

Abstract
We report the first X-ray crystal structure of ent-kaur-16-ene synthase from Bradyrhizobium japonicum, together with the results of a site-directed mutagenesis investigation into catalytic activity. The structure is very similar to that of the α domains of modern plant terpene cyclases, a result that is of interest since it has been proposed that many plant terpene cyclases may have arisen from bacterial diterpene cyclases. The ent-copalyl diphosphate substrate binds to a hydrophobic pocket near a cluster of Asp and Arg residues that are essential for catalysis, with the carbocations formed on ionization being protected by Leu, Tyr and Phe residues. A bisphosphonate inhibitor binds to the same site. In the kaurene synthase from the moss Physcomitrella patens, 16-α-hydroxy-ent-kaurane as well as kaurene are produced since Leu and Tyr in the P. patens kaurene synthase active site are replaced by smaller residues enabling carbocation quenching by water. Overall, the results represent the first structure determination of a bacterial diterpene cyclase, providing insights into catalytic activity, as well as structural comparisons with diverse terpene synthases and cyclases which clearly separate the terpene cyclases from other terpene synthases having highly α-helical structures.
Terpenes (or terpenoids) represent the largest class of small molecules on the planet1 and most are synthesized by enzymes that contain one or more of six main types of fold: α, β, γ, δ, ε and ζ2. About 20% of terpenes are diterpenes, molecules that contain a core of 20 carbon atoms, and most of these are made by plants and fungi. Diterpenes have, however, also been found in bacteria3,4,5,6,7,8,9,10,11, and several of these compounds have activity as anti-infective and anti-cancer drug leads, virulence factors, as well as plant growth hormones. There are two different classes of terpene cyclases, Class I and Class II, defined by the amino acid motifs that are essential for catalysis12,13,14. Class I cyclases contain two conserved motifs, DDXXD, and (N,D)DXX(S,T,G)XXX(E,D) (hereafter referred to as NTE), in which the boldface residues generally bind to the three Mg2+ that facilitate ionization of an isoprenoid diphosphate group, generating a reactive carbocation intermediate. In contrast, Class II cyclases typically contain only a DXDD motif in which the boldface aspartic acid protonates an isoprenoid double bond or an oxirane to, once again, generate a reactive carbocation intermediate. In some cases, both Class I as well as Class II motifs are present and these molecules are bi-functional or in one reported case, tri-functional15. For example, abietadiene synthase from Abies grandis (AgABS) is bi-functional and converts geranylgeranyl diphosphate (GGPP) to copalyl diphosphate (CPP) in a Class II reaction, then CPP is converted to abietadiene in a Class I reaction, Figure 1a. Similarly, the moss Physcomitrella patens utilizes a tri-functional enzyme to convert GGPP to ent-CPP in a Class II reaction, then ent-CPP is converted to ent-kaur-16-ene (20%) and 16α-hydroxy-ent-kaurane (80%) via a Class I reaction with, in the case of kauranol, carbocation quenching16, Figure 1b.
Figure 1. Reactions catalyzed by AgABS, PpCPPS/KS and BjKS, and proposed catalytic mechanism for the BjKS reaction.

(a) Abietadiene is formed from GGPP by a bi-functional terpene cyclase abietadiene synthase, in Abies grandis. (b) Formation of ent-CPP, ent-kaurene and kauranol from GGPP catalyzed by Physcomitrella patens CPPS/KS. (c) Two proteins, ent-CPPS and ent-KS, are used to produce ent-kaurene in the soil bacterium, Bradyrhizodium japonicum. Proposed carbocation intermediates are shown.
Here, we report the first structure of a (Class I) bacterial diterpene cyclase, ent-kaurene synthase, from the soil bacterium Bradyrhizobium japonicum (BjKS; gene # blr2150)4. BjKS catalyzes the cyclization of ent-CPP which is produced by a separate (Class II) enzyme, ent-copalyl diphosphate synthase (ent-CPPS; blr2149), from GGPP (Figure 1c), and one possible mechanism of action17 for the ent-kaurene synthase reactions is shown in Figure 1c. The ent-KS reactions have been proposed to proceed via initial diphosphate loss from ent-CPP (catalyzed by Mg2+) followed by cyclization to form the 8-carbonium ion, the pimaren-8-yl cation, which then undergoes secondary cyclization with Wagner-Meerwein migration of C-12 to C-16 to form the ent-kauran-16-yl cation. This cation can then loose a proton to form ent-kaur-16-ene, or in P. patens the ent-kauranyl cation can also be quenched by a water molecule to form 16-α-hydroxy-ent-kaurane, Figure 1b16.
Structurally, Class I proteins contain a highly α-helical catalytic α domain while Class II proteins contain two domains, βγ, and with bi-functional species such as abietadiene synthase, all three domains (αβγ) are present18. In previous work19 we proposed that these αβγ domain proteins might have arisen by the fusion of the genes of α + βγ proteins in ancestral, perhaps soil dwelling, bacteria, so determining the structures of bacterial α (and βγ) cyclases is of interest not only from a mechanism of action perspective, but also from an evolutionary one. Is, for example, the structure of BjKS (a predicted19 α domain Class I protein) very similar to that of the α domains seen in modern plant terpene cyclases? How does ent-CPP bind? To investigate these and other questions we determined the structure of BjKS in the presence of its ent-CPP substrate, as well as apo- and inhibitor-bound structures, and we used site-directed mutagenesis to probe catalytic activity. In addition, we compared the BjKS structure with the structures of diverse other terpene cyclases and synthases, to see to what extent there were structural similarities among the different species.
