

Abstract
Quantitative mass spectrometry has become central to the field of proteomics and metabolomics. Selected reaction monitoring is a widely used method for the absolute quantification of proteins and metabolites. This method renders high specificity using several product ions measured simultaneously. With growing interest in quantification of molecular species in complex biological samples, confident identification and quantitation has been of particular concern. A method to confirm purity or contamination of product ion spectra has become necessary for achieving accurate and precise quantification. Ion abundance ratio assessments were introduced to alleviate some of these issues. Ion abundance ratios are based on the consistent relative abundance (RA) of specific product ions with respect to the total abundance of all product ions. To date, no standardized method of implementing ion abundance ratios has been established. Thresholds by which product ion contamination is confirmed vary widely and are often arbitrary. This study sought to establish criteria by which the relative abundance of product ions can be evaluated in an absolute quantification experiment. These findings suggest that evaluation of the absolute ion abundance for any given transition is necessary in order to effectively implement RA thresholds. Overall, the variation of the RA value was observed to be relatively constant beyond an absolute threshold ion abundance. Finally, these RA values were observed to fluctuate significantly over a 3 year period, suggesting that these values should be assessed as close as possible to the time at which data is collected for quantification.
Keywords: Ion Abundance Ratios, Relative Abundance, Selected-Reaction Monitoring, Protein Quantification, PC-IDMS
Introduction
The relevance and use of LC-MS/MS for absolute quantification continues to be at the forefront of biological mass spectrometry. Improvements in methodology have allowed for proteins and metabolites to be routinely quantified using internal standards1-5. Increasing sample complexity and scale has put more demand on instruments and investigators to provide high-throughput quantification. This demand has required optimization of targeted LC-MS/MS methods in order to allow data to be collected and analyzed rapidly and reliably6,7. An obstacle that has become a major concern in targeted proteomics is the confidence in the specificity by which a peptide or other biological molecule is identified as well as the accuracy and precision of the absolute value obtained for a given experiment. With biological samples often containing many isobaric species, the coupling of LC to the mass spectrometer has become a powerful tool, allowing separation and quantification over a hundred proteins in a single run using gradient elution6,8. For high-throughput quantification the gradient is often minimized to reduce analysis time. However, this time advantage must be balanced with the ability to efficiently separate isobaric species of interest as a faster gradient elution further increases the probability of co-eluting contamination of product ion spectra9. This is of particular concern when using low resolving power instruments such as triple quadrupole instruments, which are the most common implementation for selected reaction monitoring (SRM) and absolute quantification assays.
Protein cleavage-isotope dilution mass spectrometry (PC-IDMS) is widely used for the absolute quantification of peptides and proteins1,3,6,10,11. A PC-IDMS assay typically involves triple quadrupole technology and selected reaction monitoring. A known amount of isotopically labeled tryptic peptide is spiked into a sample containing native tryptic peptides. This mixture is then analyzed by LC-MS/MS in which a list of precursor ions are fragmented at their optimized collision energies and their corresponding product ion abundances monitored. The resulting peak area of each transition can then be integrated and the ratio of the native peptide signal to the stable isotope labeled peptide can then be used to quantify the peptide/protein of interest. The accuracy with which a species can be quantified is highly dependent on the purity of these transitions.
Fundamentally, measurements require a signal-to-noise ratio ≥10 to obtain a useful signal which can reliably quantified. The way this noise is determined is often based on the variation in the absolute signal between replicate injections, which could be attributed to other factors including electrospray variability. Additionally, the nature of advanced instrumentation, modern electron multipliers and the specificity of tandem mass spectrometry using triple quadrupole technology has greatly reduced noise due to electronic sources as well as much of the chemical background noise12. Thus, it seems appropriate to simplify the measurement and focus on the role of absolute abundance. Furthermore, signal-to-noise is often evaluated in a single sample but not revisited in subsequent samples used for quantitative analysis. This source of variation is often overlooked when collecting large data sets; however, it is crucial to assessing whether each individual piece of data is appropriate for quantification. There are many methods to separate noise and chemical background from signal due to the analyte of interest13-15. Many of these procedures are quite cumbersome, non-automated and impractical for large data sets. To date, evaluation of product ion signal purity by a standardized method has remained elusive in the field of mass spectrometry.
Recently, novel bioinformatic tools have been shown to aid in the optimization of SRM assays16. Methods for evaluation of product ion purity have been of particular interest for quantitative mass spectrometry. Ion abundance ratio assessments were introduced to alleviate some of the issues regarding confirmation of specificity and contamination of product ions6,17-19. The method allows the experimenter to compare experimental data to an unlabeled or stable-isotope labeled internal standard for which the respective abundance of each product ion from a given precursor at particular collision energy, is accepted as the true value. If the experimental values deviate significantly from this true value, the product ion is deemed “contaminated” and is not used for quantification. This allows one to ensure that the product ions used for peak area integration and quantification are the species of interest, preventing potential bias in the quantification. The thresholds by which these RA values are implemented to evaluate experimental data in order to determine whether a value is significantly different, vary widely. Many of these established thresholds are empirical or arbitrary. Previous work has based these thresholds on the RA value of the transition being evaluated, however6, there is little analytical justification that this provides adequate information for evaluation of product ion purity.
Data collected over a three year period reveals that these ion ratios and their variance are not constant over time. Development of a systematic and analytical approach to implementing RA thresholds, which can be applied to transitions based on their absolute peak area, is presented. This approach was applied and validated in an absolute quantification experiment using protein obtained from stem differentiating xylem tissue of the model woody plant Populus trichocarpa. This study demonstrates the critical importance of using appropriate RA thresholds to evaluate experimental data as well as the frequent reassessment of RA value during large scale studies, which compare absolute quantification data over long periods of time.
