

Abstract
Phage antibody technology can be used to generate human antibodies to essentially any antigen. Many therapeutic target antigens are cell surface receptors, which can be challenging targets for antibody generation. In addition, for many therapeutic applications, one needs antibodies that not only bind the cell surface receptor but that also are internalized into the cell upon binding. This allows use of the antibody to deliver a range of payloads into the cell to achieve a therapeutic effect. In this chapter we describe how human phage antibody libraries can be selected directly on tumor cell lines to generate antibodies that bind cell surface receptors and which upon binding are rapidly internalized into the cell. Specific protocols show how to: 1) directly select cell binding and internalizing antibodies from human phage antibody libraries; 2) screen the phage antibodies in a high throughput flow cytometry assay for binding to the tumor cell line used for selection; 3) identify the antigen bound by the phage antibody using immunoprecipitation and mass spectrometry; and 4) direct cell binding and internalizing selections to a specific tumor antigen by sequential selection on a tumor cell line followed by selection on yeast displaying the target tumor antigen on the yeast surface.
Keywords: Phage antibody, antibody internalization, targeted drug delivery, cell selection, flow cytometry, yeast display
Introduction
Selective expression of receptors on the surface of cancer cells has been exploited for the development of targeted cancer therapies. Antibodies targeting HER2 (Herceptin, Genentech) [1], EGFR (Cetuximab, ImClone) [2], CD20 (Rituxan, Genentech) [3], and CD52 (Alemtuzumab, Genzyme) [4] have been approved by the FDA for the treatment of cancer. These antibodies are ‘naked’ IgG and thus work either by directly interfering with normal growth factor signaling, induction of apoptosis or via elicitation of antibody dependent cellular cytotoxicity (ADCC) or complement dependent cytotoxicity (CDC) by the Fc portion of the antibody. Additional antibodies that interfere with normal receptor signaling are also in clinical development. At the same time, the next generation of “armed” tumor-specific antibodies and antibody fragments have been developed and are in pre-clincal development and some have entered clinical trials. Armed antibodies have enhanced effector activity, either via engineered Fc receptors or differential glycosylation that more efficiently active ADCC and CDC or by fusions to toxins, radioactive molecules, chemotherapeutic agents, or nucleic acids for targeted delivery [5–11]. For toxin, chemotherapy, or nucleic acid delivery, it is essential that the antibody not only bind to the cell surface receptor, but that the antibody and its fusion partner undergo endocytosis so that the payload is delivered into the cytosol.
While chimeric antibodies with mouse variable regions and human constant regions are in clinical use, antibodies that are currently entering clinical trials are either humanized or fully human in sequence, in order to reduce immunogenicity [12]. Human antibodies are currently generated from either mice transgenic for the human immunoglobulin locus or from human antibody gene diversity libraries and display technologies [13]. Large human antibody gene diversity libraries displayed on filamentous phage as either single chain Fv (scFv) or Fab antibody fragments have proven to be a reliable source of human antibodies to any purified protein antigen [14–16].
Purified protein is not always available for proteins that are difficult to express, for example multi-pass membrane proteins such as G-coupled protein receptors (GPCRs). In addition, some pure proteins are poor mimics of the protein conformation present on the cell surface. In some instances, it is possible to directly select peptides and antibody fragments binding cell-surface receptors from filamentous phage libraries by incubation of phage libraries with the target cell line [17–21]. This has led to a marked increase in the number of potential cell targeting molecules. However, the isolation of cell type specific antibodies from large phage antibody libraries has proven challenging because selections often result in the generation of cross reactive antibodies binding to frequently expressed cell surface proteins [22].