Results and Discussion
Structure of apo-ent-kaurene synthase
We cloned, expressed and purified ent-kaurene synthase from B. japonicum then crystallized it and solved its structure using a selenomethionine-substituted (SeMet) protein (Supplementary Table S1). Full data acquisition and crystallographic details for two native (i.e. apo) crystals are given in Table 1. These crystals were found in the same drop. The final models were refined at a resolution of 1.9–2.0 Å. There are four molecules (chains A, B, C, D) in the asymmetric unit of the apo-1 crystal and two in the asymmetric unit of the apo-2 crystal. Chain A and chain B in the apo-1 structure contained the most complete densities (residues 3–290 for chain A, 3–281 for chain B), while there was a missing loop (residues ~211–221) in both chains C and D in the apo-1 structure, and this loop was also missing (i.e. disordered) in all other structures (Table S2). The structures of the monomers of apo-1 and apo-2 are very similar with a 0.52–0.77 Å Cα rmsd for any apo-1/apo-2 chain comparison. A stereo-view of chain A in the apo-1 structure (PDB ID code 4KT9) is shown in Figure 2a, from which it can be seen that the structure consists primarily of twelve α helices, as found in many other terpene cyclases, as well as synthases such as farnesyl diphosphate synthase. Both apo-1 and apo-2 crystal structures contain dimers (two dimers/4 chains in apo-1, one dimer in apo-2). In the apo-1 chain A, B structure (the most complete structure) there is a cluster of side-chains of hydrophobic residues (M235, Y232 and F240, cyan spheres) from both chains at one end of the dimer interface, Figure 2b, with a buried surface area of ~1000 Å3. Further up (green spheres), the E244 side-chain interacts with the N-terminus of the helix starting at Y155 in the other subunit in the dimer, and salt bridges exist between the E157(A) and R250(B) side-chains, and between E157(B) and R250(A) (purple spheres). These interactions all stabilize the dimer, at least in the crystal. To determine whether dimers are present in solution, we used size exclusion chromatography coupled with multi-angle light scattering (SEC/MALS). The SEC/MALS result (Figure 2c) indicates a molecular weight of 66.4 ± 0.3 kDa for the symmetric elution peak with a poly-dispersity value of 1.000, indicating that BjKS forms (and is presumably active as) a dimer in solution (the theoretical dimer MW is 66.76 kDa). However, as can be seen in Figure 2b the predicted catalytic motifs (DDXXD and NTE) are ~20Å from the dimer interface and unlike some other prenyl synthases20, there is no chain inter-digitation. This suggests that the presence of a dimer is not essential for catalysis.
Table 1. Data collection and refinement statistics for KS crystals.
| Apo-1 | Apo-2 | D75C + CPP | WT + BPH-629 | |
|---|---|---|---|---|
| Data collection | ||||
| Resolution (Å) | 25.0 − 2.0 | 25.0 − 1.9 | 25.0 − 1.8 | 25.0 − 2.0 |
| (2.1 − 2.0) | (2.0 − 1.9) | (1.9 − 1.8) | (2.1 − 2.0) | |
| Space group | P21 | P41 | P41 | P41 |
| Unit-cell a, b, c (Å) | 65.0, 130.1, | 65.8, 65.8, | 65.8, 65.8, | 65.8, 65.8, |
| 66.4 | 135.2 | 136.2 | 135.7 | |
| β (°) | 95.6 | |||
| No. of unique reflections | 72234 (6728) | 43643 (4390) | 51614 (5123) | 38697 (3888) |
| Redundancy | 3.0 (2.7) | 5.6 (5.8) | 4.9 (4.4) | 3.4 (3.3) |
| Completeness (%) | 98.0 (91.5) | 99.8 (100.0) | 99.5 (98.9) | 99.4 (100.0) |
| Mean I/σ(I) | 18.1 (3.1) | 42.9 (4.5) | 42.3 (3.6) | 29.2 (3.3) |
| Rmerge (%) | 5.6 (20.7) | 5.9 (46.7) | 3.5 (37.2) | 3.9 (46.3) |
| Refinement | ||||
| No. of chain(s) | 4 | 2 | 2 | 2 |
| No. of reflections | 70362 (6036) | 42679 (3982) | 50837 (3310) | 37623 (3499) |
| Rwork (95% of data) | 0.176 (0.230) | 0.191 (0.251) | 0.206 (0.28) | 0.197 (0.279) |
| Rfree (5% of data) | 0.220 (0.276) | 0.232 (0.287) | 0.239 (0.309) | 0.235 (0.291) |
| R.m.s.d. bonds (Å) | 0.020 | 0.010 | 0.008 | 0.010 |
| R.m.s.d. angles (°) | 1.7 | 1.3 | 1.2 | 1.3 |
| Ramachandran plot (%) | ||||
| Most favored (%) | 97.3 | 96.7 | 97.5 | 96.6 |
| Allowed (%) | 2.2 | 2.8 | 2.1 | 2.8 |
| Disallowed (%) | 0.5 | 0.5 | 0.4 | 0.6 |
| Mean B (Å2)/atoms | ||||
| Protein atoms | 27.1/8766 | 38.0/4310 | 39.8/4129 | 46.4/4300 |
| Ligand atoms | 90.0/58 | 154.0/30 | ||
| Water molecules | 38.2/651 | 51.2/624 | 50.8/415 | 57.4/360 |
| PDB ID code | 4KT9 | 4W4R | 3WBV | 4W4S |
Values in parentheses are for the outer-most resolution shells.