Experimental
Materials
Unless otherwise stated, all reagents were purchased from Sigma-Aldrich (St. Louis, MO). All solvents were HPLC-grade from Honeywell Burdick & Jackson (Muskegon, MI)
SDX protein extraction and Filter Aided Sample Preparation
Stem differentiating xylem tissue was collected from three six-month-old Populus trichocarpa (genotype Nisqually-1), grown in a greenhouse as previously described20. Protein extracts were prepared from each tree, by grinding 3 g of SDX in liquid nitrogen then homogenizing the cells (2 min, on ice) in 15 mL of extraction buffer containing: 50 mM Bis-Tris (pH 8.0), 20 mM sodium ascorbate, 0.4 M sucrose, 100 mM NaCl, 5 mM DTT, and 10% (w/w) polyvinylpolypyrrolidone. After removing the cell debris by centrifugation (3,000 × g, 4 °C, 15 min – twice), the protein concentration was measured using a Coomassie Plus Bradford assay (Thermo Scientific, Rockford, IL) prior to storage at -80°C.
Filter-aided sample preparation was performed as described previously6. All solutions comprised of 50 mM Tris buffer at pH 8. Protein extracts were incubated for 30 min. at 56°C in a 2-fold dilution of 8 M Urea and 100 mM dithiothreitol to denature and reduce the protein. The sample was then alkylated using 200 mM iodoacetamide for 1 hour at 37°C. 100 μg of each sample was then added to a 10 kDa MW cutoff filter (EMD Millipore, Billerica, MA). Samples were washed 3× for 15 minutes at 14,000 × g with digestion buffer containing 2 M Urea and 10 mM CaCl2. Samples were then digested with 20 μg of bovine unmodified trypsin in 100 μL for 8 hours at 37°C. Additionally, 2 nmoles of a SIL peptide cocktail containing all peptides being measured were added concurrent with digestion. Digestion was then quenched using 50 μL of 1% formic acid and 0.0001% zwittergent 3-16 (Calbiochem, La Jolla, CA). Peptides were then eluted by centrifugation at 14,000 × g for 15 min.. 400 μL of 1% formic acid and 0.0001% zwittergent was added and centrifuged again to ensure adequate recovery. Peptides were then stored at -80°C prior to analysis.
Stable Isotope-labeled Peptide Standards and Transition Characterization
Stable isotope-labeled (SIL) C13N15 peptides were synthesized by the Mayo Clinic Proteomics Research Center (Rochester, MN). All 23 peptides were used as received with the exception of C3H3.125-134 and CCoAOMT3.217-232. These two cysteine containing peptides were carbamidomethylated as previously described6. SIL peptides stock solutions were produced by dissolving ∼2 mg in water and aliquots of 36 pmol/vial were dried down and stored at -20°C. Absolute concentrations were previously confirmed by spectrophotometry using Scope's method6,21. Collision energy optimization experiments were performed previously for the 6 most abundant product ions for each peptide as described using Skyline (v.1.4.0.4222) in which methods were developed, testing a range of collision energies and choosing the energy for maximum total product ion abundance for each peptide6,22,23.
LC-MS/MS Analysis
Liquid chromatography was performed using a nanoLC-2D system equipped with an AS1 autosampler and a cHiPLC-nanoflex system (Eksigent, Dublin, CA), which was coupled to a TSQ Vantage Triple stage quadrupole mass spectrometer (Thermo Scientific, San Jose, CA) using a 10 μm i.d. SilicaTip ESI emitter (New Objective, Woburn, MA). Selected reaction monitoring (SRM) analysis was performed as previously described in the development of a multiplexed assay for quantification of monolignol-pathway enzymes6. The entire list of peptides and transitions is provided (Table S1) along with their optimized collision energies. Mobile phases A and B were composed of water/acetonitrile/formic acid (98/2/0.2% and 2/98/0.2%, respectively). Each sample was loaded and desalted using a 9 μL metered injection of a 5 μL sample using 100% mobile phase A at 1.5 μL/min. Subsequently, peptides were eluted at 400 nL/min using a 22 min ramp from 5 to 38.5% mobile phase B, after which the column was washed with 95% mobile phase B for 5 min prior to re-equilibrating both columns to initial conditions. The column eluent was ionized using a 1400 V ESI potential and a capillary temperature of 200°C. SRM was performed using the EZ Method with a defined cycle time of 1.5 s, chromatographic filter of 30 s, Q1 and Q3 peak width (full-width half maximum) of 0.7 Da and a collision pressure of 1.5 mTorr Argon. Triplicate injection LC-MS/MS analysis of a fresh 20 nM SIL peptide mixture, prepared from a dried aliquot stored at -20°C, was performed at 5 separate time points (September 2011, August 2011, August 2012, May 2013 and May 2014) to characterize the relative abundance (RA) of each particular transition. Absolute signal was examined to ensure appropriate ion intensity for RA calculations. The same peptide mixture was then diluted (5×, 10×, 20×, and 50×) and injected to purposefully and systematically vary the absolute abundance of all transitions. SDX protein samples were analyzed by replicate injection LC-MS/MS using the same method of analysis with the exception of different peptide targets as well as implementation of a scheduled method for monitoring of transitions.
Raw LC-MS/MS data files were imported into Skyline where peak integration was performed after adjusting retention time windows to be consistent between injections. Reports were then exported into Excel, which contained peak area information. The peak area output for Skyline subtracts out the area due to background. The relative abundance of each transition was then determined. Data collected from analysis of SDX proteins was imported into an excel template where transition thresholds were implemented and manual verification of transition purity was performed. Data collected from diluted SIL mixtures was used to determine the deviation of the RA as a function of absolute peak area. Variance in RA values obtained from triplicate injections from 9/2011 to 12/2011, 2011 to 2012, 2011 to 2013 and 2011 to 2014 were compared using a two-tailed F-Test at α = 0.05. Additionally, a two tailed (unequal variance) T-test (α = 0.05) was implemented to compare mean RA values and RSD values.
