linkers A and B at 365 nm, which exhibit lower molar absorptivity than (undegraded) A and B.
Table 1.
Molar absorptivity of o-NB linkers A–E and degraded products at λ=365 nm, 405 nm and 436 nm and kapp at λ=370 nm.
| o-NB group | ε365 (M−1cm−1) | ε405 (M−1cm−1) | ε436 (M−1cm−1) | kapp/I0 × 104(cm2/mW·s) | τ, min |
|---|---|---|---|---|---|
| A degraded | 3420 ± 165 | 729 ± 42 | 55.6 ± 3.8 | 0.26 ± 0.014 | 65 ± 3.5 |
| 1925 ± 22 | 1269 ± 2 | 692 ± 2 | |||
| B degraded | 3500 ± 57 | 845 ± 16 | 77.8 ± 2.6 | 3.3 ± 0.022 | 5.0 ± 0.03 |
| 2637 ± 32 | 1610 ± 31 | 685 ± 9 | |||
| C degraded | 1975 ± 299 | 86.2 ± 3.8 | 4.15 ± 0.21 | 0.42 ± 0.035 | 40 ± 3.3 |
| 2201 ± 26 | 815.3 ± 18.6 | 515.7 ± 6.7 | |||
| D degraded | 2509 ± 93 | 161 ± 6 | 2.65 ± 2.08 | 8.3 ± 0.14 | 2.0 ± 0.03 |
| 4061 ± 234 | 1608 ± 59 | 358 ± 16 | |||
| E degraded | 146.7 ± 3.6 | 4.70 ± 0.26 | 0.35 ± 0.21 | 2.6 ± 0.24 | 6.5 ± 0.6 |
| 507.7 ± 6.8 | 166.7 ± 6.1 | 84.7 ± 4.2 |
We incorporated each linker into a PEG macromer (Chart 2); we first esterified the benzylic alcohol of linkers A-D using acryloyl chloride and triethylamine in THF. We subsequently converted the carboxylic acid of the o-NB to an acid chloride using PCl5, which we used to esterify PEG 4K (supporting information Scheme S1–S2). Although both 3-hydroxymethyl-2-nitrobenzoic acid and 1,3-dihydroxymethyl-2-nitrobenzene (E) can be used to prepare the final macromer, the overall yield using linker E is substantially higher than that obtained using 3-hydroxymethyl-2-nitrobenzoic acid (~20% versus ~5% over four steps). To obtain the macromer incorporating linker E, we monoesterified 1,3-di(hydroxymethyl)-2-nitrobenzene with acryloyl chloride (limiting reagent). We used the remaining benzyl alcohol to ring-open succinic anhydride, yielding a carboxylic acid, which we converted to the acid chloride and coupled to PEG as described for linkers A-D above (supporting information Scheme S3).
Chart 2.

PEG-based macromers incorporating photodegradable o-NB linkers A–E.
We copolymerized each macromer (Mn~4500) with PEG-375 acrylate, using redox initiation (APS/TEMED) to form a hydrogel (50 μm thickness) between the plates of a rheometer modified to allow in situ light exposure, and measured the decrease of the normalized elastic portion (G′~G) of the complex shear modulus of each hydrogel as a function of exposure time at λ=370 nm (UVLED, Figure 1a). Figure 1b shows the consumption of photodegradable group as a function of exposure time; the slope is used to determine kapp. Equation 1 gives kapp, where ϕ is quantum yield, ε is molar absorptivity, I0 is light irradiance (intensity), λ is wavelength, NA is Avogadro's number, h is Planck's constant, and c is the speed of light.
Figure 1.
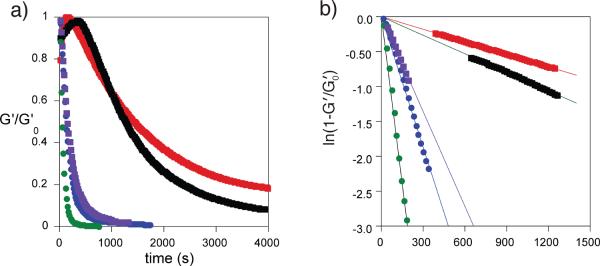
Kinetics of photodegradation. a) Relative decrease in the elastic portion of the shear modulus (G′) as a fraction of exposure time for hydrogels formed from macromers containing linkers A – E. b) Consumption of the o-NB group as a function of exposure time for linkers A – E. λ=370 nm, I0=10 mW/cm2.