The ability of phage displaying short peptides to undergo receptor-mediated endocytosis into cells [18, 23] indicated that phage libraries might be selected not only for cell binding but also for internalization into mammalian or other target cells. Such an approach would be especially useful for generating antibodies which could deliver drugs, toxins, or nucleic acids into a cell for therapeutic applications. A number of years ago, we developed a methodology which allows direct selection of internalizing phage antibodies by incubating phage libraries directly with the target cells [24]. Using a model system employing an anti-HER 2 scFv and HER2 expressing cells, we showed that anti-HER2 phage could be endocytosed by HER2 expressing cells and that cellular uptake of phage required both the targeting scFv and expression of the receptor on cells (Figure 1). We also showed that enrichment ratios were greater when phage were recovered from within the cell compared to recovery from the cell surface. Enrichment ratios were also higher when the phage were capable of crosslinking the HER2 receptor, rather than merely binding (Figure 1)[24]. Cross linking could be made to occur when either bivalent antibody fragments, such as diabodies, were displayed in a phagemid system, or when monovalent scFv antibody fragments were displayed using a multivalent phage vector. Thus phage antibody libraries constructed using phage vector systems [25, 26] may prove more useful for generation of internalizing antibodies than antibody fragments displayed using phagemid systems.
Figure 1. Phage antibodies are endocytosed into ErbB2 expressing cells.

Top panel shows different phage antibody constructs studied. Bottom panel shows immunofluorescent microscopy, staining for phage major coat protein pVIII. Panels A–D phage are displayed in a phagemid vector where there is a single scFv/phage. Panel E phage are displayed in a phage vector with 3–5 copies of scFv/phage. A. Control phage antibody (binds BoNT). B. C6.5 anti-ErbB2 scFv. C. Higher affinity ML3-9 anti-ErbB2 scFv. D. Dimeric C6.5 diabody. E. C6.5 scFv displayed multivalently in true phage vector.
We have used the approach to generate human scFv antibodies to HER2 and EGFR by selecting a non-immune scFv human phagemid antibody library for internalization into cancer cell lines that overexpress EGFR or HER2 or cells transfected with the target receptor[27, 28] (Figure 2). The scFv are rapidly internalized into cancer cell lines that overexpress the target receptor [29] and can be used to construct receptor targeted drugs or nucleic acids such as anti-HER2 and anti-EGFR immunoliposomes [10, 11, 23, 30–32]. We have also generated panels of antibodies to prostate and breast cancer cell surface proteins by selecting phage and phagemid antibody libraries on prostate and breast cancer cell lines [33, 34] and again shown that such antibodies can be used for intracellular delivery [35].
Figure 2. Cartoon of the approach used to select internalizing phage antibodies.

A. A phage antibody library is first depleted of common cell surface binders by incubating with a large number of cells lacking the antigens of interest. The depleted phage antibodies are allowed to bind cells at 4°C. B. Cells are then washed to remove unbound phage. C. Cells are then warmed to 37°C to allow endocytosis to occur. D. Surface bound phage are then stripped from the cell surface using low pH buffer. E. Cells containing endocytosed phage are lysed and the lysate used to infect E. coli to produce phage for the next round of selection. F. Selections are repeated for 2–3 rounds
A major challenge when selecting phage antibody libraries directly on cells is identifying the antigen bound by the internalizing antibody. In some instances, we have been able to identify the antigen by using the antibody to immunoprecipitate the antigen followed by mass spectrometry sequencing [34, 36]. In other instances, it has not proven possible to identify the antigen recognized by the internalizing antibody. To overcome this limitation, we recently reported the use of yeast displayed tumor antigens to direct selections to a specific tumor antigen (Figure 3) [37]. In this approach, phage antibodies are first selected for internalization on a tumor cell line known to express the antigen of interest, followed by selection for binding to the yeast displayed antigen. Using this approach, we generated antibodies to EphA2 and CD44 that internalize into breast cancer cell lines [37].
Figure 3. Cartoon of selections to direct internalizing antibodies to a specific target antigen.

A. The phage library is selected for two rounds for internalization into a mammalian cell. B. The polyclonal phage from the 2nd round of selection is applied to yeast displaying the target antigen. C. Unbound phage are removed by washing. D. Bound phage are eluted by incubating yeast with low pH. E. Eluted phage are used to infect E. coli to amplify phage for the next round of selection. F. Selections are repeated for 2–3 rounds.