Defining Relative Abundance
The RA of a transition has been previously defined as the sum of all transitions measured for a given precursor minus the transition of interest, divided by the transition of interest6 (equation 1). This results in lesser abundant transitions having a greater RA value. This was formatted for convenience to generate only RA values > 1; however, this is counterintuitive as transitions with higher RA values are less abundant. We can perform a simple inverse manipulation to the previously defined relative abundance calculation such that the RA value is a ratio of the transition of interest divided by, the sum of all transitions minus the transition of interest (equation 2). In this manner more abundant transitions will have a higher RA value and lesser abundant, a lower RA value. Subtraction of the transition of interest from the sum total area in order to calculate the relative abundance of a transition was originally performed in order to prevent “contaminated” transitions from biasing both the numerator and denominator of the calculation, making it easier to flag transitions as being “contaminated”, particularly those transitions which comprise a large portion of the total area as this will dominate both the numerator and denominator. Alternatively, if we do not subtract the transition of interest from the total area (equation 3), our method becomes slightly less sensitive detecting a change in peak area (Figure S1). However, this effect is not significant in the practice of analyzing large quantitative data sets as the data here will show. Additionally, applying a fixed threshold when subtracting the area of the transition results in a bias against higher abundant transitions which will have tighter thresholds for allowed change in peak area (Figure S1). This is important since a change of 1% in percent peak area for a transition which comprises 10% of the total peak area will result in a 10% quantitative bias. A transition which comprises 90% of the total area would have to change by 9% in order to reach the same quantitative bias. Nevertheless, for relative abundance thresholds to be effective in flagging “contaminated” transitions, the peak area of a transition should be allowed to effect the quantitative bias of an experiment equally regardless of its relative abundance. If we exclude subtraction of the transition of interest in the denominator this results in a consistent allowed variation in the percent peak area for all transitions regardless of the proportion of total area for which they account (equation 3). For these reasons we have chosen to redefine relative abundance as the area of the transition divided by the sum total area of all transitions. This will be the “new” RA calculation referred to for all of the following experiments and the “old” method being that which was previously published (equation 1).
| (1) |
| (2) |
| (3) |
Previous work6 established thresholds for RA values based on binning RA values with transitions of higher RA (based on equation 1) having higher thresholds and transitions of lower RA having lower thresholds. These dynamic thresholds were done in order to prevent any single transition from causing a bias of greater than 10% in the quantitation. This method makes many assumptions and is not necessarily the most effective and practical approach. While it is true that the relative abundance of any single transition should be consistent for a given precursor at a particular collision energy, the absolute abundance is not. Transitions with a high RA may have a low absolute abundance. This would result in a higher variation than the same transition with a higher absolute abundance. Similarly, a transition with a low relative abundance may have a high absolute abundance. This would result in less variation than expected. Thus, this would result in researchers unintentionally and inappropriately allowing for more or less variability in their measurements.
Results and Discussion
Examining the Dynamic of Relative Abundance over Time
The absolute relative abundance values from triplicate injection of 24 peptides yielded significantly different results from 2011, 2012, 2013 and 2014 compared to their established “expected” values in 2011 for several peptides and their corresponding transitions. Figure 1 shows the relative abundance of each transition for peptide AAGIDSGFFELPK from a single injection of SIL peptide mixture LC-MS/MS analysis from 2011 and 2013. A significant decrease (α = 0.05) in relative abundance of y2 (244.1656+) and a4 (292.2093+) was observed. Meanwhile, a corresponding significant increase (α = 0.05) in the relative abundance of y10 (1167.5681+) and y9 (1052.5411+) was also observed.
Figure 1.
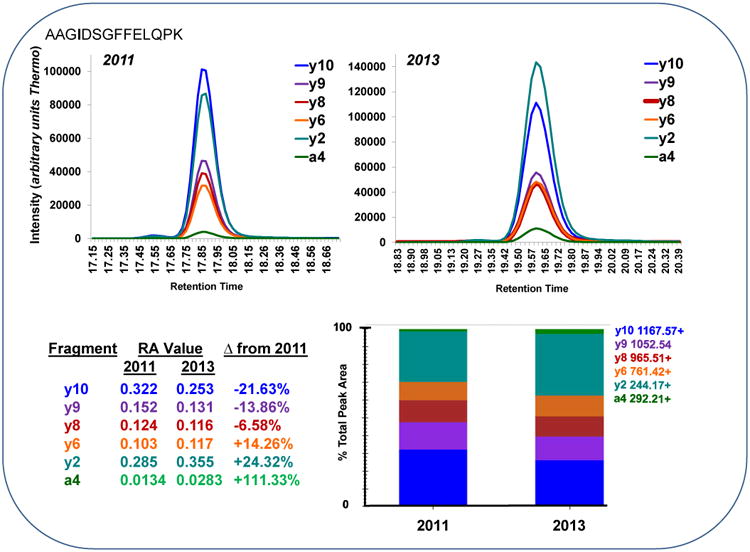
An extracted ion chromatogram is shown for the peptide AAGIDSGFFELPK for six transitions (a4, y2, y6, y8, y9 and y10) from a single LC-MS/MS run from 2011 and 2013. A significant increase (α = 0.05) in relative abundance of y2 (244.1656+) and a4 (292.2093+) was observed. Meanwhile, a corresponding significant decrease (α = 0.05) in the relative abundance of y10 (1167.5681+) and y9 (1052.5411+) was also observed.
Figure 2A shows a summary of the significant differences observed for the peptides and transitions analyzed from 2011 to 2012, 2013, and 2014. Of the 21 peptides analyzed by LC-MS/MS in 2012, 50% of these had a transition with a significantly different RA value and ∼11% of all transitions experienced a statistically significant change with an average change in the RA value of 15%. From the data analyzed in 2013, ∼82% of peptides had at least 1 transition with a significantly different RA value and 42% of all transitions experienced a significant change with an average change of ∼12%. In 2014 ∼96% of peptides analyzed had a transition with a significantly different RA value and 64% of all transitions had changed significantly with an average change of ∼12%.
Figure 2.

A: Data is compared from 2011 through 2014. LC-MS/MS analysis of 20 nM peptide mixture, n=3 injections of 24 peptides total. Data shown from December of 2011 revealed significant changes in the RA value for the majority of peptides and transitions with no significant changes to report in the variance. The majority of peptides measured in 2012-2014 exhibited a significantly higher variance (α = 0.05) than in 2011. The majority of transitions in 2012 also exhibited a higher variance. The number of peptides and transitions with a significantly different RA value (α = 0.05) from 2011 increased from 2012 to 2014 as fewer peptides and transitions experienced a higher variance. The average deviation for RA values that changed significantly was similar from year to year with an approximate change of 10% per year.
