

Abstract
Objectives
Recent genome-wide association studies have identified several novel susceptibility genes for systemic lupus erythematosus (SLE) and IgA nephropathy (IgAN). Since both lupus nephritis (LN) and IgAN are autoimmune kidney diseases, they may share common disease mechanisms that overlap with genetic susceptibility. To test this hypothesis, we sought to identify genetic variants associated with IgAN in LN.
Methods
In the first stage, 500 LN patients, 240 SLE patients without LN, and 500 healthy controls were enrolled. Fifteen reported SNPs with top association signals with IgAN were selected for further testing in LN. Three independent cohorts from Beijing, Shanghai and Hong Kong were included as replicates. We also analyzed the functional significance of identified non-coding variants on regulatory motifs and gene expression.
Results
Except for associations with HLA gene polymorphisms, genetic variants of MTMR3 in 22q12 showed associations with LN (rs9983A with P = 2.07×10−3; OR 1.61; 95% CI 1.19–2.19) compared to healthy controls in the first stage. In replications, associations were replicated and reinforced with northern (LN vs. non-LN patients, P = 0.01) but not southern Han Chinese, although significant genetic heterogeneity was observed. In silico analyses predicted conservative and regulatory features of rs9983. In expression analysis, we observed lower MTMR3 transcription levels in blood samples with rs9983A and renal biopsies from LN and IgAN.
Conclusions
Our results suggested that the MTMR3 gene was shared between IgAN and LN in the northern Chinese, further highlighting the role of autophagy in SLE. However, widespread replication of these experiments, fine mapping, and functional assays are required to establish this connection.
Keywords: IgA nephropathy, lupus nephritis, MTMR3, SLE
Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune disease characterized by diverse clinical phenotypes and the presence of multiple antibodies. The disease is variable in presentation and outcome among individuals and across different ancestral populations (1). Despite the variability, a strong genetic component has been suggested, and over 40 loci have been associated with susceptibility to SLE to date (2). These identified SLE susceptibility loci are predominantly common variants that have been confirmed among multiple ancestries, suggesting that there are shared pathogenic mechanisms in disease etiology. However, genetic studies that will enhance our understanding of the links between genotype and specific disease manifestations are still on the horizon. Such studies will surely have a significant impact on our views about disease pathogenesis and targeted therapy.
Lupus nephritis (LN), a major disease manifestation of SLE that has a substantial impact on survival and quality of life, disproportionately affects ethnic minorities and is an independent indicator of poor prognosis. Susceptibility to LN is the end-result of complex interactions between organ-specific reactions and polymorphic genetic factors involved in the regulation of immune responses. However, a clear etiology for this disease is currently obscure. Associated genes that have been identified thus far include C1Q, C4A/B, FCGR2A, TNFSF4, ITGAM, ACP5 and NDASE1L3, but the modest effects of these loci can only account for a small proportion of the heritability of SLE, and many of them were not directly pinpointed by current genome-wide association studies (GWAS) (2). Thus identifying more susceptibility loci will enhance the link between genotype and phenotype in SLE.
As is the case for many other forms of nephritis, organ-specific reactions within the kidney are common causes for end-stage organ damage. Recent genetic studies have largely supported the presence of shared genetics in immune-related diseases. From this point of view, we investigated genetic variants associated with Immunoglobulin A nephropathy (IgAN) in lupus nephritis, as these two diseases are the most common forms of nephritis with an autoimmunity component, and there is well-established evidence of the co-occurrence of SLE and IgAN (3–5).
Subjects and Methods
Study population
In the first stage, a total 1,000 Chinese people of Han ethnicity living in Beijing were enrolled from the renal division of the Peking University First Hospital, including 500 patients with lupus nephritis (31.9 ± 11.2 years; female/male ratio 6:1) and 500 healthy blood donors (40.0 ± 8.6 years) as controls. All of the patients with lupus nephritis met the revised SLE criteria of the American College of Rheumatology (ACR) (6) and were confirmed by renal biopsy using light microscopy, immunofluorescence, and electron microscopy. To further determine whether any observed associations were LN specific, a second cohort containing 240 SLE patients without LN (35.7 ± 13.3 years; female/male ratio 13:1) was enrolled from the dermatological department. All patients fulfilled the criteria of the ACR (6). None of the patients had proteinuria, abnormal urinary sediment, or renal biopsy evidence of nephropathy.
