

Abstract
Objective
Obesity-related glomerulopathy is characterized initially by glomerular hyperfiltration with hypertrophy and then development of proteinuria. Putative mechanisms include endothelial dysfunction and filtration barrier injury due to oxidant stress and immune activation. There has been recent interest in targeting dipeptidyl peptidase 4 (DPP4) enzyme due to increasing role in non-enzymatic cellular processes.
Methods
We utilized the Zucker obese (ZO) rat, (aged 8 weeks) fed a normal chow or diet containing the DPP4 inhibitor linagliptin for 8 weeks (83mg/kg rat chow).
Results
Compared to lean controls, there were increases in plasma DPP4 activity along with proteinuria in ZO rats. ZO rats further displayed increases in glomerular size and podocyte foot process effacement. These findings occurred in parallel with decreased endothelial stromal-derived factor-1α (SDF-1α), increased oxidant markers, and tyrosine phosphorylation of nephrin and serine phosphorylation of the mammalian target of rapamycin (mTOR). DPP4 inhibition improved proteinuria along with filtration barrier remodeling, circulating and kidney tissue DPP4 activity, increased active glucagon like peptide–1 (GLP-1) as well as SDF-1α, and improved oxidant markers and the podocyte-specific protein nephrin.
Conclusions
These data support a role for DPP4 in glomerular filtration function and targeting DPP4 with inhibition improves oxidant stress-related glomerulopathy and associated proteinuria.
Keywords: SDF-1α, linagliptin, NADPH oxidase, DPP4 activity, obesity
Introduction
The rates of overweight and obesity have increased over the past 3 decades (1, 2) and coincide with a growing incidence in chronic kidney disease (CKD) (3). In this regard, obesity has emerged as an independent risk factor for CKD [4] that is characterized initially by glomerular hyperfiltration with subsequent development of hypertrophy and proteinuria (5, 6). The putative mechanisms for obesity-related hypertrophy and proteinuria include activation of growth pathways and endothelial dysfunction/injury to the filtration barrier. The filtration barrier is composed of three layers; the endothelium, a basement membrane, and visceral epithelial podocytes. Damage to the filtration barrier such as that can occur with oxidant stress and dysfunctional immune activation leads to loss of protein (e.g. proteinuria). However, the mechanisms underlying initiation and progression of obesity-related kidney disease are not well understood.
Recently, there has been increasing interest in the dipeptidyl peptidase 4 (DPP4) enzyme as a novel target to improve kidney injury (7, 8). DPP4 is an exopeptidase expressed at high levels in glomerular epithelial and endothelial cells, kidney proximal tubule cells (PTCs), and immune cells such as T-cells (a.k.a. CD26) (9, 10). The DPP4 enzyme degrades substrates related to signal transduction, such as glucagon-like peptide (GLP)-1 and immune function (9, 10). Under conditions of obesity and insulin resistance, circulating and tissue DPP4 activity, including that in the kidney, may be increased independent to that of the contribution of diabetes (11–14). Recent work suggests that DPP4 inhibition is protective against ischemic-reperfusion injury in the kidney (15) and reduces inflammation as well as oxidant stress in the kidney, heart and in the endothelium of diabetic animal models (16–18). Moreover, in another model of diabetic nephropathy such as the Zucker diabetic fatty (ZDF) rat, DPP4 inhibition improved selective markers of inflammation and oxidant stress in addition to improvements in glomerulosclerosis and albuminuria (17). However, little is known regarding the role of DPP4 inhibition on obesity-related kidney disease in non-diabetic conditions.
In this regard, the Zucker obese (ZO) rat has been widely used as a model of obesity-related kidney disease or glomerulopathy with progressive increases in glomerular size and proteinuria (19, 20). In this model, proteinuria is initially due to underlying endothelial dysfunction and injury to the glomerular filtration barrier with loss of endothelial fenestrae and podocyte foot process effacement (19, 20). Filtration barrier injury is thought to be attributable to inflammation and oxidative stress and alteration in growth pathways such as the mammalian target of rapamycin (mTOR) (21). In contrast to diabetic kidney disease, obesity-related kidney disease does not manifest severe diabetic changes such as GBM thickening. To address the impact of DPP4 inhibition on kidney tissue injury in an obesity model we used linagliptin, a DPP4 inhibitor that has greater selectivity than other peptidase inhibitors (22), and it has an in vitro IC50 of approximately 1 nM indicating a high level of potency as a DPP4 inhibitor (23). Due to the tissue specific effects of linagliptin, we hypothesized that treatment with linagliptin over two months would attenuate development of glomerulopathy and filtration barrier injury in the ZO rat through improvements in oxidative stress.
Methods
Methods details for previously described procedures are presented in the supplemental file.