B: Transitions with significantly different (α = 0.05) relative abundance values from 2011-2012 and 2011-2013 (n=52 data points) were plotted versus their corresponding product ion m/z. Lower m/z values showed an increase in relative abundance while higher m/z values decreased in relative abundance. The R2 of -0.48 represents a fair inverse correlation between m/z and the observed deviation from expected RA.
Of all transitions experiencing a significant change from 2011 to 2012/13, those which RA value decreased, tended to be product ions of higher m/z values while the transitions experiencing an increase in their RA value, tended to be product ions of lower m/z values (Figure 2B). It should be noted that when we include all the data from 2011-14 that this correlation is not seen, suggesting many other variables are influencing these observed changes. This observed correlation does suggest that a portion of the observed change in RA values could be due to a decrease in higher m/z product ion transmission through Q3. Other potential factors were investigated; however, transitions with significantly different RA values were not biased towards a particular precursor m/z range or to the difference in precursor to product m/z of the transition.
In order to further investigate the time scale with which RA values fluctuate data obtained 3 months apart in 2011 was also compared. Of the data collected in Dec. 2011, 96% of peptides had a transition which changed significantly from Sept 2011 and ∼67% of all transitions had significantly changed with an average change of ∼10%. Overall, this suggests that the magnitude of change in RA is relatively consistent across time points (∼10%). At any point the proportion of these which are significant seems to depend on the magnitude of the variance at a given time as this will impact which particular transitions were determined to have a significant change in their RA. Thus, it is plausible that some of these changing RA values could simply be an artifact of changing in variability of the measurements. In addition, the data also suggest that observed changes in RA values can occur within a period of time as short as 3 months however, these observed changes in RA values do not seem to become significantly greater from a 3 month interval as compared to a year. Nonetheless, significant changes in the RA values from 2011-2014 (Table S3) suggests that to be an effective metric for validation of experimental data, the RA values must be reassessed frequently to be a relevant, evaluative tool.
In addition to the change in the absolute RA value, an increased variance was common for the RA values for peptides in 2011, 2012, 2013 and 2014 (Figure 2A). Between the three month span in 2011 none of the transitions had an increased variance. The variance of some transitions actually decreased significantly. Meanwhile, the majority of transitions experienced a higher variance in 2012 (73%). In 2013, only 38% of transitions analyzed had a significantly higher variance. In 2014, 34% percent of transitions had a significantly higher variance. These data suggest that variance in the RA values can decrease or increase from year to year but overall will tend towards higher variance over time.
Several potential variables inherent to the peptides analyzed were investigated in order to determine whether these possible sources of variability were analyte specific or systematic. To ensure that the change in RA and variation of the measurements was not simply a result of differing absolute signals over the 3 years, the percent change in peak area of each transition was plotted against the change in percent relative standard deviation (RSD) (Figure S2). No relationship between the two was observed implying that these observed changes are not due to a change in absolute abundance of the data used to calculate RA values. Additionally, the mean relative standard deviation in the RA value for all transitions from 2011-2014 were calculated, grouped and plotted according to their precursor m/z (Figure S3). It was observed that transitions do vary in terms of their RSD as well as in the variability of their RSD from year to year. Within a peptide transitions can vary as well suggesting that this variation is likely transition specific. This could be related to variability in the kinetics of product ion formation for each particular transition as this variability will be unique to the transition in question at particular collision energy.
Further investigation of this variability with respect to the peptide level was also investigated. The relative standard deviation of all peptides was calculated and compared to peptide specific properties. Factors investigated included the hydrophobicity of the peptide, molecular weight, isoelectric point as well as m/z of the precursor. No relationship was observed between these peptide specific properties and the overall variance of the peptide. However, it is interesting to note that the peptide with the highest m/z and lowest PI, “IYPTVDPTMDPDYAEYLK”, also had the highest mean RSD as well as the RSD with the highest variance of all peptides investigated. This suggests that a portion of the variability could certainly be peptide specific in addition to being transition specific although it is unclear from the data what particular peptide/transition properties are influencing the variability in the relative abundance of particular product ions.
The lack of distinct patterns in the data suggest that there are many factors which are interacting and influential in dictating the observed variability in the RA of particular product ions in a given experiment at a given time. Overall fluctuation in performance of the mass spectrometer and dynamic internal instrument conditions are a likely source of this variability, particularly in the collision cell and in the transmission of product ions to the detector as these are the most probable variables which will influence the abundance of particular product ions. It should be noted that the quadrupoles in the instrument were cleaned and the electron multiplier was replaced prior to collecting the 2014 data. This seemed to have a slight effect on decreasing the observed variance as compared with 2012-2013. The variance was however, still significantly higher than that which was observed originally in 2011. This suggests factors other than charging and/or contamination are likely involved with this changing variance. These could be related to any aspect effected by overall instrument age and usage which could cause changes in the effective collision energy and transmission of product ions to the detector. Irrespective of the amount of time or the number of samples run in between experiments, it would be beneficial to the experimenter to reassess the RA and its variance as frequently and as close as possible to the time at which experimental data is collected in order to determine the expected threshold by which RA values should be evaluated in experimental data. Obtaining an accurate expected RA value and overall variance is crucial for confirming contamination or purity of transitions in MS/MS spectra. If the variance is underestimated or overestimated, this would lead to the inadvertent exclusion of useful data or inclusion of contaminated data respectively.
Relative Abundance and Absolute Abundance
Data collected from diluted peptide mixtures was analyzed for deviations from current expected transition RA values (2013) and plotted versus absolute peak area for each data point (Figure 3A). The deviation from the expected RA value was substantially greater for data points with lower absolute ion abundance. The observed trend suggested that this variation is reduced at higher absolute abundance. The y11++ ion 564.8486 m/z from the peptide DGIVSLGGPHIPLK (shown in red) was examined at various peak area values (Figure 3A). The deviation of the same transition varied widely depending on the absolute ion abundance. At low absolute ion abundance, the RA of the transition was much higher than expected (43%). At higher absolute ion abundance the RA value deviated very little from the expected value (1%). Higher absolute ion abundance was not a guarantee that the RA of the transition would fall near the expected value. While less common at intermediate absolute abundance values, the RA could still deviate quite significantly as it did for the y11++ ion (40%). This unexpected observed deviation at higher absolute ion abundances emphasizes the importance of including RA thresholds during data analysis as they are still capable of deviating quite significantly, which could greatly impact quantitation.