To replicate any genetic associations, three other independent SLE cohorts from Beijing (Peking University People’s Hospital; 378 LN and 316 non-LN patients with SLE) (7, 8), Shanghai (Shanghai Renji Hospital; 313 LN and 321 non-LN patients) (9), and Hong Kong (The University of Hong Kong, 222 LN and 367 non-LN patients) (9) were enrolled. All patients were diagnosed by local rheumatologists and fulfilled the classification criteria of the ACR (6).
All subjects were of Chinese Han ancestry and were separated into northern or southern Han by ancestral home based on self-report. All study subjects provided an informed consent to participate in this genetic study and institutional review boards or local ethics committees approved this study.
SNP selection and genotyping
The IgAN susceptibility loci discovered in genome-wide association studies (GWAS) were primarily from five genomic regions: the MHC region at 6p21, the DEFA locus at 8p23, the TNFSF13 locus at 17p23, the HORMAD2 locus at 22q12 and the CFH/CFHR locus at 1q32 (10, 11). Thus all of the SNPs reported as top association signals in GWASs of IgAN were selected for further testing in LN. In total, 15 SNPs, including 8 SNPs within the MHC loci and seven SNPs outside the MHC region, were genotyped. In the discovery cohort from Beijing, genotyping in LN patients was performed with an Illumina Solexa HiSeq 2000 platform, and selected SNPs checked in the SLE patients without LN were genotyped using TaqMan allele discrimination assays (Applied Biosystems, Foster City, California, USA). For replicates, genotypes were determined using TaqMan allele discrimination assays (Beijing and Shanghai) or were directly retrieved from the original GWAS data (Hong Kong) (9).
Bioinformatic analysis
Variant effects on regulatory motifs were investigated using the HaploReg database (http://www.broadinstitute.org/mammals/haploreg/haploreg.php) and quantified as the difference of LOD (alt) – LOD (ref). A negative result indicated that the predicted relative affinity was higher for the reference sequence, while a positive result indicated that the predicted relative affinity was higher for the alternative. MiRNA target site was predicted by PolymiRTS (Polymorphism in microRNAs and their TargetSites) Database 3.0 (http://compbio.uthsc.edu/miRSNP/). To explore whether the identified SNPs had an expression quantitative trait loci (eQTLs) effect, associations between sequence variation and gene expression were determined. The sequence variation and gene expression profiling data were from the following datasets: lymphoblastoid cell lines from 726 HapMap3 individuals, three tissue types (adipose, LCL and skin) derived from a subset of 160 MuTHER healthy female twins, lymphoblastoid cell lines from the MRCA/MRCE dataset (405 and 550 samples for each platform), and non-transformed peripheral blood samples from the Blood eQTL dataset (5,311 individuals). Differential gene expression was checked in LN (E-GEOD-32592) and IgAN (E-GEOD-37460) and compared with healthy controls from large-scale genome-wide gene expression analyses conducted in renal biopsies (ArrayExpress Archive database; http://www.ebi.ac.uk/arrayexpress/).
Statistical analyses
Only SNPs meeting the quality control criteria of less than 1% overall missing data were included. Testing for deviations from Hardy-Weinberg equilibrium (HWE) was conducted using a χ2 goodness-of-fit test separately for cases and controls in different cohorts. Genotype and allele frequencies were compared between LN cases and controls (healthy controls or non-LN controls) using the χ2 test. Power was calculated using the Power and Sample Size Calculations software (http://biostat.mc.vanderbilt.edu/PowerSampleSize). Principal component analysis was performed with the SNP & Variation Suite v7.7.4 (Golden Helix, Inc., Bozeman, MT; www.goldenhelix.com) using genotype data from an ongoing GWAS on the Immunochip platform (Supplementary Figure 1 and 2). Statistical analyses were performed with SPSS12.0 software (SPSS Inc., Chicago, IL) or the comprehensive meta analysis computer program (Biosta, Englewood, New Jersey, USA). A two-tailed P-value of less than 0.05 was considered statistically significant.