Experimental parameters
Male ZO and age-matched Zucker Lean (ZL) rats were purchased from Charles River, Inc and housed in a 12 hour light/dark altered room. Animals were cared for in accordance with National Institutes of Health guidelines. All procedures were approved and performed in accordance with the Institutional Animal Care and Use Committee of the University of Missouri. Linagliptin (BI 1356; (R)-8-(3-aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin2-ylmethyl)-3,7-dihydro-purine-2,6-dione) was administered orally by mixing drug with rat chow (24). The final concentration of linagliptin in chow was 83 mg LGT•kg−1 a concentration chosen to achieve a dose and plasma level of approximately 4 mg•kg−1•day−1 and 50–100 nM, respectively (24). Rats were divided into four groups to include ZL control (ZL-C), ZL treated with linagliptin (ZL-L), ZO control (ZO-C) and ZO treated with linagliptin (ZO-L). Rats were weighed immediately prior to the start of the experiment (8 weeks of age) and every week thereafter until the end of the experiment (16 weeks of age) to monitor weight gain.
Proteinuria measurements
Both creatinine and protein concentrations in urine were analyzed on an automated clinical chemistry analyzer (AU680; Olympus America Inc., Centerville, PA) using commercially available assays (25).
Protein expression semi-quantitation
Preparation of Whole Tissue Extracts
30–50 mg of kidney tissue was homogenized in ice-cold buffer containing 1% triton X-100, 100 mM NaCl, 20 mM Tris pH 7.5, 2.0 mM EDTA, 10 mM MgCl2, 10 mM NaF, 40 mM β-glycerol phosphate, 1 mM PMSF, 2 mM sodium orthovanadate, protease inhibitor cocktail tablet (Roche Diagnostics, Indianapolis IN), 10 μg/ml leupeptin, 7 μg/ml pepstatin. Homogenates were treated with 1% SDS (Triton-X insoluble fraction) and boiled and centrifuged to collect the supernatant. Protein concentrations were determined using a BCA protein quantitation kit (Thermo Scientific, Rockford, IL).
Immunoblotting
Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes and blocked for 1 hr at RT. Primary antibodies used in the study were DPP4 (ProteinTech, Chicago IL), p-Tyr1217-nephrin (Abcam, Cambridge MA), nephrin (kind gift from Puneet Garg, Ann Arbor MI), p-Ser2448-mTOR, p-Ser2481-mTOR, and mTOR, (Cell Signaling, Danvers MA). Antibody binding was detected by chemiluminescence and images recorded using a Bio-Rad ChemiDoc XRS image analysis system (Bio-Rad Laboratories, Santa Cruz, CA). Protein band density quantitation was performed using Image Lab software (Bio-Rad Laboratories).
Immunohistochemistry
Kidney cortical tissue was harvested and prepared as previously described (25). Primary antibodies used were p-Ser2448-mTOR (Ab109268) and mTOR (Ab2732), both from Abcam (Cambridge MA), SDF-1α (Novus Biologicals, Littleton CO). Briefly, 4-μm sections were incubated with primary antibodies overnight at room temperature. Then slides were washed and incubated with 1:300 secondary antibodies for 4 hrs and viewed with a biphoton confocal laser-scanning microscope and signal intensities quantified by MetaVue as average gray-scale intensities.
DPP4 activity and active and total GLP-1 assay
As previously described (24), blood was collected in EDTA tubes and plasma was stored at −80C. Kidney whole tissue extracts were prepared in sucrose buffer as described before (19, 25). Sucrose buffer and not SDS containing buffer was used to prevent enzyme dissociation/degradation. Further details on DPP4 activity are provided in the supplemental file. GLP-1 active and total in plasma was performed by Mesoscale discovery metabolite assay as described earlier (26).
Polyclonal antibody generation versus intact SDF-1α
Antigens were generated by conjugating terminus specific peptides (KPVSLHHHC) to keyhole limpet hemocyanin (KLH, Pierce, Rockford, IL, USA). Rabbits were immunized at Pineda Antibody Service, Berlin, Germany and polyclonal antibodies were purified using peptide-affinity chromatography.
Total SDF-1α assay
Luminex assay was performed with the matched antibody pair, anti-human/mouse SDF1α mab (clone 79018) from R&D (MAB350) and as a detector goat anti-human/mouse SDF1α (BAF310 from R&D). Samples were then run on Luminex100.
Measurement of NADPH oxidase activity
NADPH oxidase activity was determined in plasma membrane fractions as described before (19, 25) and details are in the supplemental file.
Reactive oxygen species (ROS) formation
Accumulation of ROS in whole kidney tissue was measured by chemiluminescense assay as described before (19) and details are in the supplemental file.
3-Nitrotyrosine
3-nitrotyrosine staining of kidney sections was done as described previously (25) and details are in the supplemental file.
Quantification of Glomeruli Size
Five μm sections of paraffin embedded cortical tissue from different treatments groups were stained with Verhoeff van Gieson (VVG) stain. Briefly, images from all cross sections of cortical tissue (minimum 10 glomeruli per animal cut at the vascular pole) were randomly captured, and the glomerular tuft area was quantified with MetaVue. Samples from five rats from each of the four treatment groups were analyzed.
Ultrastructure analysis with transmission electron microscopy
Details of kidney tissue preparation, sectioning, staining and viewing are as previously described (19, 25). A JOEL 1200-EX transmission electron microscope (TEM) was utilized to review three fields randomly chosen per rat to obtain three 60,000 X images/LV.
Statistical Analysis
Results are reported as the mean ± SE. Two way ANOVA and post hoc t-tests (Holm-Sidak) were performed to examine differences in outcomes between control and linagliptin treated ZL and ZO groups. A p value italic>0.05 was considered significant.