Figure 3.
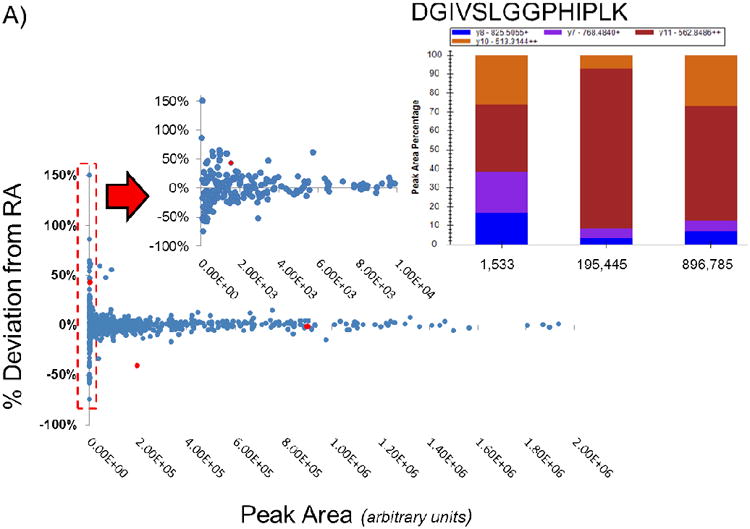
A: 684 data points gathered from injecting varying amounts of a 21 SIL peptide mixture are plotted based on the absolute peak area against the percent deviation from their expected RA value (2013). At low absolute abundance the deviation from the expected RA is highly variable. As the absolute abundance increases, an observed decrease in the deviation from the expected RA value is observed. The peptide DGIVSLGGPHIPLK is shown as an example of a peptide which has the same y11++ at different absolute abundances and with varying deviations from the expected RA value.
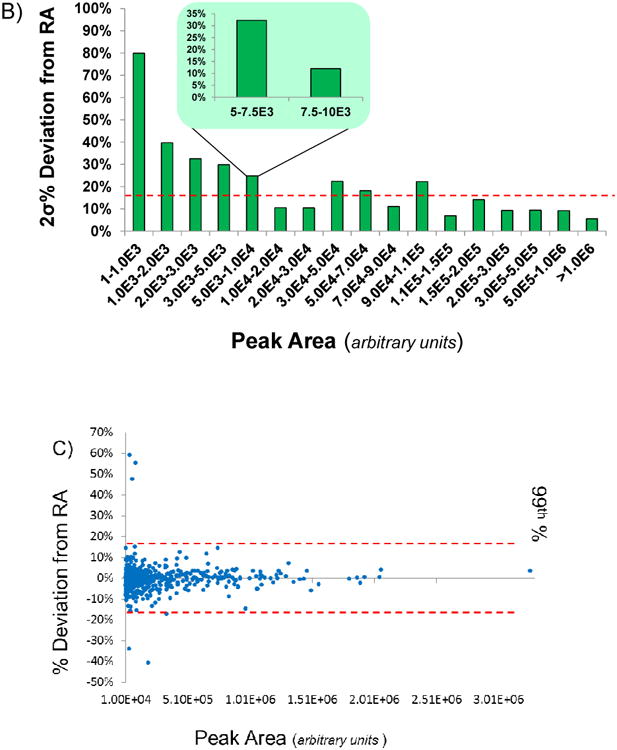
B: Data points gathered from injecting varying amounts of standard peptide mixture were binned based on their absolute area and plotted according to 2σ of each bin. At an absolute peak area above 10,000, the deviation of the RA value is observed to reach a constant of approximately 10-20%. Within the range of 5,000 – 10,000 the standard deviation of the measurements drop significantly at 7,500. The final threshold of 17.24% is indicated by the dotted line.
C: Data points with a peak area greater than 10,000 are examined in this plot. The 99th percentile of the data is shown by the red dotted line at 17.24%.
The data was further sorted and binned (ensuring a minimum of 30 data points in each bin) based on absolute peak area and 2σ of the data was examined (Figure 3B). The 2σ deviation from expected RA values was significantly greater for data points with peak areas less than 1,000. For data points between 1,000 and 5,000 this deviation decreased from 40% to 30%. Beyond a peak area of 10,000, variation in the data was relatively constant (2σ ∼10-20%), excluding two (3.0E4 - 4.0E4 and 9.0E4 - 1.1E5) that were biased by a few outliers. Further binning of the 5,000-10,000 bin resulted in an observed distinct change in the percent deviation from expected RA. Between 5,000-7,500 2σ was ∼30%. Between 7,500 and 10,000 this decreased to 12%. This suggested that a minimum absolute peak area is critically important in order to effectively determine expected RA values/thresholds which can be applied regardless of the RA value of the transition. The present data suggest that beyond a minimum peak area, the threshold variation allowed for any RA value should be the same. Below this minimum peak area, transitions can be included provided that they meet RA thresholds however, these points will be more inherent to variability due to their abundance.
In order to investigate an appropriate value for determining RA thresholds, the data with peak areas >10,000 was further examined (Figure 3C). The standard deviation of all data points with a peak area >10,000 was calculated to be 6.68% and the 99th percentile was 17.24%. This suggests that 17.24% would be an appropriate threshold. Additionally, the outliers discussed previously which were present in the higher absolute abundance bins become more obvious in this plot where the data appears to be tightly distributed between + or − 15% of the expected RA value. This also implies that the absolute abundance and threshold RA values are an adequate metric for determining the presence of variation in the data outside of 15-20% and that all transitions are subject to this specific variation. These data indicate that RA thresholds implemented should be no less than 15% and no greater than 20%, regardless of the RA value of any given transition to ensure inclusion of all useful data while excluding contaminated transitions. The data supports the hypothesis that relative product ion abundance variation is largely dependent on the absolute ion abundance.