Figure 2. Structure of BjKS.

(a) Stereoview of (apo-1) BjKS (PDB ID code 4KT9) showing highly α-helical structure. (b) Dimer interface in B. japonicum ent-kaurene synthase. Hydrophobic residues (M235, Y232 and F240, cyan spheres) form a hydrophobic core; E244 side-chains interacts with the N-terminus of the helix starting at Y155 in the other subunit (green spheres); salt bridges exist between the E157(A) and R250(B) side-chains, and between E157(B) and R250(A) (purple spheres). The two catalytic motifs (DDXXD, red and NTE, orange) are also shown. (c) SEC-MALS results.
Comparisons between kaurene synthase and other terpene cyclases/synthases
In previous work19, we used purely computational methods to predict the structure of (α-domain) BjKS (as well as βγ domain BjCPPS) which led to the idea that modern plant terpene cyclases might have arisen by the fusion of the genes of ancestral (bacterial) α and βγ domain proteins. It is thus of particular interest to now compare the actual BjKS structure with those of other, modern terpene cyclases (and synthases, which also have highly α-helical structures).
The BjKS structure is generally similar to that found in the α domains (αc19) of terpene cyclases from bacteria, fungi as well as plants: some representative examples are shown in Figure 3. Figure 3a shows a superposition between BjKS (red; PDB ID 4KT9) and pentalenene synthase (green; PDB ID 1HM7)21 from the bacterium Streptomyces UC5319 where there is a 2.7 Å Cα rmsd (root mean square deviation) between Cα atoms over 204 residues; Figure 3b shows a superposition with 1, 8-cineole synthase (PDB ID 2J5C) from the flowering plant Salvia fruticosa (Sf-CinS1, blue), an “αβ” domain protein19 in which the Cα rmsd in 2.4 Å over 195 residues, while Figure 3c shows a superposition with abietadiene synthase from the gymnosperm Abies grandis (AgABS; PDB ID 3S9V), an αβγ domain protein18.
Figure 3. Superimposition of the overall structure of BjKS (red; PDB ID 4KT9) with other terpene cyclases.

(a) Superimposition of BjKS with pentalenene synthase from Streptomyces UC5319 which has only an α domain (α, green; 1HM7); (b) Superimposition of BjKS with 1, 8-cineole synthase from Salvia fruticosa which has two domains (αβ; blue; 2J5C); (c) Superimposition of BjKS with abietadiene synthase from Abies grandis which has three domains (αβγ; yellow; 3S9V); (d) Homology tree (based on α domain Q-scores22,23) showing clustering of BjKS with the plant α domain proteins. Note that the α domains in the terpene cyclase (top) are distinct from the α domains in synthases such as FPPS, and have previously been referred to as “αc” domains19.
As can be seen in Figure 3c, the BjKS structure (red) is similar to the α domain of AgABS (yellow), having a 2.6 Å Cα rmsd over 224 residues. The Cα rmsd value for the three other αβγ domain proteins: taxadiene synthase (PDB ID 3P5R); ent-copalyl diphosphate synthase (PDB ID 3PYB) and bisabolene synthase (PDB ID 3SDV) are 2.6 Å/201 residues, 3.5 Å/169 residues, and 2.9 Å/226 residues for taxadiene synthase, ent-copalyl diphosphate synthase and (the sesquiterpene) bisabolene synthase, respectively, and the closest similarity to abietadiene synthase is likely due to the fact that AgABS produces a C20 species that is structurally more similar to ent-kaurene than are the products of the three other αβγ proteins. Remarkably, however, there is only a 17% sequence identity between BjKS and AgABS.
We next sought to see how the BjKS structure compared with a broader range of α, β, γ, δ, ε, and ζ-fold proteins by using their Q-scores22. The Q-score attempts to minimize difficulties than may arise by use of e.g. just Cα rmsd values when the numbers of residues in target and query proteins, are different. The Q-score is defined in Equation (1)22,23:
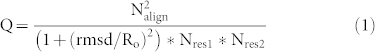 |
where Nalign is the number of aligned residues, determined here by using the secondary structure matching (SSM) program22; Nres1 and Nres2 are the total numbers of residues in the two aligned proteins whose structures are to be compared, rmsd in the Cα root mean square deviation of the Cα residues in the two proteins, and Ro is an empirical parameter, set (by the program) to 3 Å22,23. The Q-score is 1 if two structures are identical, but obviously decreases as the rmsd increases, Nalign decreases, or if Nres1 or Nres2 are much larger than Nalign. As expected, Figure S1, the use of complete structures results in a clustering of BjKS with other bacterial and fungal cyclases (which are all small proteins, as is BjKS), while the plant αβ or αβγ proteins are very distant because of the large Nres in these systems. The Q-scores used to construct Figure S1 are shown in Table S3. However, since we are primarily interested in structural comparisons between α domains, we next constructed a second tree using just the highly α-helical domains in a broad range of terpene cyclases and synthases. As shown in Figure 3d (data in Table S4), we now see that BjKS clusters with the plant α domains (in αβγ proteins), while the fungal cyclases are separate, as are other α proteins such as farnesyl diphosphate synthase and geranylgeranyl diphosphate synthase (yellow, Figure 3d). The αβγ domain protein ent-CPPS from the plant Arabidopsis thaliana is more distant from the three other αβγ domain plant proteins due, we propose, to the fact that the α domain in ent-CPPS is not catalytic in this protein, so its structure is less likely to be conserved. The BjKS structure thus most closely resembles the α domain structures in modern plant terpene cyclases, and as noted previously4,19, in B. japonicum, the ent-CPPS and ent-KS genes overlap by a single nucleotide4, a potential route to the origin of bi-functional species. Indeed, very primitive plants such as mosses do contain such bi- (or tri-) functional species, as discussed below.