Results
DPP4 inhibition suppresses DPP4 activity and increases SDF-1α in the kidney, as well as increases active SDF-1α and active GLP-1 in the plasma
Very recently, we established that linagliptin (83 mg/kg chow) suppressed DPP4 activity in the plasma by >80% in ZO rats (24). At this dose, we demonstrated improvements in cardiac diastolic function and blood pressure despite minimal changes in body weight or insulin sensitivity. In the current study, kidney cortical tissue DPP4 activity was suppressed by 50% in ZL rats and ~75% in ZO rats although there was no significant increase in the ZO when compared to the ZL rats (Fig 1A). To then determine whether the reduction in renal DPP4 activity was related to protein expression, we examined kidney protein extracts and found no significant change in DPP4 expression with linagliptin treatment (Fig 1B).
Figure 1. DPP4 inhibition in the Zucker obese (ZO) rat improves.

A) DPP4 kidney tissue activity but not B) expression as determined by western blot in the Zucker obese (ZO) rat. DPP4 inhibition also increases substrates C) circulating active GLP1 and D) SDF1-α determined by immunostaining with corresponding measures of intensity on the right. E) Representative images of SDF-1α immunostaining with corresponding measures of intensity to the right. F) Circulating levels of serum SDF-1α. Data are represented by means ± sterr. *, p< 0.05, when ZO controls (ZO-C) are compared to Zucker lean controls (ZL-C); †, p< 0.05 when linagliptin treated Zucker obese rats (ZO-L) are compared to ZO-C. For Western blot, equal loading was ensured via quantitation of Ponceau S.
In plasma, an important substrate of DPP4, GLP-1, trended towards an increase in the ZO rat as determined by both active and total GLP-1 levels (Fig 1C & 1D). However, linagliptin treated ZO and ZL rats had a ~6-fold increase in active GLP-1 levels when compared to untreated controls (Fig 1C). DPP4 is an exopeptidase and degrades other substrates with Xaa-Ala/Pro at the N-terminus and therefore has the potential to mediate pleiotropic effects independent of GLP-1. In this regard, another known substrate, SDF-1α, is a chemokine responsible for recruitment of endothelial progenitor cells and endothelial function (27). The expression of SDF-1α was localized to the glomerulus as well as the distal tubule on immunostaining (Fig 1E). There were decreases in glomerular intensities of SDF-1 α in the ZO compared to ZL controls, a difference which was abolished with DPP4 inhibition (Fig 1E). Similar to GLP-1, linagliptin increased plasma SDF-1α (Fig 1F).
DPP4 inhibition improves obesity induced glomerular filtration barrier injury in the ZO rat
DPP4 is expressed in glomerular endothelial and epithelial cells in the kidney (10). The endothelium comprises a part of the glomerular filtration barrier and injury over time can lead to proteinuria. Similar to previous work in the ZO (5, 17, 19), there were significant increases in proteinuria in the ZO compared to ZL rats (pbold>0.05) and proteinuria was ameliorated with linagliptin (Fig 2A). On ultrastructural analysis utilizing TEM, ZO rats exhibited loss of endothelial fenestrae along with podocyte effacement and loss of slit pore diaphragm which were restored by linagliptin treatment (Fig 2B). To determine the underlying proteins involved in the integrity of the glomerular filtration barrier, we examined expression of the podocyte-specific protein nephrin. Phosphorylation of nephrin is indicative of podocyte effacement in certain conditions such as passive Heymann nephritis or 27A antibody injection (28). In this regard, the increases in proteinuria and ultrastructural changes to the filtration barrier in ZO rats were accompanied by increases in Tyr1217 phosphorylation of nephrin, and this was attenuated with linagliptin treatment (Fig 2C).
Figure 2. DPP4 inhibition improves filtration barrier injury in the Zucker obese (ZO) rat.
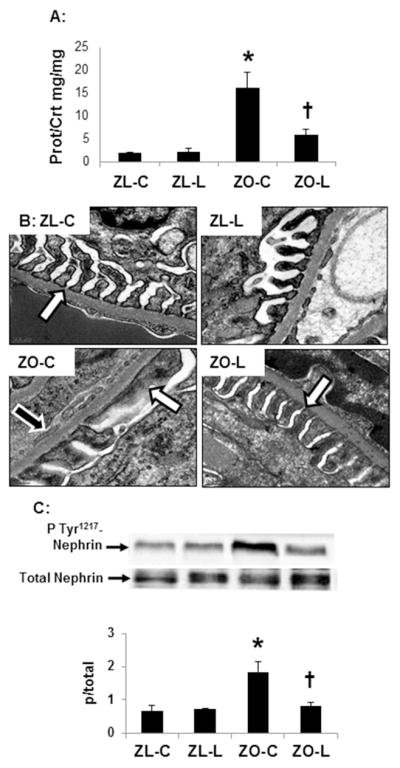
A) Measures of proteinuria. B) Ultrastructural analysis of the glomerular filtration barrier utilizing transmission electron microscopy. Images depict podocyte foot process effacement in the ZO rat with loss of the slit-pore membrane (white arrows) and endothelial fenestrae (black arrows) compared to ZL controls and linagliptin treated ZO rats. C) Western blot analysis of the ratio of phosphorylated (pTyr1217) to total of the podocyte-specific protein nephrin with quantitative analysis below. Data are represented by means ± sterr. *, p< 0.05, when ZO controls (ZO-C) are compared to zucker lean controls (ZL-C); †, p< 0.05 when linagliptin treated zucker obese rats (ZO-L) are compared to ZO-C. For Western blot, equal loading was ensured via quantitation of Ponceau S.