For the case of absolute abundance vs. deviation from expected RA value there was no observable pattern between peptide/transitions and their deviation from expected RA values. Overall, all peptides and transitions behaved similarly in their observed variance. While data from 2011-2014 does suggest that certain peptides/transitions appeared to have differing degrees of variance, this did not appear to effect the overall variance as a function of absolute abundance. It would be interesting to determine whether the overall variance for the same peptides/transitions change significantly over time. Peptide/transition specific data suggests this will change. Again, it is likely that this variation is specific and subject to variability itself for a given instrument and a given time.
Implementation of Relative Abundance Thresholds for Absolute Quantification Experiments
Data obtained in 2012 from the quantification of 23 peptides from 23 lignin proteins in 3 wild-type poplar trees was used as a template for comparing previous and newly established relative abundance thresholds. Previous thresholds were dependent on the RA value (equation 1) with RA values > 9 having a 50% threshold, RA values between 1 and 9 having a threshold of 25% and RA values < 1 having a 15% threshold based on the previously defined RA calculation. Alternatively, newly developed thresholds were set at 17.24% based on the data collected and implemented only for transitions having a peak area > 10,000. Transitions with peak areas <10,000 were included in the data providing they met the RA thresholds. For the following data discussed (collected 10/2012), the original RA values (2011) were used as the expected value for all peptides and transitions analyzed.
Table 1 shows the results of absolute quantification data processed using the two different methods across replicate injection of 3 samples. The relative standard deviation for previously used (old) RA thresholds was compared to that obtained by using new RA thresholds. Overall, the RSD was not significantly different between methods. Using both methods the %RSD was ∼5.5% with a similar standard deviation of ∼8.6% (Figure 4A). The most notable difference between the methods was in the number of quantifiable points. For example, in the case of the peptide DGIVSLGGPHIPLK, the data point was omitted from the WT-2 data using the old method but was included in the new method. For this peptide, the threshold of 15% for was used for one of the transitions in the old method, preventing the quantification of the peptide. The new RA calculation with a threshold of 17.24% allowed for use of this transition as well as quantification of the peptide DGIVSLGGPHIPLK and resulted in an RSD of 10.21%. Using new RA thresholds, all but one of the 69 data points was quantified by replicate analysis (Figure 4B). Using old RA thresholds, 7 data points were not quantifiable by replicate analysis. This implies that the new method of calculating RA values is appropriate for quantitation of large data sets. Additionally, the thresholds used are not too stringent and do not exclude useful data, allowing for quantification of data which by previous methods would be flagged as “not quantifiable”. Many of the additional points quantifiable by the new RA calculation/thresholds had an RSD less than the biological variation between wild type trees (Table S1). The data is not only quantifiable but is also useful in being able to determine biological differences between the samples. With respect to LC-MS based quantitative assays our goal is to obtain an RSD of 10% however, the biological variation is often greater as shown here. As a result, it is often not the necessary to achieve this level of agreement in the data in order to obtain biologically significant information. More pertinent to these current studies is the ability to analyze large quantitative data sets, remove transitions which increase the variability in our measurements without manual inspection of the data while still maintaining the ability to quantify all useful data.
Table 1.
Percent RSD is shown for 23 peptides using “Old” and “New” methods of implementing RA thresholds for replicated injections of 3 wild type samples from stem differentiating xylem tissue of Populus trichocarpa.
| Peptide | %RSD WT-1 | %RSD WT-2 | %RSD WT-3 | |||
|---|---|---|---|---|---|---|
| Old | New | Old | New | Old | New | |
| FDIGTLLGLIEK | 0.03% | 0.03% | 5.80% | 5.80% | 2.86% | 2.86% |
| FEIGSLLGLIEK | 7.26% | 8.64% | 1.22% | 0.18% | 2.03% | 1.19% |
| VCTLELFSPK | 10.67% | 5.95% | 9.38% | 0.96% | 3.20% | 3.98% |
| DYFVDER | 14.84% | 2.31% | 60.05% | 55.72% | 21.83% | 16.56% |
| DYFVEER | NQ | 14.27% | NQ | 38.97% | 39.04% | 12.64% |
| GGILGLGGVGHMGVK | 0.46% | 0.46% | 0.06% | 0.06% | 4.08% | 4.08% |
| FLEPGVPDFK | 0.15% | 1.46% | 1.18% | 1.55% | 1.77% | 1.77% |
| FMKPGVPDFK | 2.95% | 2.95% | 5.51% | 5.51% | 8.88% | 8.88% |
| FLKPGVPDFK | 14.81% | 12.85% | 0.85% | 0.85% | NQ | 2.54% |
| ENYELGLPVIQK | 0.14% | 0.14% | 2.26% | 2.41% | 2.22% | 1.33% |
| VGGLIGYDNTLWNGSVVAPPDAPMR | 6.55% | 7.41% | 2.25% | 3.08% | 7.59% | 7.59% |
| VGGLIGYDNTLWNGSVVAPADAPMR | 4.92% | 4.92% | 2.52% | 2.52% | 4.68% | 4.68% |
| FLSMLLK | 3.35% | 4.09% | 0.63% | 0.63% | 0.96% | 0.96% |
| DLGFEFTPVK | 1.57% | 1.18% | 1.30% | 0.41% | 2.55% | 5.23% |
| AGPGAFLSTSEIASHLPTK | 4.10% | 4.10% | 2.68% | 2.68% | 2.06% | 2.06% |
| SALDFLELQPDLSALVR | 0.78% | 2.06% | 0.63% | 1.04% | 0.47% | 0.08% |
| SALDYLELQPDLSALVR | 2.86% | 1.16% | 3.35% | 3.75% | 1.61% | 1.09% |
| EELGTGLLTGEK | 8.78% | 11.65% | 11.37% | 14.42% | NQ | 17.92% |
| EELGTILLTGEK | 6.33% | 0.47% | 4.38% | 3.28% | 1.77% | 2.80% |
| EELGTVLLTGEK | 2.66% | 5.60% | 3.55% | 0.25% | 6.79% | 5.31% |
| IGSFEEELK | 4.70% | 5.19% | NQ | 14.48% | NQ | NQ |
| DGIVSLGGPHIPLK | 11.17% | 0.50% | NQ | 10.21% | 7.19% | 0.38% |
| AFEIIEDLR | 0.07% | 0.07% | 0.33% | 0.33% | 0.53% | 0.53% |
Figure 4.