The variation in kapp/I0 with structure is summarized in Table 1. As expected, decreasing the number of aryl ethers results in an increase in kapp/I0. Changing the functionality at the benzylic position from a secondary benzyl ester (B and D) to a primary benzyl ester (A, C and E) impacts kapp more significantly. Linker D, with one aryl ether and a secondary benzyl ester, exhibits the fastest degradation of the series. After normalizing to intensity (kapp/I0) at a fixed wavelength (λ), the rate of photodegradation should depend on the product of the molar absorptivity at that wavelength and the quantum yield (quantum product, ϕε). Very low molar absorptivity at a particular wavelength implies little to no photodegradation unless the quantum yield is high. Linkers B and E have similar kapp (3.3×10−3 and 2.6×10−3s−1, respectively) but very different ε (3500 and 146.7 M−1cm−1, respectively), indicating a significant difference in quantum yield for these two structures.
Our series of photodegradable hydrogels incorporating o-NB linkers offers several advantages to the previously reported system utilizing linker B alone. For example, the dose of light required to degrade the hydrogel may be substantially reduced by using the faster degrading linker D. Additionally, the depth of light penetration into the hydrogel increases if linker E is used, as the molar absorptivity of this linker is an order of magnitude lower than that of linkers A-D. Greater penetration of light into the hydrogel allows for bulk erosion, in contrast to the surface erosion achieved for linkers with higher absorptivity. Moreover, combinations of these linkers can be used achieve degradation in an orthogonal manner. Recently, small-molecule wavelength-orthogonal systems including surfaces24 and caged neurotransmitters17 have been reported, but photoresponsive biomaterials25, particularly wavelength-orthogonal biomaterials, are still an emerging research area.
One potential application of photodegradable hydrogels is the encapsulation and on-demand release of therapeutic cells, such as stem cells. Sequential cell introduction and growth is essential in both embryo development26 and proper wound healing.27,28 The ability to deliver or recruit multiple cell types in a staged or sequential manner is therefore critical to wound healing and tissue regeneration, yet remains a major challenge.
Many researchers have reported the combined29 and/or sequential30,31 delivery of growth factors, which represents an indirect route to generating the different cells. That is, the growth factors must arrive at the site and act on progenitor cells or otherwise recruit host cells at an appropriate time to generate the parenchymal cells. There are some limitations to this approach. For example, growth factors may degrade during delivery, and/or may act on cells other than their intended targets in vivo. Delivering cells directly to the injury site is an alternative, however intravenously delivered cells are associated with low engraftment rates, limiting the effectiveness of such procedures. Therefore, there is a need for new cell delivery therapies.
To address this need, researchers have developed technologies such as erodible hydrogels32 and stem cell patches33. Despite these advances, no material-based platform for sequentially delivering stem cells in response to environmental or external triggers exists. Several groups have reported techniques for the release of cells from a surface, such as photoablation of a substrate to release adherent cells.34 Kloxin et al. reported the migration of encapsulated fibrosarcoma cells within channels photopatterned into a hydrogel, however, no cells were released from the gel nor was cell viability determined.2
In order to demonstrate the utility of our system to deliver stem cells, we copolymerized macromer B with PEG-375 acrylate (as above) in the presence of human mesenchymal stem cells (hMSCs). We quantified the viability of the encapsulated cells via a LIVE/DEAD® assay; 87±3% cells survive encapsulation. We exposed the hydrogels to light (λ=365 nm, I0=10 mW/cm2) for three intervals of ten minutes, allowing the gels to equilibrate in media after each interval. We collected the media after each interval and assessed the viability of the released cells. Light exposure has no apparent effect on hMSC viability, as 91±2% hMSCs were viable post-release, successfully demonstrating that this system can be used for on-demand delivery of therapeutic cells.
While the entrapment and on-demand release of cells has many applications in tissue engineering and regenerative medicine, the controlled release of two or more cell populations in a staged manner may provide synergistic behavior and therapeutic benefit not induced by the release of a single cell type or the simultaneous release of multiple cell types. Exploiting the differences in reactivity of the o-NB groups should allow biased release of a cell population encapsulated in one gel over another gel. To demonstrate this, we chose the two o-NB linkers with the fastest degradation times, B and D, which is the most challenging system in which to observe bias. We encapsulated hMSCs expressing red fluorescent protein (RFP-hMSCs) in hydrogels incorporating macromer B and green fluorescent protein (GFP-hMSCs) into hydrogels incorporating macromer D. The hydrogels were fabricated in direct contact with one another (Figure 2a). The interface where the two hydrogels meet is directly observable, as is the partitioning of RFP-hMSCs and GFP-hMSCs (Figure 2b). The entire construct was exposed to light (λ=365 nm, I0=10 mW/cm2) for three intervals of ten minutes, and the released cells collected and counted after each interval (Figure 2c). GFP-hMSCs are released at a faster rate than RFP-hMSCs, demonstrating the biased release of one cell population over another in a system exposed to a single light program. The ratio of the GFP-hMSC to RFP-hMSC release rates, RGFP/RRFP=2.36±0.68, matches well with the ratio of the apparent degradation rate constants of the two linkers, D and B, kappD/kappB=2.47.