DPP4 inhibition attenuates obesity-related glomerulomegaly in the ZO rat
In parallel with our proteinuria findings, there were increases in glomerular size in ZO compared to ZL rats that were improved with linagliptin in ZO rats (Fig 3A). Along with notable glomerulomegaly in ZO controls on light microscopy there was marked podocyte hypertrophy on ultrastructural TEM analysis (Fig 3B). Compared to ZL control rats, the ZO controls demonstrated marked hypertrophy and increase of variable electron dense intracytoplasmic proteinaceous and glycogen inclusions in the peripherally located podocytes. These findings were attenuated in the linagliptin treated ZO rats (Fig 3B). Cell growth and hypertrophy is dependent on serine (Ser)/threonine (Thr) kinases that regulate cell growth and protein translation such as the mammalian target of rapamycin (mTOR) pathway. In parallel with our light and ultrastructural observations in the ZO rat, there were increases in phosphorylation of mTOR in the kidney cortex (p-Ser2448-mTOR and p-Ser2481-mTOR; Fig 3C, immunoblots and immunohistochemistry). Moreover, consistent with our structural observations there were significant reductions in glomerular mTOR (p)/activation with linagliptin treatment (Fig 3C lower panel).
Figure 3. DPP4 inhibition improves obesity-related glomerulomegaly in the Zucker obese (ZO) rat.

A) Representative light micrographs of Verhoeff-van gieson stained kidney cortical sections, which stains collagen pink, with corresponding measures of glomerular area to the right. B) Ultrastructural analysis using transmission electron microscopy demonstrates marked epithelial podocyte cell hypertrophy and increase of variable electron dense intracytoplasmic proteinaceous and glycogen inclusions (arrow) in the body of podocytes in the ZO control (ZO-C). These findings were absent in linagliptin treated ZO rats (ZO-L) that were similar to Zucker lean controls (ZL-C). Magnification x800; bar = 2 μm. C) Western blot analysis of mTOR growth kinases with quantitative analysis to the right. D) Representative images of Ser2448 phosphorylated mTOR immunostaining with corresponding measures to the right. Data are represented by means ± sterr. *, p< 0.05, when ZO controls (ZO-C) are compared to Zucker lean controls (ZL-C); †, p< 0.05 when linagliptin treated Zucker obese rats (ZO-L) are compared to ZO-C. For Western blot, equal loading was ensured via quantitation of Ponceau S.
DPP4 inhibition improves oxidant stress
A major source of ROS in the kidney is derived from the activation of NADPH oxidase enzyme. We and others have reported increased NADPH oxidase activity and expression of NADPH oxidase subunits the ZO kidney (19). In the current study, there were increases in NADPH oxidase activation (Fig 4A) as well as ROS production (Fig 4B) in ZO when compared to ZL rats; findings that were improved with linagliptin treatment in the ZO. We noted a paradoxical increase in NADPH oxidase activity in linagliptin treated ZL rats; however, this did not contribute to ROS accumulation. 3-nitrotyrosine (3-NT) is a marker for peroxynitrite formation and largely thought to be a marker for NO-dependent nitrative stress. Similar to our NADPH oxidase and ROS observations, renal 3-NT was also increased in the ZO compared to ZL and this was attenuated with linagliptin (Fig 4C).
Figure 4. DPP4 inhibition improves oxidant stress in the Zucker obese (ZO) rat.

A) NADPH oxidase enzyme activity. B) Chemiluminescence determination of total ROS. C) Representative images of 3-nitrotyrosine immunostaining as a marker for nitroso-stress with corresponding measures of intensity to the right. Data are represented by means ± sterr. *, p< 0.05, when ZO controls (ZO-C) are compared to Zucker lean controls (ZL-C); †, p< 0.05 when linagliptin treated Zucker obese rats (ZO-L) are compared to ZO-C.
Discussion
The current observation that DPP4 inhibition improved obesity-related glomerulopathy, including glomerular filtration barrier injury and proteinuria in the ZO rat, suggest that targeting DPP4 may have a beneficial effect on the initial stages of obesity-related kidney disease. In this regard, obesity-related kidney disease parallels the initial stages of diabetic kidney disease and our finding that the ZO rat developed glomerular hypertrophy, loss of filtration barrier integrity with loss of endothelial fenestrae and segmental podocyte effacement, and proteinuria support what has previously been observed in the ZO as well as the Zucker diabetic rat (5, 17, 19, 20). In contrast to diabetic kidney disease, obesity-related kidney disease may have less severe glomerular injury with more subtle kidney lesions such as focal and segmental podocyte effacement. Although reduction in blood pressure may be partially responsible for the observed reductions in glomerular injury, there is evidence in this study for non-hemodynamic benefits of DPP4 inhibition (14). Indeed, worsening of glomerular structural integrity occurred contemporaneously with reductions in SDF-1α, increases in the redox-sensitive growth kinase mTOR, and increases in NADPH oxidase, ROS production and 3-NT content suggest the glomerular filtration barrier is particularly susceptible to redox homeostasis. The observation that DPP4 inhibition normalized SDF-1α, the podocyte-specific nephrin, and markers of oxidant stress in the ZO kidney support a unique role for DPP4 inhibition on glomerular integrity in obesity-related kidney disease.