A: Comparative plots showing the relative standard deviation for 23 peptides in 3 samples of wild-type P. trichocarpa stem differentiating xylem tissue for two methods of implementing RA thresholds. This plot shows a comparison of “Old” and “New” RA calculation methods with no significant difference between the RSD of the measurements made.
B: The percent of quantifiable data points was shown to increase using the new RA calculation and thresholds. Six additional data points out of sixty-nine were quantifiable using new methods.
In order to demonstrate the effectiveness of this new method in identifying contaminated transitions within a quantitative data set, a peptide with a visible contamination was selected for comparative analysis. Figure 5 shows an extracted ion chromatogram for the peptide EELGTGLLTGEK with distinct contamination of the y9+ transition. Table 2 shows the percent change in RA values for each transition using old and new RA calculation methods along with their corresponding thresholds. The new RA calculation correctly identifies the y9+ contamination while the old method incorrectly identifies the y7+ transition as being contaminated. This provides validation for this new approach to calculating RA values and thresholds. Additionally, it also highlights the shortcoming of the previously used method which results in a bias against higher abundant transitions as it did in this case, incorrectly identifying the y7+ ion, the most abundant transition which comprised 32% of the total area.
Figure 5.

An extracted ion chromatogram of all transitions corresponding to the peptide EELGTGLLTGEK for the WT-2 sample with visual contamination of the y9+ is shown.
Table 2.
The percent change in RA values for all 6 fragment ions from the peptide EELGTGLLTGEK are shown along with the corresponding thresholds for old and new RA calculation methods. The new RA calculation method shows the correctly identified y9+ fragment as being the contaminated transition while the old method incorrectly identifies the y7+ ion as the contaminated transition.
| Old Method RA | |||||
| Fragment | % Peak Area | Theoretical RA | Experimental RA | Δ%RA | Threshold |
| y9+ | 28% | 2.540 | 1.919 | -24.45% | 25.00% |
| y7+ | 32% | 2.119 | 2.686 | 26.76% | 25.00% |
| y6+ | 4.7% | 20.268 | 22.847 | 12.72% | 50.00% |
| y5+ | 12.5% | 7.000 | 7.444 | 6.34% | 25.00% |
| y4+ | 12.7% | 6.878 | 6.568 | -4.50% | 25.00% |
| b2+ | 9.8% | 9.208 | 9.679 | 5.12% | 50.00% |
| New Method RA | |||||
| Fragment | % Peak Area | Theoretical RA | Experimental RA | Δ%RA | Threshold |
| y9+ | 28% | 0.28 | 0.343 | 21.28% | 17.24% |
| y7+ | 32% | 0.32 | 0.271 | -15.38% | 17.24% |
| y6+ | 4.7% | 0.047 | 0.042 | -10.81% | 17.24% |
| y5+ | 12.5% | 0.125 | 0.118 | -5.26% | 17.24% |
| y4+ | 12.7% | 0.127 | 0.132 | 4.09% | 17.24% |
| b2+ | 9.8% | 0.098 | 0.094 | -4.41% | 17.24% |
Conclusion
Development of methodologies for the automated processing of SRM data as well as assigning a statistical probability to the identification and quantification of targets of interest, remains a work in progress. Through this study we have demonstrated an analytical approach to implementing thresholds for the estimation of relative abundance of product ions. In examining the variation in the relative abundance of 21 peptides we were able to demonstrate a strong correlation and dependence on absolute peak area. These results suggest a minimum peak area (in our case >10,000) is necessary in order to obtain the most accurate determination of the RA value for a transition as well as the expected variability in a given experiment. The variation of data above this minimum peak area exhibited similar behavior with a consistent standard deviation and 99% of the data collected was observed to be within 17.24% of the expected RA value. The data supports that this is an effective and analytical approach to determine RA thresholds and that 17.24% is an appropriate threshold to apply to our quantitative data. Furthermore, the significant change observed in the relative abundance for many of the transitions from 2011 - 2014 emphasizes the importance of reevaluating relative abundance values prior to each quantitative experiment when conducting large scale and long term absolute quantification experiments in order to ensure the most appropriate RA values and expected variation of those values is being implemented. Finally, in applying these thresholds to absolute quantification data obtained from a complex biological sample, these thresholds can be used to successfully quantify peptides/proteins as well as filter out contaminated transitions. Following this simple analytical approach will result in a more automated, consistent and systematic method for processing absolute quantification data. Overall, the study herein contributes to the development of analytical tools for verification of product ion purity in absolute quantification.
Supplementary Material
Acknowledgments
A special thanks to Quanzi Li, Jie Liu, Jack P. Wang, Rui Shi, and Ying-Chung Lin for the preparing SDX protein extracts utilized in this study as well as Zhichang Yang for collecting the LC-MS/MS quantitative data from these samples. The authors gratefully acknowledge the NIH/NCSU Molecular Biotechnology Training Program (Grant 5T32GM00-8776-08) and The National Science Foundation Plant Genome Research Program (Grant DBI-0922391) for funding this work.