Figure 2.

Wavelength-biased release of encapsulated cells. a) Macromers containing either B (left of interface) or D (right of interface) were used to encapsulate RFG-expressing hMSCs or GFP-expressing hMSCs. b) The interface between the two gels is directly observable in optical microscopy. c) The rate of release of GFP cells from gel containing linker D is faster than the release of RFP-expressing cells from a gel containing linker B: this is consistent with ratio of the apparent rate constants of degradation (kappD/kappB=2.47; RGFP/RRFP=2.36±0.68).
CONCLUSIONS
We have synthesized and characterized a series of macromers incorporating o-NB groups that photolyze over a broad range of rates. Decreasing the number of aryl ethers on the o-NB group increases kapp, and changing the functionality at the benzylic site dramatically increases kapp. Hydrogels formed from these macromers can be used to encapsulate and release hMSCs without compromising cell viability. By exploiting the differences in reactivity of different o-NB linkers, we quantitatively demonstrated the biased release of one cell population over another. Balancing the ratio of the rate constants of degradation, the number of cells encapsulated within different gels, and the light program should allow complex, multistaged delivery of multiple cells types with great precision. This approach has important applications in tissue engineering and regenerative medicine, where the controlled, sequential delivery of different cell populations from a single device may allow regeneration of complex tissues that cannot be repaired using a single cell type.
Supplementary Material
Scheme 3.
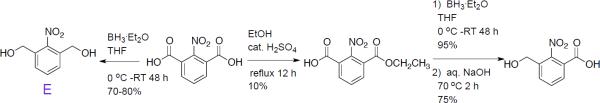
Synthesis of 3-hydroxymethyl-2-nitrobenzoic acid, and 1,3-di(hydroxymethyl)-2-nitrobenzene (E).
ACKNOWLEDGMENT
This work was funded by UCLA HSSEAS Start-up funds and the National Institutes of Health through the NIH Director's New Innovator Award Program, 1-DP2-OD008533. We gratefully acknowledge J. Huynh, Matthew Mattson and Prof. J. A. Kornfield for access to and assistance with photorheology experiments. Some of the materials employed in this work were provided by the Texas A&M Health Science Center College of Medicine Institute for Regenerative Medicine at Scott & White through a grant from NCRR of the NIH, Grant #P40RR017447.
ABBREVIATIONS
- o-NB
ortho-nitro benzyl
- kapp
apparent rate constant
- hMSCs
human mesenchymal stem cells
- PEG
poly(ethylene glycol)
- APS
ammonium persulfate
- TEMED
N,N,N′,N′-tetramethylethylene diamine
- Mn
number average molecular weight
- PCl5
phosphorous pentachloride
Footnotes
Supporting Information. Experimental details for synthesis and characterization of linkers A-E and their macromers, degradation kinetics, cell encapsulation and release, reaction schemes for macromer synthesis, absorbance spectra. This material is available free of charge via the Internet at http://pubs.acs.org
A.K. is a co-inventor on U.S. Patent Application No. 11/374,471 which includes compounds described in this report.