Site-directed mutagenesis and enzyme inhibition
We next sought to investigate the catalytic mechanism of BjKS. We first used the BjKS and AgABS sequences to generate a set of 354 homologous proteins (using the JPRED3 program24) which was then used as input to the SCORECONS server25 which ranks residues in terms of their essential nature: 1.000 is essential, 0 is non-essential. The top 10 most essential residues are shown in Figure 4a. We show in Figure 4b the locations of the top 10 non-structural (i.e. not Gly or Pro) residues that are most conserved, from which it can be seen that D75, D76, D79, R204 and D208 (red spheres) are close to the catalytic site seen in other terpene cyclases while the other residues are more distant. We elected to mutate D75, D79 and R204 (to D75A, D75C, D79C and R204A) to test whether they were essential for catalysis since D75 and D79 are part of the highly conserved (Figure S2) DDXXD motif found in many Class I cyclases, and R204 is also highly conserved, as part of the RLX(N,D)DXX(S,TR,G)XXX(E,D) (Figure S2) motif. As can be seen in Figure 4c and Table S5, mutation of D75, D79 or R204 (to Ala or Cys) greatly reduced activity to, on average, 1.7 ± 1.6%.
Figure 4. Essential residues, catalysis and inhibition.

(a) SCORECONS25 results. (b) Highly conserved residues; residues in red were mutated to A or C. (c) Activity of wild-type and mutant proteins. (d) Inhibition of BjKS by the bisphosphonate BPH-629.
We also sought to obtain a BjKS inhibitor, since such compounds might be leads for plant growth hormones targeting gibberellin biosynthesis, as well as providing information on the structure of the active site. The structures of several bisphosphonates bound to the α domain in the αβγ protein bisabolene synthase have been reported26, so we chose to investigate a bisphosphonate inhibitor, BPH-629 (Figure 4d), which has a C19O side-chain, comparable in size to the side-chain in ent-CPP. We found that BPH-629 inhibited BjKS with an IC50 = 9.5 μM, Figure 4d and we report its structure in the following section.
Substrate and ligand-bound structures
In other articles27 there have been reports of the binding of CPP isosteres (containing nitrogen) to terpene cyclases, but there have been no reports of ent-CPP (or CPP) bound to a terpene cyclase (or synthase). We therefore next determined the structure of the substrate ent-CPP (Figure 1) bound to the BjKS D75C mutant, in addition to determining the structure of the bisphosphonate inhibitor, BPH-629. Full data acquisition and crystallographic details are given in Table 1.
We obtained an ent-CPP structure by soaking apo-1 crystals with ent-CPP, while BPH-629 bound crystals were obtained by co-crystallization. Electron density results (for L71, D/C75, R204 and N207 residues) in all three structures are shown in Figures 5a–c (Fo-Fc omit maps, contoured at 2σ). These results clearly indicate that ent-CPP and BPH-629 and not e.g. tartrate or PEG from the crystallization buffer, bind to the protein. More complete ligand densities (Fo-Fc omit maps, contoured at 1σ (grey) and 3σ (red)) are shown in Figures 5d, e. The cyclic structures are well defined, although density in weaker for the diphosphate/bisphosphonate moieties, due perhaps to some phosphate/phosphonate group disorder, since Mg2+ was not bound.
Figure 5. Electron density results for BjKS.

In (a)–(c) both protein residues (L71, D/C75, R204 and N207) as well as the ligands (ent-CPP, BPH-629) are shown as Fo-Fc omit maps, contoured at 2σ. (a) apo-BjKS. (b) ent-CPP soaked. (c) BPH-629 co-crystal. (d) ent-CPP Fo-Fc map, contoured at 1σ (grey) and 3σ (red). (e) as (d) but BPH-629 ligand.
The complete BjKS structure with ent-CPP bound is shown in Figures 6a, and in Figure 6b, the BjKS-ent CPP structure is shown superimposed on that of 2-F-geranylgeranyl diphosphate (FGG) bound to the α domain of taxadiene synthase (TXS; PDB ID 3P5R). The 3 Mg2+ are from the taxadiene synthase structure. Three Asp residues (D76, D79 and D208) in BjKS are located in very similar positions to the corresponding Asp residues in TXS (Cα rmsd of 0.8 Å for these 3 Asp residues), and might be expected to coordinate to a [Mg2+]3 cluster which was, however, not observed in our BjKS crystals. This may be due to use of a D75C mutant, or to the high tartrate concentration (~1 M) used in BjKS crystallization, since tartrate can chelate Mg2+. The hydrophobic side-chain of ent-CPP is buried in a hydrophobic pocket composed of I36, L68, L71, F72, Y136, I166, A167, Y168 and N207, shown as the orange surface in Figure 6b. In this conformation, the exo-methylene group is poised (~4 Å distance) to interact with the carbocation formed on diphosphate ionization, forming the first initial reactive intermediate, the pimaren-8-yl ion (Figure 1).