DPP4 expression is localized to the podocytes, endothelial cells and the proximal tubule brush border in the kidney (10, 14). Similarly, GLP-1 receptors are expressed on podocytes and endothelial cells (29, 30). The observation that DPP4 inhibition reduced circulating active GLP-1 level in the ZO rat along with marked inhibition of DPP4 enzyme activity in kidney tissue lysates support a role for DPP4 actions in the kidney and on glomerular function. Although DPP4-dependent effects do contribute to glycemic control, DPP4 is known to have a number of substrates not related to GLP-1 regulation that may account for the beneficial effect on the filtration barrier (30, 31). In this regard, recent data suggest that the DPP4 enzyme regulates other substrates such as neuropeptide Y and SDF-1α (30–32). SDF-1α is constitutively expressed in stromal cells, dendritic cells as well as endothelial cells and is responsible for the recruitment of endothelial progenitor cells that differentiate into mature endothelial cells and promote vascular repair and improve endothelial function (31, 32). Accordingly, the finding that DPP4 inhibition increased glomerular expression of SDF-1α suggests a role for DPP4 in regulating SDF-1α in the glomerular endothelium, beyond its actions on GLP-1. In addition, we observed an increase in total SDF-1α levels in the serum of both ZL and ZO rats treated with linagliptin suggesting that DPP4 inhibition increases systemic as well as kidney active SDF-1α levels.
The glomerular filtration barrier is composed of endothelium, basement membrane and podocytes and injury results in loss of endothelial fenestrae and podocyte foot process effacement. The mechanisms proposed for loss of endothelial fenestrae include sFlt1 overexpression or decreased VEGF expression. Podocytes cover the outside of the basement membrane and form interdigitating foot processes that serve as a final barrier against urinary protein loss. Nephrin is an integral membrane protein in the podocyte that interacts with other structural proteins such as podocin and CD2AP in forming the slit pore diaphragm. Modulation of the slit pore diaphragm underlies podocyte effacement which in turn precipitates proteinuria. Podocyte effacement can result from loss of nephrin as occurs in congenital nephrotic syndrome of the Finnish type or decrease in phosphorylation of nephrin seen in minimal change disease (28). However, nephrin phosphorylation is increased in passive Heymann nephritis or 27A antibody injection models of podocyte effacement (28). Hyperphosphorylation of nephrin could disrupt nephrin-podocin interaction or it could lead to unstable nephrin protein and loss from the slit pore diaphragm resulting in podocyte effacement. In addition, nephrin is critical to signaling and lamellipodial modulation via tyrosine phosphorylation at the COOH terminus and hyperphosphorylation may be a dynamic feature in the adaptation to effacement. In this regard, there have been limited data on the function of DPP4 on the podocyte, yet one study suggests DPP4 is present in both podocytes and the endothelium (10).
Our ultrastructural observations of foot process effacement were also accompanied by podocyte hypertrophy as well as mesangial cell expansion, findings typically associated with obesity (5, 6). There has been much interest in growth kinases in the kidney in recent years and specifically the mTOR (21, 33). mTOR is an evolutionarily conserved nutrient sensing kinase that is highly regulated by insulin, carbohydrates and other nutrients (33) that is recently shown to induce mesangial expansion (34). In this regard, mTOR-dependent mesangial expansion occurs through effects on protein transcription and translation through the repressor protein 4E-binding protein (4EBP) and the 70-kD ribosomal S6 kinases (p70S6K) (33–35). The observation that increased phosphorylation of mTOR in the ZO renal cortical tissue was accompanied by glomerular expansion and podocyte hypertrophy, support this notion. Moreover, DPP4 inhibition decreased phosphorylation of mTOR in the glomeruli suggesting that DPP4 may regulate mTOR signaling in the kidney in cell types other than mesangial cells. In this respect, the reduction in glomerular size is likely due to the effects of DPP4 inhibition on mTOR/S6K signaling related growth.