Results
As can be seen from Table 1, of the 15 SNPs reported to be associated with IgAN, 5 (5/15) were observed to be associated with LN in our current study (P < 0.05). The significant signals were from two regions, namely the 6p21 HLA-DRB1-DQA1-DQB1 region (rs660895G with P = 8.97×10−5 and OR 0.61 (95% CI 0.48–0.79); rs2856717T with P = 1.09×10−5 and OR 1.55 (95% CI 1.28–1.89); and rs9275596C with P = 1.22×10−5 and OR 1.58 (95% CI 1.29–1.94)), and the 22q12 HORMAD2-MTMR3-LIF-OSM-GATSL3-SF3A1 region (rs12537T with P = 3.66×10−2 and OR 0.81 (95% CI 0.66–0.98) and rs9983A with P = 2.07×10−3 and OR 1.61 (95% CI 1.19–2.19)). Three other SNPs (3/15; rs9357155, rs2071543 and rs2412971) showing marginal associations (P < 0.1) were also from the 6p21 PSMB8-PSMB9-TAP1-TAP2 region and the 22q12 HORMAD2-MTMR3-LIF-OSM-GATSL3-SF3A1 region. This data indicated that the associations between genetic variants within 6p21, 22q12 and IgAN were also observed in LN (8/15).
Table 1.
Association of Immunoglobulin A nephropathy (IgAN) susceptibility loci with lupus nephritis (LN) in the Beijing discovery cohort.
| SNP | Chr. | Bp | Candidate Gene | Minor Allele | Frequency (Case/Control %) | P | OR (95% CI) in LN | OR in IgAN | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| rs6677604 | 1 | 194953541 | CFH | A | 7.36/7.45 | 0.94 | (11) | ||
| rs2523946 | 6 | 30049922 | HLA-A | C | 50.30/48.59 | 0.45 | (10) | ||
| rs660895 | 6 | 32685358 | HLA-DRB1 | G | 12.60/19.01 | 8.97×10−5 | 0.61 (0.48–0.79) | 1.34 | (10) |
| rs2856717 | 6 | 32778286 | HLA-DQB1 | T | 33.06/24.14 | 1.09×10−5 | 1.55 (1.28–1.89) | 0.73 | (11) |
| rs1794275 | 6 | 32779226 | HLA-DQB1 | T | 14.68/12.90 | 0.25 | (10) | ||
| rs9275596 | 6 | 32789609 | HLA-DQB1 | C | 29.33/20.82 | 1.22×10−5 | 1.58 (1.29–1.94) | 0.63 | (11) |
| rs9357155 | 6 | 32917826 | PSMB8 | A | 23.08/19.62 | 0.06 | 1.23 | 0.71 | (11) |
| rs2071543 | 6 | 32919607 | PSMB8 | A | 24.60/20.93 | 0.05 | 1.23 | 0.73 | (11) |
| rs3129269 | 6 | 33205592 | HLA-DPB2 | T | 25.81/26.16 | 0.86 | (11) | ||
| rs2738058 | 8 | 6810195 | DEFA | G | 33.87/31.79 | 0.32 | (10) | ||
| rs3803800 | 17 | 7403693 | TNFSF13 | A | 30.54/32.60 | 0.33 | (10) | ||
| rs12537 | 22 | 28753460 | MTMR3 | T | 25.10/29.28 | 3.66×10−2 | 0.81 (0.66–0.98) | 0.78 | (10) |
| rs9983 | 22 | 28753744 | MTMR3 | A | 11.49/7.45 | 2.07×10−3 | 1.61 (1.19–2.19) | 1.18 | (11) |
| rs2412971 | 22 | 28824371 | HORMAD2 | A | 36.39/40.24 | 0.08 | 0.85 | 0.75 | (11) |
| rs2412973 | 22 | 28859631 | HORMAD2 | A | 36.79/40.14 | 0.13 | (11) |
Only associations with statistical significance or marginal significance are presented with OR values. Ninety-five percent confidence intervals are shown for significant associations.