Figure 6. BjKS ent-CPP and BPH-629 structures.

(a) Stereo-view of ent-CPP bound structure (PDB ID 3WBV). (b) Stereo-view of the structure of ent-CPP (yellow) bound to BjKS superimposed on 2-F-geranylgeranyl diphosphate (FGG, cyan) bound to the α domain of taxadiene synthase (TXS) (PDB ID code 3P5R). The highly conserved “DDXXD” Asps of BjKS are indicated. D75 is constructed based on C75 in the mutant protein. The 3 Mg2+ are from the TXS structure and are not observed in BjKS. The hydrophobic side-chain of ent-CPP is buried in a hydrophobic pocket composed of I36, L68, L71, F72, Y136, Y168 and N207, shown as an orange surface. (c) Stereo-view of ent-CPP (yellow) and BPH-629 (pink) in the BjKS active site. Hydrophobic residues surround the diterpene's decahydronaphthalene and the bisphosphonate's dibenzofuran side-chains.
We also obtained the structure of BPH-629 bound to BjKS at 2.34 Å resolution (PDB ID code 4W4S), finding that the inhibitor occupies the same site as does ent-CPP, Figure 6c. Full data acquisition and crystallographic details are given in Table 1. The bisphosphonate has hydrogen bonds from the bisphosphonate group to D75, S165, R204 and N207, but just as with the ent-CPP complex, there is no Mg2+ observed. The lack of a [Mg2+]3 cluster in this case cannot be due to use of a D75C mutant (as used with the ent-CPP structure) since wild type protein was employed for the bisphosphonate structure, so again it seems likely that the very high tartrate levels in the crystallization buffer may be responsible for Mg2+ chelation.
Structural basis for formation of ent-kaurene and kauranol in P. patens
The results described above delineate the active site in BjKS together with several residues (D75, D79 and R204) that are essential for catalysis. These residues comprise part of the conserved DDXXD/NTE motifs involved in diphosphate loss to form carbocation transition states/reactive intermediates (Figure 1c) that need to be protected from solvent attack in BjKS, raising the question: which residues provide this protection? As noted above, in the moss Physcomitrella patens, there is in fact solvent attack in the PpCPPS/KS protein16 which results in the formation of only 20% ent-kaurene but 80% 16-α-hydroxy-ent-kaurane, and Kawaide et al.15 identified a key residue (A710, corresponding to L140 in BjKS) that enabled water access, quenching the kauranyl cation. A comparison between the two protein structures would be informative since it might help clarify the BjKS mechanism of action, but the structure of the P. patens protein has not been reported. There is, however, a 45% sequence identity between PpCPPS/KS and the A. grandis abietadiene synthase, and a Phyre228 structure prediction of the tri-functional P. patens protein is shown in Figure 7a. As expected, the protein is predicted to have an αβγ domain structure and the 45% conserved residues, shown in red in Figure 7a, are distributed over all three domains. When the P. patens active site structure prediction is superimposed on the BjKS structure (with ent-CPP from the BjKS structure), Figure 7b it is clear that BjKS contains larger residues (Y136 and L140) adjacent the ent-CPP ligand-binding site than those found in P. patens (which has Ala instead of Leu, and Leu instead of Tyr), and these larger residues (Figure 7b) prevent water from quenching the kauranyl cation (Figure 1). The increase in side-chain volume is expected to be ~100 Å3 (based on amino-acid volumes), and such steric differences might be particularly important in the presence of Mg2+, because Class I terpenoid synthases typically undergo a structural transition from an open to a closed active-site conformation after Mg2+ binding to the substrate's diphosphate group, helping protect the reactive carbocation intermediates from premature quenching by bulk solvent.
Figure 7. Comparisons between BjKS structure and the tri-functional P. patens CPPS/KS structural model.

(a) Phyre228 homology model of Physcomitrella patens ent-CPPS/ent-KS showing the 45% of residues (in red) that are identical to those in abietadiene synthase. The conserved residues are uniformly dispersed over the α, β and γ domains. (b) ent-CPP (yellow) bound to BjKS (PDB ID 3WBV) superimposed on corresponding P. patens KS residues. The large BjKS residues adjacent ent-CPP are in cyan. The smaller P. patens residues are in magenta and permit water quenching of the kauranyl-16-yl cation. (c) Stereo view of residues that are proposed to stabilize the carbocation intermediates in BjKS (PDB ID 3WBV). Distances between ent-CPP C12 and Y136 O, ent-CPP C12 and L140 Cδ, ent-CPP C8 and F72 C are 5.1 Å, 6.2 Å and 4.2 Å, respectively.
In addition to Y136 and L140 protecting the carbocation intermediates in BjKS, we propose that F72 plays a similar role since F72 is very close to the ent-CPP substrate, as shown in Figure 7c. The phenyl group in F72 could also be involved in cation-π stabilization of one or more reactive intermediates, a widely used catalytic strategy adopted by terpenoid cyclases29,30,31. Based on the x-ray structural results and structure comparison, it thus appears that Y136 and L140 can protect carbocations from solvent attack on one side of the intermediate(s), while F72 protects from solvent attack from the other face, as shown in Figure 7c.