There is accumulating data suggesting that mTOR is redox-sensitive and the pathways that govern hypertrophy and hyperfiltration in obesity-related glomerulopathy are susceptible to oxidant injury (5, 6, 35, 36). In this regard, ROS are generated by endothelial and epithelial cells within the kidney and mediate a wide range of cellular functions. In the early stages of obesity-induced hyperfiltration, ROS contribute to endothelial dysfunction through reductions in bioavailable nitric oxide, impaired endothelial growth or repair, and increased activation of adhesion and inflammatory cytokines that occur independent of elevations in blood pressure or hyperglycemia (37, 38). The effects on impaired endothelial function subsequently contribute to podocyte injury and inflammation in the mesangium, mediated by TNF-α and MCP-1 pathways (20, 37, 38). Our observations that NADPH oxidase activity and ROS production were increased in the ZO kidney are consistent with this notion. Moreover, the increased glomerular intensities of 3-NT are consistent with the notion of an early stage of obesity-induced glomerular injury. DPP4 inhibition has been shown to reduce circulating serum malondialdehyde in a model of renal ischemia-reperfusion injury and kidney tissue malondialdehyde levels in an endothelial nitric oxide synthase (eNOS) knockout mouse (15, 39). A recent report demonstrated that deletion of DPP4 in isolated cardiomyocytes protected cells from H2O2-induced oxidant injury (40). Thus, our data support an anti-oxidative role for DPP4 inhibition mediated by a reduction in NADPH oxidase-dependent ROS generation resulting in improved lipid peroxidation and marked reductions in glomerular 3-NT content. Although we have observed increased NADPH oxidase by linagliptin in control ZL rats, this was not accompanied by increased accumulation of ROS or peroxynitrite suggesting either more efficient removal of superoxide through up regulation of antioxidant pathways or that NADPH oxidase is increased as a compensatory mechanism in response to decreased ROS.
In summary, the findings of this investigation suggest for the first time a role for DPP4 in glomerular filtration function and that targeted inhibition of DPP4 improves obesity-related glomerulopathy and filtration barrier injury due to oxidant stress. Our finding that DPP4 inhibition improved oxidant stress in this early stage of kidney injury is particularly noteworthy. Although a reduction in blood pressure may be partially responsible for kidney improvement, our data suggest contribution of non-hemodynamic factors that are regulated by DPP4 (14).
‘What is already known about this subject’
Obesity-related glomerulopathy is characterized initially by glomerular hyperfiltration with glomerular hypertrophy and then development of proteinuria.
Putative mechanisms include endothelial dysfunction and injury to the filtration barrier due to oxidant stress and immune activation.
In this regard, there has been recent interest in targeting DPP4 to reduce oxidant stress, attenuate proteinuria and improve vascular function.
‘What this study adds’
The role that the DPP4 enzyme has on glomerular function and progression of obesity-related glomerulopathy is not known.
Our data support a role for DPP4 enzyme activity in glomerular filtration function and targeting DPP4 with inhibition improves obesity-related glomerulopathy and proteinuria, in part through reductions in oxidant stress.
Acknowledgments
This research was supported by an investigator initiated grant from Boehringer Ingelheim Pharma to VGD; NIH HL73101 and HL1079100 to JRS and AG040638 to AWC, the Veterans Affairs Merit System (0018) for JRS and CDA-2 for AWC, and the American Society of Nephrology–Association of Specialty Professors (ASN-ASP) Development Grant in Geriatric Nephrology to AWC was supported by a T. Franklin Williams Scholarship Award, funding provided by Atlantic Philanthropies Inc, the John A. Hartford Foundation. Research support was also provided to RN by Dialysis Clinics Inc.
Footnotes
Author contributions: RN, JH, AA, MRH, AM, WK, TH and TK conducted the experiments; RN, AWC, VGD and JRH conceived study design and data interpretation. All authors were involved in writing the paper and had final approval of the submitted and published versions.
Conflicts of Interest statement: VGD reports having received investigator initiated funding from Boehringer Ingelheim, otherwise the authors have nothing to report. TK reports that he is an employee of Boehringer Ingelheim.
References
- 1.Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295(13):1549–55. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 2.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303(3):235–41. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 3.Iseki K, Ikemiya Y, Kinjo K, Inoue T, Iseki C, Takishita S. Body mass index and the risk of development of end-stage renal disease in a screened cohort. Kidney Int. 2004;65(5):1870–6. doi: 10.1111/j.1523-1755.2004.00582.x. [DOI] [PubMed] [Google Scholar]