References
- 1.Williams DK, Muddiman DC. Absolute quantification of C-reactive protein in human plasma derived from patients with epithelial ovarian cancer utilizing protein cleavage isotope dilution mass spectrometry. J Proteome Res. 2009;8:1085–90. doi: 10.1021/pr800922p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrews Kingon GL, Petitte JN, Muddiman DC, Hawkridge AM. Multi-peptide nLC-PC-IDMS-SRM-based assay for the quantification of biomarkers in the chicken ovarian cancer model. Methods. 2013;61:323–30. doi: 10.1016/j.ymeth.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnidge DR, Goodmanson MK, Klee GG, Muddiman DC. Absolute quantification of the model biomarker prostate-specific antigen in serum by LC-Ms/MS using protein cleavage and isotope dilution mass spectrometry. J Proteome Res. 2004;3:644–52. doi: 10.1021/pr049963d. [DOI] [PubMed] [Google Scholar]
- 4.Shuford CM, Poteat MD, Buchwalter DB, Muddiman DC. Absolute quantification of free glutathione and cysteine in aquatic insects using isotope dilution and selected reaction monitoring. Anal Bioanal Chem. 2012;402:357–66. doi: 10.1007/s00216-011-5416-2. [DOI] [PubMed] [Google Scholar]
- 5.Hembrough T, Thyparambil S, Liao WL, Darfler MM, Abdo J, Bengali KM, Hewitt SM, Bender RA, Krizman DB, Burrows J. Application of selected reaction monitoring for multiplex quantification of clinically validated biomarkers in formalin-fixed, paraffin-embedded tumor tissue. J Mol Diagn. 2013;15:454–65. doi: 10.1016/j.jmoldx.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Shuford CM, Li Q, Sun YH, Chen HC, Wang J, Shi R, Sederoff RR, Chiang VL, Muddiman DC. Comprehensive quantification of monolignol-pathway enzymes in populus trichocarpa by protein cleavage isotope dilution mass spectrometry. J Proteome Res. 2012;11:3390–404. doi: 10.1021/pr300205a. [DOI] [PubMed] [Google Scholar]
- 7.Loziuk PL, Wang J, Li Q, Sederoff RR, Chiang VL, Muddiman DC. Understanding the role of proteolytic digestion on discovery and targeted proteomic measurements using liquid chromatography tandem mass spectrometry and design of experiments. J Proteome Res. 2013;12:5820–9. doi: 10.1021/pr4008442. [DOI] [PubMed] [Google Scholar]
- 8.Percy AJ, Chambers AG, Yang J, Hardie DB, Borchers CH. Advances in multiplexed MRM-based protein biomarker quantitation toward clinical utility. Biochimica et biophysica acta. 2013 doi: 10.1016/j.bbapap.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Sherman J, McKay MJ, Ashman K, Molloy MP. How specific is my SRM?: The issue of precursor and product ion redundancy. Proteomics. 2009;9:1120–3. doi: 10.1002/pmic.200800577. [DOI] [PubMed] [Google Scholar]
- 10.Heudi O, Barteau S, Zimmer D, Schmidt J, Bill K, Lehmann N, Bauer C, Kretz O. Towards absolute quantification of therapeutic monoclonal antibody in serum by LC-MS/MS using isotope-labeled antibody standard and protein cleavage isotope dilution mass spectrometry. Anal Chem. 2008;80:4200–7. doi: 10.1021/ac800205s. [DOI] [PubMed] [Google Scholar]
- 11.Arsene CG, Ohlendorf R, Burkitt W, Pritchard C, Henrion A, O'Connor G, Bunk DM, Guttler B. Protein quantification by isotope dilution mass spectrometry of proteolytic fragments: cleavage rate and accuracy. Anal Chem. 2008;80:4154–60. doi: 10.1021/ac7024738. [DOI] [PubMed] [Google Scholar]
- 12.Boyd B, Basic C, Bethem R. Trace quantitative analysis by mass spectrometry. John Wiley & Sons; Chichester, West Sussex, England ; Hoboken, N.J.: 2008. [Google Scholar]
- 13.Marengo E, Robotti E, Gosetti F, Zerbinati O, Gennaro MC. Evaluation of signal and noise and identification of a suitable target function in the tuning of an ESI ion trap mass spectrometer by multivariate pattern recognition tools. J Am Soc Mass Spectrom. 2009;20:1859–67. doi: 10.1016/j.jasms.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Payne TG, Southam AD, Arvanitis TN, Viant MR. A signal filtering method for improved quantification and noise discrimination in fourier transform ion cyclotron resonance mass spectrometry-based metabolomics data. J Am Soc Mass Spectrom. 2009;20:1087–95. doi: 10.1016/j.jasms.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Wang SC, Huang CM, Chiang SM. Improving signal-to-noise ratios of liquid chromatography-tandem mass spectrometry peaks using noise frequency spectrum modification between two consecutive matched-filtering procedures. J Chromatogr A. 2007;1161:192–7. doi: 10.1016/j.chroma.2007.05.080. [DOI] [PubMed] [Google Scholar]
- 16.Brusniak MY, Chu CS, Kusebauch U, Sartain MJ, Watts JD, Moritz RL. An assessment of current bioinformatic solutions for analyzing LC-MS data acquired by selected reaction monitoring technology. Proteomics. 2012;12:1176–84. doi: 10.1002/pmic.201100571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Zeeuw RA. Substance identification: the weak link in analytical toxicology. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;811:3–12. doi: 10.1016/j.jchromb.2004.07.043. [DOI] [PubMed] [Google Scholar]
- 18.Kushnir MM, Rockwood AL, Nelson GJ, Yue B, Urry FM. Assessing analytical specificity in quantitative analysis using tandem mass spectrometry. Clin Biochem. 2005;38:319–27. doi: 10.1016/j.clinbiochem.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Sauvage FL, Gaulier JM, Lachatre G, Marquet P. Pitfalls and prevention strategies for liquid chromatography-tandem mass spectrometry in the selected reaction-monitoring mode for drug analysis. Clin Chem. 2008;54:1519–27. doi: 10.1373/clinchem.2008.105478. [DOI] [PubMed] [Google Scholar]
- 20.Shi R, Sun YH, Li Q, Heber S, Sederoff R, Chiang VL. Towards a systems approach for lignin biosynthesis in Populus trichocarpa: transcript abundance and specificity of the monolignol biosynthetic genes. Plant Cell Physiol. 2010;51:144–63. doi: 10.1093/pcp/pcp175. [DOI] [PubMed] [Google Scholar]
- 21.Scopes RK. Measurement of protein by spectrophotometry at 205 nm. Anal Biochem. 1974;59:277–82. doi: 10.1016/0003-2697(74)90034-7. [DOI] [PubMed] [Google Scholar]
- 22.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–8. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maclean B, Tomazela DM, Abbatiello SE, Zhang S, Whiteaker JR, Paulovich AG, Carr SA, Maccoss MJ. Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal Chem. 2010;82:10116–24. doi: 10.1021/ac102179j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.