Of interest, the non-MHC association between SNP rs9983 and LN was still significant after multiple corrections (corrected P = 3.11×10−2 using the Bonferroni method on 15 SNPs). This significant association was also corroborated by associations at neighboring SNPs. When the associations were further checked in SLE patients without LN, a marginally significant association could be observed in LN patients when compared with non-LN patients (p = 0.06). The minor allele frequency in non-LN patients with SLE was more similar to that of normal controls (8.4% vs. 7.45%; Table 2) when compared with LN patients (11.49%).
Table 2.
Selected associations between SNPs within MTMR3 and SLE patients with or without lupus nephritis (LN) in the Beijing discovery cohort.
| LN vs. Healthy control (500/500)
|
LN vs. Non-LN control (500/240)
|
SLE vs. Healthy Control (740/500)
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Minor Allele | MAF | P | OR (95% CI) | MAF | P | OR (95% CI) | MAF | P | OR (95% CI) |
| rs12537 | T | 25.10/29.28 | 3.66×10−2 | 0.81 (0.66–0.98) | 25.10/25.21 | 0.96 | 0.99 (0.77–1.28) | 25.14/29.28 | 2.30×10−2 | 0.81 (0.68–0.97) |
| rs9983 | A | 11.49/7.45 | 2.07×10−3 | 1.61 (1.19–2.19) | 11.49/8.40 | 0.06 | 1.42 (0.97–2.07) | 10.49/7.45 | 1.05×10−2 | 1.46 (1.10–1.95) |
In our current study, we had powers of 0.89 and 0.96 for the detection of a 1.5-fold increased risk in northern Han and southern Han populations, respectively, assuming an α-level of 0.05. With replications and meta analysis from one additional SLE cohort from the north of China and two SLE cohorts from the south of China, the genetic associations was replicated in the northern Han cohort (combined P = 0.01), but not the southern Han cohorts (Figure 1 and Supplementary Table 1). We observed substantial heterogeneity between the studies (P = 0.02). The distribution of rs9983A in northern and southern Han Chinese populations was similar to data from the Human Genome Diversity Project (HGDP) and GWAS data derived from northern and southern Han Chinese (Supplementary Figure 3) (10, 11). When positive selection for the human genome was checked, rs9983 appeared to exhibit a significant selection signal (iHS >2) in HapMap Asian population using the integrated haplotype score (iHS) test (Supplementary Table 2).
Figure 1. The overall and pooled ORs with a 95% CI for overall and subgroup analysis testing of the association between rs9983 and lupus nephritis.
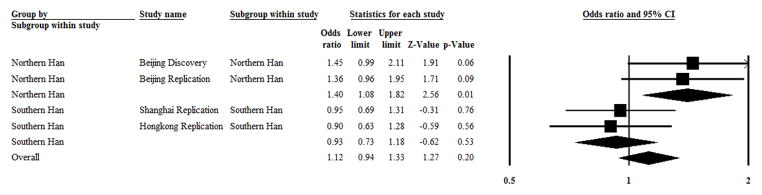
In a subgroup of northern Han Chinese, 878 SLE patients with lupus nephritis, and 556 SLE patients without lupus nephritis as a control were enrolled. I2 = 0 and P = 0.83 in test for heterogeneity, and Z = 2.56 and P = 0.01 in a test for overall effect.
In a subgroup of southern Han Chinese, 535 SLE patients with lupus nephritis and 688 SLE patients without lupus nephritis as a control were enrolled. I2 = 0, P = 0.83 in a test for heterogeneity, and Z = −0.62 and P = 0.53 in a test for overall effect.