- 4.Kurella M, Lo JC, Chertow GM. Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. J Am Soc Nephrol. 2005;16(7):2134–40. doi: 10.1681/ASN.2005010106. [DOI] [PubMed] [Google Scholar]
- 5.Sowers JR, Whaley-Connell A, Hayden MR. The Role of Overweight and Obesity in the Cardiorenal Syndrome. Cardiorenal Med. 2011;1(1):5–12. doi: 10.1159/000322822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wahba IM, Mak RH. Obesity and obesity-initiated metabolic syndrome: mechanistic links to chronic kidney disease. Clin J Am Soc Nephrol. 2007;2(3):550–62. doi: 10.2215/CJN.04071206. [DOI] [PubMed] [Google Scholar]
- 7.Aroor A, McKarns S, Nistala R, DeMarco V, Gardner M, Garcia-Touza M, et al. DPP-4 inhibitors as Therapeutic Modulators of Immune Cell Function and Associated Cardiovascular and Renal Insulin Resistance in Obesity and Diabetes. Cardiorenal Med. 2013;3(1):48–56. doi: 10.1159/000348756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hocher B, Reichetzeder C, Alter ML. Renal and Cardiac Effects of DPP4 Inhibitors - from Preclinical Development to Clinical Research. Kidney Blood Press Res. 2012;36(1):65–84. doi: 10.1159/000339028. [DOI] [PubMed] [Google Scholar]
- 9.Lambeir AM, Durinx C, Scharpe S, De Meester I. Dipeptidyl-peptidase IV from bench to bedside: an update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit Rev Clin Lab Sci. 2003;40:209–294. doi: 10.1080/713609354. [DOI] [PubMed] [Google Scholar]
- 10.Dekan G, Miettinen A, Schnabel E, Farquhar MG. Binding of monoclonal antibodies to glomerular endothelium, slit membranes, and epithelium after in vivo injection. Localization of antigens and bound IgGs by immunoelectron microscopy. Am J Pathol. 1990;137(4):913–27. [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Campitelli J, Hu G, Lin Y, Luo J, Xue C. Increase in DPP-IV in the intestine, liver and kidney of the rat treated with high fat diet and streptozotocin. Life Sci. 2007;81(4):272–9. doi: 10.1016/j.lfs.2007.04.040. [DOI] [PubMed] [Google Scholar]
- 12.Lugari R, Dei CA, Ugolotti D, Barilli AL, Camellini C, Ganzerla GC, et al. Glucagon-like peptide 1 (GLP-1) secretion and plasma dipeptidyl peptidase IV (DPP-IV) activity in morbidly obese patients undergoing biliopancreatic diversion. Horm Metab Res. 2004;36(2):111–5. doi: 10.1055/s-2004-814222. [DOI] [PubMed] [Google Scholar]
- 13.Lamers D, Famulla S, Wronkowitz N, Hartwig S, Lehr S, Ouwens DM, et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes. 2011;60(7):1917–25. doi: 10.2337/db10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nistala R, Habibi J, Lastra G, Manrique C, Aroor AR, Hayden MR, Garro M, Meuth A, Johnson M, Whaley-Connell A, Sowers JR. Prevention of obesity induced renal injury in male mice by DPP4 inhibition. Endocrinology. 2014 Apr 8;:en20131920. doi: 10.1210/en.2013-1920. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glorie LL, Verhulst A, Matheeussen V, Baerts L, Magielse J, Hermans N, D’Haese PC, De Meester I, De Beuf A. DPP4 inhibition improves functional outcome after renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2012;303(5):F681–8. doi: 10.1152/ajprenal.00075.2012. [DOI] [PubMed] [Google Scholar]
- 16.Alter ML, Ott IM, von Websky K, Tsuprykov O, Sharkovska Y, Krause-Relle K, et al. DPP-4 inhibition on top of angiotensin receptor blockade offers a new therapeutic approach for diabetic nephropathy. Kidney Blood Press Res. 2012;36(1):119–30. doi: 10.1159/000341487. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Landheer S, van Gilst WH, van Amerongen A, Hammes HP, Henning RH, et al. Attenuation of renovascular damage in Zucker diabetic fatty rat by NWT-03, an egg protein hydrolysate with ACE- and DPP4-inhibitory Activity. PLoS One. 2012;7(10):e46781. doi: 10.1371/journal.pone.0046781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ku HC, Chen WP, Su MJ. DPP4 deficiency exerts protective effect against H2O2 induced oxidative stress in isolated cardiomyocytes. PLoS One. 2013;8(1):e54518. doi: 10.1371/journal.pone.0054518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whaley-Connell A, Demarco VG, Lastra G, Manrique C, Nistala R, Cooper SA, et al. Insulin resistance, oxidative stress, and podocyte injury: role of rosuvastatin modulation of filtration barrier injury. Am J Nephrol. 2008;28(1):67–75. doi: 10.1159/000109394. [DOI] [PubMed] [Google Scholar]
- 20.Hayashi K, Kanda T, Homma K, Tokuyama H, Okubo K, Takamatsu I, et al. Altered renal microvascular response in Zucker obese rats. Metabolism. 2002;51(12):1553–61. doi: 10.1053/meta.2002.36311. [DOI] [PubMed] [Google Scholar]
- 21.Zdychová J, Kazdová L, Pelikanová T, Lindsley JN, Anderson S, Komers R. Renal activity of Akt kinase in obese Zucker rats. Exp Biol Med. 2008;233(10):1231–41. doi: 10.3181/0801-RM-29. [DOI] [PubMed] [Google Scholar]
- 22.Thomas L, Eckhardt M, Langkopf E, Tadayyon M, Himmelsbach F, Mark M. (R)-8-(3-amino-piperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (BI 1356), a novel xanthine-based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase-4 inhibitors. J Pharmacol Exp Ther. 2008;325(1):175–82. doi: 10.1124/jpet.107.135723. [DOI] [PubMed] [Google Scholar]