Methods
Cloning, expression and purification of ent-kaurene synthase from Bradyrhizobium japonicum (BjKS)
The gene encoding KS from Bradyrhizobium japonicum USDA 110 DNA was amplified by polymerase chain reaction (PCR) with a forward primer 5′-GGTATTGAGGGTCGCGAGAATCTTTATTTTCAGGGCGCTGGTGCTGGTGCTATGAT-3′ and a reverse primer 5′-AGAGGAGAGTTAGAGCCGTTATTAGGCCGGCGCGCGCTGGCCTCCCCTCAC-3′, then cloned into the pET32 Xa/LIC vector. A tobacco etch virus protease (TEV protease) cutting sequence (ENLYFQG) was added before the KS gene. The recombinant plasmids were transformed to E. coli BL21trxB (DE3) and protein was induced with 0.5 mM isopropyl-thiogalactopyranoside (IPTG) at 16°C for 20 hours. The cell lysate was harvested by centrifugation and the resulting supernatant loaded onto a Ni-NTA column equilibrated in buffer containing 25 mM Tris, pH 7.5, 150 mM NaCl, and 20 mM imidazole. His-tagged KS was eluted using an imidazole gradient (20–250 mM). The protein solution was then dialyzed against a buffer containing 25 mM Tris, pH 7.5, 150 mM NaCl and subjected to TEV protease digestion, to remove the thioredoxin and His tags. The mixture was then again passed through a Ni-NTA column and untagged KS eluted with a 5 mM imidazole-containing buffer. Protein was further purified by FPLC using a DEAE column, 25 mM Tris, pH 8.0, and a 0–500 mM NaCl gradient. The protein eluted at ~170 mM NaCl and was then concentrated to 10 mg/mL in 25 mM Tris, pH 7.5, and 150 mM NaCl. Expression of SeMet KS was carried out as described previously32 and the purification procedures were the same as those used for the wild-type enzyme, but the DEAE column was omitted. The protein was then concentrated to 6.4 mg/mL in 25 mM Tris, pH 7.5 and 150 mM NaCl.
Size exclusion chromatography-multi-angle light scattering (SEC/MALS)
The molecular weight of KS was determined by static light scattering (SLS) using a Wyatt Dawn Heleos II multi-angle light scattering detector (Wyatt Technology). This was coupled to an AKTA Purifier UPC10 FPLC protein purification system equipped with a WTC-030S5 size-exclusion column (Wyatt Technologies). 2.5 mg/mL KS (100 μL) was applied to the size-exclusion column with a buffer containing 25 mM Tris (pH 7.5), 150 mM NaCl, 10 mM DTT and 0.02% NaN3 using a flow rate of 0.5 mL/min. BSA (2 mg/mL) was used for the system calibration. The molecular weights of the individual peaks in the size-exclusion chromatogram were determined from the SLS results in conjunction with refractive index measurements (Wyatt Optilab rEX, connected downstream of the LS detector). A standard value of the refractive index, dn/dc = 0.185 mL/g, was used for the proteins and the buffer viscosity η = 1.0408 cP at 25°C, was used. The value of the reference refractive index, 1.3462, was taken directly from the measurement of the Wyatt Optilab rEX when only buffer was passing through the reference cell.
Crystallization, data collection and structure determination of BjKS
The wild-type KS protein containing 1 mM MgCl2 and 10 mM DTT was first crystallized by using the Index Kit (Hampton Research, Aliso Viejo, CA) and the sitting-drop vapor diffusion method. The reservoir solution (No. 26) contained 1.1 M ammonium tartrate dibasic, pH 7.0. Better crystals were obtained by optimizing the reservoir composition to 0.7 M ammonium tartrate dibasic, pH 7.0 and 2–4% w/v PEG4000. Two different crystals having different space groups (P21 for apo-1; P41 for apo-2) appeared in the same crystallization condition drop. Crystallization conditions for SeMet KS were 0.8 M ammonium tartrate dibasic, pH 7.0. For the inhibitor BPH-629 complex, the KS protein solution containing 1 mM MgCl2 and 10 mM DTT was co-crystallized with 2.5 mM BPH-629 under the same conditions as the wild-type protein. To obtain the KS-CPP structure, the D75C mutant crystal was prepared under the same crystallization conditions and then soaked with the cryo-protectant containing 5 mM CPP for 5 hours. All crystals were prepared at 4°C for 12 hours and were then transferred to room temperature. They reached suitable size for X-ray diffraction in 2–3 days. The cryo-protectant was 1.0 M ammonium tartrate dibasic, pH 7.0, 10% w/v PEG4000 and 20% glycerol.
X-ray diffraction data sets were processed by using the HKL2000 program33. Prior to structure refinement, 5% randomly selected reflections were set aside for calculating Rfree as a monitor of model quality34. The multiple-wavelength anomalous diffraction (MAD) data sets of the selenium-containing derivatives were collected at wavelengths of 0.9789 Å (peak), 0.9790 Å (inflection) and 0.9611 Å (remote). Using SOLVE and RESOLVE35, we obtained the figure of merit values of 0.52, Z-scores from 82.58 to 90.28, and up to 430 auto-built amino acid residues. The BPH-629 and CPP complex structures were solved by using molecular replacement. All following structural refinements were carried out by using Coot36 and CNS37. Data collection and refinement statistics are summarized in Table 1 and Supplementary Table S1. All graphics for the protein structures as well as electrostatic surface calculation were prepared by using the PyMOL program.