REFERENCES
- (1).Drury JL, Mooney DJ. Biomaterials. 2003;24:4337. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- (2).Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kloxin AM, Tibbitt MW, Kasko AM, Fairbairn JA, Anseth KS. Advanced Materials. 2010;22:61. doi: 10.1002/adma.200900917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wong DY, Griffin DR, Reed J, Kasko AM. Macromolecules. 2010;43:2824. [Google Scholar]
- (5).Kloxin AM, Benton JA, Anseth KS. Biomaterials. 2010;31:1. doi: 10.1016/j.biomaterials.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bryant SJ, Nuttelman CR, Anseth KS. J Biomat Sci-Polym E. 2000;11:439. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- (7).Forman J, Dietrich M, Monroe WT. Photoch Photobio Sci. 2007;6:649. doi: 10.1039/b616979a. [DOI] [PubMed] [Google Scholar]
- (8).Denk W. P Natl Acad Sci USA. 1994;91:6629. doi: 10.1073/pnas.91.14.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hagen V, Benndorf K, Kaupp UB, Pavlos CM, Xu H, Toscano JP, Hess GP, Gillespie DC, Kim G, Kandler K. Control of Cellular Activity. Wiley-VCH Verlag GmbH & Co. KGaA; 2005. [Google Scholar]
- (10).Lendvai B, Szabo SI, Barth AI, Zelles T, Vizi ES. Advanced Drug Delivery Reviews. 2006;58:841. doi: 10.1016/j.addr.2006.07.018. [DOI] [PubMed] [Google Scholar]
- (11).Pettit DL, Wang SSH, Gee KR, Augustine GJ. Neuron. 1997;19:465. doi: 10.1016/s0896-6273(00)80361-x. [DOI] [PubMed] [Google Scholar]
- (12).Wilcox M, Viola RW, Johnson KW, Billington AP, Carpenter BK, McCray JA, Guzikowski Aa. P., Hess GP. J Org Chem. 1990;55:1585. [Google Scholar]
- (13).Zhao H, Sterner ES, Coughlin EB, Theato P. Macromolecules. 2012;45:1723. [Google Scholar]
- (14).Aujard I, Benbrahim C, Gouget M, Ruel O, Baudin JB, Neveu P, Jullien L. Chemistry-a European Journal. 2006;12:6865. doi: 10.1002/chem.200501393. [DOI] [PubMed] [Google Scholar]
- (15).Hasan A, Stengele KP, Giegrich H, Cornwell P, Isham KR, Sachleben RA, Pfleiderer W, Foote RS. Tetrahedron. 1997;53:4247. [Google Scholar]
- (16).Zhao YR, Zheng Q, Dakin K, Xu K, Martinez ML, Li WH. J Am Chem Soc. 2004;126:4653. doi: 10.1021/ja036958m. [DOI] [PubMed] [Google Scholar]
- (17).Stanton-Humphreys MN, Taylor RDT, McDougall C, Hart ML, Brown CTA, Emptage NJ, Conway SJ. Chemical Communications. 2012;48:657. doi: 10.1039/c1cc15135e. [DOI] [PubMed] [Google Scholar]
- (18).Holmes CP, Jones DG. J Org Chem. 1995;60:2318. [Google Scholar]
- (19).Kloxin AM, Tibbitt MW, Anseth KS. Nat. Protocols. 5:1867. doi: 10.1038/nprot.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Price CC. Chemical Reviews. 1941;29:37. [Google Scholar]
- (21).Kanno HH, Chida H, Otani Y, Junsei Chemical Co. Ltd US Patent No 5,847,231. 1998
- (22).Petropoulos CC. J Polym Sci Pol Chem. 1977;15:1637. [Google Scholar]
- (23).Johnson JA, Finn MG, Koberstein JT, Turro NJ. Macromolecules. 2007;40:3589. [Google Scholar]
- (24).San Miguel V, Bochet CG, del Campo A. J Am Chem Soc. 2011;133:5380. doi: 10.1021/ja110572j. [DOI] [PubMed] [Google Scholar]
- (25).Katz JS, Burdick JA. Macromol Biosci. 2010;10:339. doi: 10.1002/mabi.200900297. [DOI] [PubMed] [Google Scholar]
- (26).Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Nature. 1996;380:435. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- (27).Echtermeyer F, Streit M, Wilcox-Adelman S, Saoncella S, Denhez F, Detmar M, Goetinck PF. J. Clin. Invest. 2001;107:R9. doi: 10.1172/JCI10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Carmeliet P. J Intern Med. 2004;255:538. doi: 10.1111/j.1365-2796.2003.01297.x. [DOI] [PubMed] [Google Scholar]
- (29).Richardson TP, Peters MC, Ennett AB, Mooney DJ. Nat Biotech. 2001;19:1029. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- (30).Tengood JE, Ridenour R, Brodsky R, Russell AJ, Little SR. Tissue Eng Pt A. 2011;17:1181. doi: 10.1089/ten.tea.2010.0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Nelson DM, Ma ZW, Leeson CE, Wagner WR. J Biomed Mater Res A. 2012;100A:776. doi: 10.1002/jbm.a.34015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bensaid W, Triffitt JT, Blanchat C, Oudina K, Sedel L, Petite H. Biomaterials. 2003;24:2497. doi: 10.1016/s0142-9612(02)00618-x. [DOI] [PubMed] [Google Scholar]
- (33).Zhang G, Wang XH, Wang ZL, Zhang JY, Suggs L. Tissue Eng. 2006;12:9. doi: 10.1089/ten.2006.12.9. [DOI] [PubMed] [Google Scholar]
- (34).Pasparakis G, Manouras T, Selimis A, Vamvakaki M, Argitis P. Angew Chem Int Edit. 2011;50:4142. doi: 10.1002/anie.201007310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.