Of the total Chinese Han individuals enrolled, 1413 were SLE patients with lupus nephritis and 1244 were SLE patients without lupus nephritis as a control. I2 = 44.7% and P = 0.02 in a test for heterogeneity, and Z = 0.62 and P = 0.54 in a test for overall effect.
A random effects model was used to pool the results because heterogeneity was present.
Notably, risk alleles for LN observed in the MHC region (OR values >1) were protective for IgAN (OR values <1), which had consistent tendencies along the entire region and were concordant with previous reports that most IgAN protective alleles were risk alleles for many autoimmune diseases. In contrast, alleles within 22q12 conferred similar risks in both IgAN and LN.
Case-only phenotype analysis of LN patients revealed no association between rs9983 alleles and other sub-phenotypes of SLE, the presence of auto-antibodies, the systemic lupus erythematosus disease activity index (SLEDAI), renal functions, and the activity index (AI) or the chronic index (CI), according to Hill’s Biopsy Index scoring criteria (2000) for lupus nephritis (data not shown).
The HaploReg database predicted a binding site motif that spans the 3′-UTR of the MTMR3 rs9983 region for binding by the transcription factors Zfp691 (zinc finger protein 691), Gfi1b (growth factor independent 1B transcription repressor), HNF6 (hepatocyte nuclear factor 6), and VDR (vitamin D 1,25-dihydroxyvitamin D3 receptor). The differences between the LOD scores for the A G alleles (reference) were +0.9 (positive), +11.9 (positive), +12 and −1.4 for Zfp691, Gfi1b, HNF6, and VDR, respectively. Therefore, this model predicted higher affinity to regulatory motifs for Zfp691, Gfi1b and HNF6 and lower for VDR for the variant A (risk) allele relative to the G allele. In addition, rs9983 was predicted to be conserved in mammals by both the GERP and SiPhy-omega programs. By further locating the set of SNPs that were in strong linkage disequilibrium (r2≥0.8) with rs9983 in Asian population among the 34 SNPs, two variants were found to be conservative and 10 variants were found to lie within regions of enhancer histone marks, 11 in regions of DNase-I hypersensitivity, one in a region of protein binding, one in a region of eQTL, and 34 in regulatory motif regions from more than one cell type, suggesting that rs9983 was located in a regulatory region (Supplementary Table 3). PolymiRTS Database 3.0 also predicted rs9983 to be putative target site for miRNA (Supplementary Table 4).
In addition, analysis of MTMR3 expression levels in various different gene expression profile databases has indicated that the transcription of the A allele of MTMR3 decreased compared to the G allele (P < 10−8) (Supplementary Table 5). With an increase in sample size, the association fit was reinforced. This was particularly true for the Blood eQTL dataset, which contained data from 5,311 individuals. Among the many e-SNPs for MTMR3 and the variants in high linkage disequilibrium (LD, r2>0.8), rs9983 showed the most significance in genotype-expression correlation (Supplementary Figure 4). Further, from genome-wide gene expression analysis from kidney biopsy, we observed that MTMR3 mRNA expression was down-regulated in glomeruli both for LN (5.92 ± 0.15 vs. 6.18 ± 0.17; p = 5.94×10−6; 32 LN patients vs. 14 controls) and IgAN (6.42 ± 0.14 vs. 6.74 ± 0.34; p = 9.64×10−5; 27 IgAN patients vs. 27 controls).