- 23.Huttner S, Graefe-Mody EU, Withopf B, Ring A, Dugi KA. Safety, tolerability, pharmacokinetics, and pharmacodynamics of single oral doses of BI 1356, an inhibitor of dipeptidyl peptidase 4, in healthy male volunteers. J Clin Pharmacol. 2008;48(10):1171–8. doi: 10.1177/0091270008323753. [DOI] [PubMed] [Google Scholar]
- 24.Aroor AR, Sowers JR, Bender SB, Nistala R, Garro M, Mugerfeld I, et al. Dipeptidylpeptidase inhibition is associated with improvement in blood pressure and diastolic function in insulin-resistant male zucker obese rats. Endocrinology. 2013;154(7):2501–13. doi: 10.1210/en.2013-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whaley-Connell A, Habibi J, Nistala R, Hayden MR, Pulakat L, Sinak C, et al. Combination of direct renin inhibition with angiotensin type 1 receptor blockade improves aldosterone but does not improve kidney injury in the transgenic Ren2 rat. Regul Pept. 2012;176(1–3):36–44. doi: 10.1016/j.regpep.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hocher B, Sharkovska Y, Mark M, Klein T, Pfab T. The novel DPP-4 inhibitors linagliptin and BI 14361 reduce infarct size after myocardial ischemia/reperfusion in rats. Int J Cardiol. 2013;167(1):87–93. doi: 10.1016/j.ijcard.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 27.Fadini GP, Boscaro E, Albiero M, Menegazzo L, Frison V, de Kreutzenberg S, et al. The oral dipeptidyl peptidase-4 inhibitor sitagliptin increases circulating endothelial progenitor cells in patients with type 2 diabetes: possible role of stromal-derived factor-1alpha. Diabetes Care. 2010;33(7):1607–9. doi: 10.2337/dc10-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones N, New LA, Fortino MA, Eremina V, Ruston J, Blasutig IM, Aoudjit L, Zou Y, Liu X, Yu GL, Takano T, Quaggin SE, Pawson T. Nck proteins maintain the adult glomerular filtration barrier. J Am Soc Nephrol. 2009;20(7):1533–43. doi: 10.1681/ASN.2009010056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kodera R, Shikata K, Kataoka HU, Takatsuka T, Miyamoto S, Sasaki M, et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia. 2011;54(4):965–78. doi: 10.1007/s00125-010-2028-x. [DOI] [PubMed] [Google Scholar]
- 30.Drucker DJ. Dipeptidyl peptidase-4 inhibition and the treatment of type 2 diabetes. Preclinical biology and mechanism of action. Diabetes Care. 2007;30:1335–1343. doi: 10.2337/dc07-0228. [DOI] [PubMed] [Google Scholar]
- 31.De La Luz Sierra M, Yang F, Narazaki M, Salvucci O, Davis D, Yarchoan R, Zhang HH, Fales H, Tosato G. Differential processing of stromal-derived factor-1alpha and stromal-derived factor-1beta explains functional diversity. Blood. 2004;103(7):2452–9. doi: 10.1182/blood-2003-08-2857. [DOI] [PubMed] [Google Scholar]
- 32.Salvucci O, Yao L, Villalba S, Sajewicz A, Pittaluga S, Tosato G. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood. 2002;99:2703–2711. doi: 10.1182/blood.v99.8.2703. [DOI] [PubMed] [Google Scholar]
- 33.Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol. 2009;20(12):2493–502. doi: 10.1681/ASN.2008111186. [DOI] [PubMed] [Google Scholar]
- 34.Dey N, Ghosh-Choudhury N, Das F, Li X, Venkatesan B, Barnes JL, Kasinath BS, Ghosh Choudhury G. PRAS40 acts as a nodal regulator of high glucose-induced TORC1 activation in glomerular mesangial cell hypertrophy. J Cell Physiol. 2010;225:27–41. doi: 10.1002/jcp.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarbassov DD, Sabatini DM. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J Biol Chem. 2005;280(47):39505–9. doi: 10.1074/jbc.M506096200. [DOI] [PubMed] [Google Scholar]
- 36.Poirier B, Lannaud-Bournoville M, Conti M. Oxidative stress occurs in absence of hyperglycaemia and inflammation in the onset of kidney lesions in normotensive obese rats. Nephrology Dialysis Transplantation. 2000;15(4):467–476. doi: 10.1093/ndt/15.4.467. [DOI] [PubMed] [Google Scholar]
- 37.Zhang M, Gao X, Wu J. Oxidized high-density lipoprotein enhances inflammatory activity in rat mesangial cells. Diabetes/Metabolism Research and Reviews. 2010;26(6):455–463. doi: 10.1002/dmrr.1102. [DOI] [PubMed] [Google Scholar]
- 38.Quigley JE, Elmarakby AA, Knight SF, Manhiani MM, Stepp DW, Olearzcyk JJ, Imig JD. Obesity induced renal oxidative stress contributes to renal injury in salt-sensitive hypertension. Clin Exp Pharmacol Physiol. 2009;36(7):724–8. doi: 10.1111/j.1440-1681.2009.05139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alter ML, Ott IM, von Websky K, Tsuprykov O, Sharkovska Y, et al. DPP-4 inhibition on top of angiotensin receptor blockade offers a new therapeutic approach for diabetic nephropathy. Kidney Blood Press Res. 2012;36(1):119–30. doi: 10.1159/000341487. [DOI] [PubMed] [Google Scholar]
- 40.Ishibashi Y, Matsui T, Maeda S, Higashimoto Y, Yamagishi SI. Advanced glycation end products evoke endothelial cell damage by stimulating soluble dipeptidyl peptidase-4 production and its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cardiovasc Diabetol. 2013;12(1):125. doi: 10.1186/1475-2840-12-125. [DOI] [PMC free article] [PubMed] [Google Scholar]