BjKS mutant preparation
The KS mutants were created by using QuickChange Site-Directed Mutagenesis Kit with KS-pET32 Xa/LIC gene as the template. The oligonucleotides were 5′-gcgctgctgttctggctctgcgattgcaacgaccttgg-3′ for D75C, 5′-ctggcgctgctgttctggctcgcggattgcaacgaccttggcctg-3′ for D75A, 5′-tggctcgacgattgcaactgccttggcctgatctcgcc-3′ for D79C, and 5′-cggctcatctccgcgataggggcgctgcagaacgatctgcatgga-3′ for R204A. The constructs were transformed to E. coli BL21trxB (DE3) for protein expression. The expression and purification of the mutant proteins were the same as those used for wild-type.
SCORECONS analysis of BjKS
A library of 10 KS sequence homologs and 342 AgABS sequence homologs were obtained by using the Jpred 3 server24. Multiple sequence alignment was carried out with the ClustalW2 server38. The aligned sequence library was then uploaded to the SCORECONS server25 to calculate the conservation scores for each residue of BjKS.
Activity assay for BjKS proteins
BjKS activity assays were carried out as described previously39. Briefly, the dephosphorylation and cyclization of ent-CPP catalyzed by BjKS was monitored by using a continuous spectrophotometric assay for diphosphate release40 in 96 well plates with 200 μL reaction mixtures containing 400 μM 2-amino-6-mercapto-7-methylpurine (MESG), 50 μM ent-CPP, 25 mM Tris-HCl (pH 7.5), 0.01% Triton X-100 and 1 mM MgCl2. The IC50 values were obtained by fitting the inhibition data to a rectangular hyperbolic dose-response function in OriginPro 8.5 (OriginLab, Northampton, MA).
Conclusions
The results presented above are of interest for several reasons. First, we report the first X-ray crystallographic structure of a bacterial diterpene cyclase. The structure is similar to that found in the α domain in plant diterpene cyclases. Second, we report the first structure of a diterpene diphosphate (ent-copalyl diphosphate) bound to a diterpene cyclase. The structure helps define the diterpene cyclase substrate-binding site by using a native substrate, and the diphosphate group is found to be close to three residues that we find (using site-directed mutagenesis) are essential for catalysis. Third, we obtained the structure of an aromatic bisphosphonate inhibitor. The bisphosphonate binds in or very close to the ent-CPP diphosphate-binding site, while the dibenzofuran side-chain occupies basically the same site as does the decahydronaphthalene ring in ent-CPP: such compounds might find use as plant growth regulators. Fourth, the results of computational modelling of a P. patens tri-functional KS together with a comparison with the BjKS structure provide strong evidence in support of the roles of L140, Y136 and F72, in carbocation protection in the bacterial enzyme.
Author Contributions
R.T.G. and E.O. designed and directed the research. W.L., Y.Z., Y.C. and J.L. purified and crystallized the proteins. C.H.H., T.P.K. and C.C.C. determined the crystal structures. C.N. and T.H. synthesized ent-CPP. X.F. and S.B. performed activity assays. R.T.G., T.P.K. and E.O. analyzed structures. I.W. and S.-T.D.H. performed the SEC-MALS analysis. E.O. wrote the manuscript with input from co-authors. All authors reviewed the manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
This work was supported by grants from the National Basic Research Program of China (2011CB710800 and 2011CBA00805), the National Natural Science Foundation of China (31200053 and 31300615), the United States Public Health Service (NIH grants GM065307 and CA158191), a Harriet A. Harlin Professorship (E.O.), and the University of Illinois/Oldfield Research Fund. X.F. was supported by a Pre-doctoral Fellowship from the American Heart Association, Midwest Affiliate (13PRE14510056). We thank the National Synchrotron Radiation Research Center of Taiwan for beam-time allocation and data collection assistance.
References
- Buckingham J. Dictionary of natural products on DVD. (CRC, FL, 2013). [Google Scholar]
- Oldfield E. & Lin F. Y. Terpene biosynthesis: Modularity rules. Angew. Chem. Int. Ed. Engl. 51, 1124–1137 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dairi T. Studies on biosynthetic genes and enzymes of isoprenoids produced by actinomycetes. J. Antibiot. (Tokyo) 58, 227–243 (2005). [DOI] [PubMed] [Google Scholar]
- Morrone D. et al. Gibberellin biosynthesis in bacteria: Separate ent-copalyl diphosphate and ent-kaurene synthases in Bradyrhizobium japonicum. FEBS Lett. 583, 475–480 (2009). [DOI] [PubMed] [Google Scholar]
- Hamano Y. et al. Functional analysis of eubacterial diterpene cyclases responsible for biosynthesis of a diterpene antibiotic, terpentecin. J. Biol. Chem. 277, 37098–37104 (2002). [DOI] [PubMed] [Google Scholar]
- Smanski M. J., Peterson R. M. & Shen B. Platensimycin and platencin biosynthesis in Streptomyces platensis, showcasing discovery and characterization of novel bacterial diterpene synthases. Methods Enzymol. (Academic Press, UT, 2012). [DOI] [PubMed] [Google Scholar]
- Komakr H. et al. Brasilicardin A, a new terpenoid antibiotic from pathogenic Nocardia brasiliensis: fermentation, isolation and biological activity. J. Antibiot. (Tokyo) 52, 13–19 (1999). [DOI] [PubMed] [Google Scholar]
- Tully R. E., van Berkum P., Lovins K. W. & Keister D. L. Identification and sequencing of a cytochrome P450 gene cluster from Bradyrhizobium japonicum. BBA-Gene Struct. Expr. 1398, 243–255 (1998). [DOI] [PubMed] [Google Scholar]