Discussion
Shared genetic studies are important for the validation of associations between common genetic variation and complex diseases, identification of disease common/specific pathways, and optimization of future therapies. In the present study, we tested genetic variants associated with IgA nephropathy in lupus nephritis and observed for the first time that variants of MTMR3 were associated with both of these common autoimmune kidney diseases. We observed significant associations between rs9983A and a risk of lupus nephritis in northern Han rather than southern Han populations. This may be because of genetic heterogeneity, as evidenced by differing allele frequencies and signals of selection (iHS >2) in Asian populations. The lack of association between another variant of MTMR3, rs12537, and lupus nephritis in replication studies may be because of its poor capacity for the tagging of rs9983 (the r2 between rs9983 and rs12537 was 0). Associations between rs9983 and lupus nephritis could be replicated in another independent northern Han cohort. As both MTMR3 and its variant rs9983 showed various possible functions, the current data provided important clues for understanding the pathogenesis of lupus nephritis. However, due to the relatively low frequency of rs9983A, further widespread replications of these studies in other Asian populations are still needed. Fine mapping of MTMR3 among different populations with different LD blocks and systematic detection of pleiotropic effects are also needed.
MTMR3 is an autophagy-related gene involved in autophagy initiation (12), and recent genetic studies have suggested that several autophagy genes, including ATG5, ATG7, IRGM, DRAM1, CDKN1B and APOL1, are susceptibility genes for SLE (7, 13–15). Therefore, autophagy may play an important role in the pathogenesis of SLE, a fact that broadens the current list of autophagy genes involved in the etiology of human diseases.
A significant difference was detected with the risk allele (the A allele) of SNP rs9983 located in the 3′-UTR of MTMR3, the presence of which was correlated with a lower expression level for that gene. This was consistent with the observation that deregulation of autophagy was common in lupus-prone mouse models and SLE patients (16). Notably, the differences in in silico analysis were very small. In eQTL analysis of tens of samples from both MuTHER and HapMap populations, no significant association was observed between various genotypes of rs9983 and MTMR3 expression. However, when hundreds of samples were included, the associations became evident and could be replicated in multiple datasets with genome-wide significance. The HaploReg database predicted a much higher affinity between the factor-binding site of the rs9983A risk allele and the transcription motifs of Zfp691, HNF6, and Gfi1b (also identified as a potential regulator of CDKN1A translocation in chronic myeloid leukemia). The higher affinity between the A allele and the transcription factor motifs could be the cause of the lower transcription levels found in in silico analysis. Because the 3′UTR plays a crucial role in gene expression by influencing mRNA stability, localization, export, and translation efficiency (inhibiting mRNA expression in most cases) (17), a change in the binding capacity between miRNA and a complementary target site, or a change in the affinity between these transcription factor motifs and the 3′-UTR may play a role in lowering MTMR3 gene expression. Of special note, the LOD scores that changed the most in affinity analysis were for HNF6 and Gfi1b. HNF6 is a transcription factor that influences a variety of cellular processes, including glucose metabolism and cell cycle regulation. CDKN1A plays a critical role in the cellular response to DNA damage, and its over-expression results in cell cycle arrest. Also, CDKN1B has been associated with SLE in recent meta analysis of GWASs in Asian populations (13). In addition, in accordance with eQTL data, lower MTMR3 levels were observed in both LN and IgAN patients in genome-wide gene expression analyses conducted in renal biopsies. These bioinformatics analyses may shed more light on the role MTMR3 plays in the pathogenesis in LN and IgAN.
In summary, the present study established a relationship between IgAN genetic variants and lupus nephritis in a northern Han population from China. The susceptibility gene MTMR3 identified in this study may suggest a role for autophagy in both SLE and IgAN, broadening our understanding of autophagy in autoimmune kidney diseases and the genetics of autophagy in SLE.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 81200524, Z.X.J.), The Foundation of Ministry of Education of China (20120001120008, Z.X.J.), the Major State Basic Research Development Program of China (973 program, No. 2012CB517700 Z.H.), the Research Fund of Beijing Municipal Science and Technology for the Outstanding PhD Program (20121000110, Z.H.), and the Natural Science Fund of China to the Innovation Research Group (81021004, Z.M.H. and Z.H.). This work also was supported by grants from the NIH (AI103399, and AR060366, S. K. N.), Major State Basic Research Development Program of China (973 program, No. 2014CB541902, S.N. and H.X.F.), China National Funds for Distinguished Young Scientists (81025016, S.N.), the National Natural Science Foundation of China (81230072, S.N.), Chinese Ministry of Health (201202008, S.N.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Conflict of interest statement
The authors have declared no conflict of interest.
References
- 1.Feldman CH, Hiraki LT, Liu J, Fischer MA, Solomon DH, Alarcon GS, et al. Epidemiology and sociodemographics of systemic lupus erythematosus and lupus nephritis among US adults with Medicaid coverage, 2000–2004. Arthritis Rheum. 2013;65(3):753–63. doi: 10.1002/art.37795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rullo OJ, Tsao BP. Recent insights into the genetic basis of systemic lupus erythematosus. Ann Rheum Dis. 2013;72 (Suppl 2):i56–61. doi: 10.1136/annrheumdis-2012-202351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corrado A, Quarta L, Di Palma AM, Gesualdo L, Cantatore FP. IgA nephropathy in systemic lupus erythematosus. Clin Exp Rheumatol 2007. 2007 May 01;25(3):467–9. [PubMed] [Google Scholar]
- 4.Horino T, Takao T, Terada Y. IgA nephropathy in a patient with systemic lupus erythematosus. Lupus. 2010;19(5):650–4. doi: 10.1177/0961203309349384. [DOI] [PubMed] [Google Scholar]
- 5.Zhou XJ, Cheng FJ, Zhu L, Lv JC, Qi YY, Hou P, et al. Association of systemic lupus erythematosus susceptibility genes with IgA nephropathy in a Chinese cohort. Clin J Am Soc Nephrol. 2014;9(4):788–97. doi: 10.2215/CJN.01860213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25(11):1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 7.Zhou XJ, Lu XL, Lv JC, Yang HZ, Qin LX, Zhao MH, et al. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis. 2011;70(7):1330–7. doi: 10.1136/ard.2010.140111. [DOI] [PubMed] [Google Scholar]
- 8.Zhou XJ, Lu XL, Nath SK, Lv JC, Zhu SN, Yang HZ, et al. Gene-gene interaction of BLK, TNFSF4, TRAF1, TNFAIP3, and REL in systemic lupus erythematosus. Arthritis Rheum. 2012;64(1):222–31. doi: 10.1002/art.33318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang W, Shen N, Ye DQ, Liu Q, Zhang Y, Qian XX, et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010;6(2):e1000841. doi: 10.1371/journal.pgen.1000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu XQ, Li M, Zhang H, Low HQ, Wei X, Wang JQ, et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat Genet. 2012;44(2):178–82. doi: 10.1038/ng.1047. [DOI] [PubMed] [Google Scholar]
- 11.Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43(4):321–7. doi: 10.1038/ng.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taguchi-Atarashi N, Hamasaki M, Matsunaga K, Omori H, Ktistakis NT, Yoshimori T, et al. Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy. Traffic. 2010;11(4):468–78. doi: 10.1111/j.1600-0854.2010.01034.x. [DOI] [PubMed] [Google Scholar]
- 13.Yang W, Tang H, Zhang Y, Tang X, Zhang J, Sun L, et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. Am J Hum Genet. 2013;92(1):41–51. doi: 10.1016/j.ajhg.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou XJ, Cheng FJ, Zhang H. Emerging View of Autophagy in Systemic Lupus Erythematosus. Int Rev Immunol. 2014 doi: 10.3109/08830185.2013.879711. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 15.Freedman BI, Langefeld CD, Andringa KK, Croker JA, Williams AH, Garner NE, et al. End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol. 2014;66(2):390–6. doi: 10.1002/art.38220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gros F, Arnold J, Page N, Decossas M, Korganow AS, Martin T, et al. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy. 2012;8(7):1113–23. doi: 10.4161/auto.20275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chatterjee S, Pal JK. Role of 5′- and 3′-untranslated regions of mRNAs in human diseases. Biol Cell. 2009;101(5):251–62. doi: 10.1042/BC20080104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.