- Smanski M. J., Peterson R. M., Huang S.-X. & Shen B. Bacterial diterpene synthases: New opportunities for mechanistic enzymology and engineered biosynthesis. Curr. Opin. Chem. Biol. 16, 132–141 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann F. M. et al. Characterization and inhibition of a class II diterpene cyclase from Mycobacterium tuberculosis: Implications for tuberculosis. J. Biol. Chem. 284, 23574–23579 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prach L., Kirby J., Keasling J. D. & Alber T. Diterpene production in Mycobacterium tuberculosis. FEBS J. 277, 3588–3595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt K. U., Schulz G. E., Corey E. J. & Liu D. R. Enzyme mechanisms for polycyclic triterpene formation. Angew. Chem. Int. Ed. Engl. 39, 2812–2833 (2000). [PubMed] [Google Scholar]
- Wendt K. U. & Schulz G. E. Isoprenoid biosynthesis: Manifold chemistry catalyzed by similar enzymes. Structure 6, 127–133 (1998). [DOI] [PubMed] [Google Scholar]
- Koksal M., Hu H., Coates R. M., Peters R. J. & Christianson D. W. Structure and mechanism of the diterpene cyclase ent-copalyl diphosphate synthase. Nat. Chem. Biol. 7, 431–433 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaide H. et al. Identification of the single amino acid involved in quenching the ent-kauranyl cation by a water molecule in ent-kaurene synthase of Physcomitrella patens. FEBS J. 278, 123–133 (2011). [DOI] [PubMed] [Google Scholar]
- von Schwartzenberg K., Schultze W. & Kassner H. The moss Physcomitrella patens releases a tetracyclic diterpene. Plant Cell Rep. 22, 780–786 (2004). [DOI] [PubMed] [Google Scholar]
- MacMillan J. & Beale M. H. Diterpene Biosynthesis. 217–243 Cane, D. E. (ed.) (Elsevier, Amsterdam, 1999). [Google Scholar]
- Zhou K. et al. Insights into diterpene cyclization from structure of bifunctional abietadiene synthase from Abies grandis. J. Biol. Chem. 287, 6840–6850 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R. et al. Diterpene cyclases and the nature of the isoprene fold. Proteins 78, 2417–2432 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T.-H. et al. Structure of a heterotetrameric geranyl pyrophosphate synthase from mint (Mentha piperita) reveals intersubunit regulation. Plant Cell 22, 454–467 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesburg C. A., Zhai G. Z., Cane D. E. & Christianson D. W. Crystal structure of pentalenene synthase: Mechanistic insights on terpenoid cyclization reactions in biology. Science 277, 1820–1824 (1997). [DOI] [PubMed] [Google Scholar]
- Krissinel E. & Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D. 60, 2256–2268 (2004). [DOI] [PubMed] [Google Scholar]
- Henrick E. & Krissinel E. Protein structure comparison service Fold at European Bioinformatics Institute. http://www.ebi.ac.uk/msd-srv/ssm/ (Date of access: 01/06/2002).
- Cole C., Barber J. D. & Barton G. J. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdar W. S. Scoring residue conservation. Proteins 48, 227–241 (2002). [DOI] [PubMed] [Google Scholar]
- McAndrew R. P. et al. Structure of a three-domain sesquiterpene synthase: A prospective target for advanced biofuels production. Structure 19, 1876–1884 (2011). [DOI] [PubMed] [Google Scholar]
- Koeksal M., Hu H., Coates R. M., Peters R. J. & Christianson D. W. Structure and mechanism of the diterpene cyclase ent-copalyl diphosphate synthase. Nat. Chem. Biol. 7, 431–433 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L. A. & Sternberg M. J. E. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 4, 363–371 (2009). [DOI] [PubMed] [Google Scholar]
- Christianson D. W. Structural biology and chemistry of the terpenoid cyclases. Chem. Rev. 106, 3412–3442 (2006). [DOI] [PubMed] [Google Scholar]
- Dougherty D. A. Cation-pi interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp. Science 271, 163–168 (1996). [DOI] [PubMed] [Google Scholar]
- Jenson C. & Jorgensen W. L. Computational investigations of carbenium ion reactions relevant to sterol biosynthesis. J. Am. Chem. Soc. 119, 10846–10854 (1997). [Google Scholar]
- Van Duyne G. D., Standaert R. F., Karplus P. A., Schreiber S. L. & Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229, 105–124 (1993). [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. & Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol., Pt A 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- Brunger A. T. Assessment of phase accuracy by cross validation: The free R value. Methods and applications. Acta Crystallogr. D. 49, 24–36 (1993). [DOI] [PubMed] [Google Scholar]
- Adams P. D. et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P. & Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- Brunger A. T. et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. 54, 905–921 (1998). [DOI] [PubMed] [Google Scholar]
- Larkin M. A. et al. Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007). [DOI] [PubMed] [Google Scholar]
- Durrant J. D. et al. Non-bisphosphonate inhibitors of isoprenoid biosynthesis identified via computer-aided drug design. Chem. Biol. Drug. Des. 78, 323–332 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb M. R. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc. Natl. Acad. Sci. U. S. A. 89, 4884–4887 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information