In the following sections, we describe in detail the specific methodologies required to generate and characterize internalizing antibodies from phage display libraries. Specifically, we provide protocols for: 1) Selecting tumor cell specific internalizing phage antibodies; 2) profiling the specificity of phage antibodies on panels of tumor cells using flow cytometry, 3) identifying the cognate antigen recognized by tumor specific antibodies using immunoprecipitation and mass spectrometry [34], and 4) generating antigen specific internalizing phage antibodies by sequential selection on tumor cell lines and yeast displayed tumor antigens [37].
Selection of tumor cell specific internalizing scFv from phage antibody libraries
Prior to positive selection on the target tumor cells, antibodies recognizing common cell surface molecules need to be removed by pre-absorbing the phage antibody library using control cells; this step is termed “depletion”. The cell line chosen for depletion should be related to the cell line used for positive selection but should lack the cell surface antigen profile that has been targeted for antibody generation. For example, when selecting to generate phage antibodies binding specifically to the basal subtype of breast cancers, we used the luminal subtype breast cancer cell line for depletion (Figure 1). Serial repetition of the depletion process for two to six times prior to positive selection on the target cell line may be more effective at removing unwanted phage antibodies. However, we typically do not use a ‘depleting’ cell line prior to the first round of selection to avoid eliminating phage antibodies which might bind receptors overexpressed on the target cell line, but which are also present at lower density on the subtracting cell line and might be removed during the depletion process. This is especially important in the first round of selection, where each individual phage antibody is present at a relatively low copy number.
For the selection protocol described below, we utilize a non-immune human scFv phage antibody library constructed in the true phage vector fd [26]. This vector results in multicopy phage display which may increase the selection efficiency [24]. Alternatively, a phagemid display antibody library may be used [15] in which case the method of phage preparation would differ (steps 12–14).
To optimize the stripping condition, the stripping buffer may need to be adjusted for different cell types to ensure that washing does not lyse the cells. Parameters to investigate inlcude pH and osmolality. We typically find that one of the four stripping buffers detailed in the materials section removes phage effectively from the cell surface without prematurely lysing the cells.
Materials
Cells for depletion of phage binding coomon cell surface antigens. For example we used luminal subtype breast cancer cell lines MDAMB453 and SUM52PE to deplete prior to selection for basal subtype breast cancer specific antibodies. Cells are grown in tissue culture flasks (Costar, T75) to a density of approximately 3–5 million cells per flask (approximately 80% confluent).
Cells for specific selection of phage antibody libraries. For example, the basal subtype breast cancer cell lines (MDAMB231 and SUM159PT, and BT549). Cells are grown in tissue culture flasks (Costar, T75) to a density of approximately 3–5 million cells per flask (approximately 80% confluent).
Cell culture media
Fetal bovine serum (FBS) (Equitech) for growing cell lines.
Phosphate buffered saline (PBS)
Cell stripping buffer 1: 100 mM glycine/150 mM NaCl, pH 2.5
Cell stripping buffer 2: 100 mM glycine/500 mM NaCl, pH 2.5
Cell stripping buffer 3: 50 mM glycine/150 mM NaCl/200mM urea/2 mg/mL polyvinylpyrrolydone (PVP), pH 2.8
Cell stripping buffer 4: 50 mM glycine/500 mM NaCl/200mM urea/2 mg/mL polyvinylpyrrolydone (PVP), pH 2.8
0.05% Trypsin-EDTA (Gibco)
100 mM triethylamine (TEA)
1 M Tris-HCl, pH 7.4
Exponentially growing E. coli TG1 (OD600 nm approximately 0.5)
2 x TY media
2 x TY media containing 50 mg/mL tetracycline
100 and 150 mm TYE plates containing 50 mg/mL tetracycline (TYE/tet)
50% glycerol
PEG/NaCl solution: 20 % (w/v) polyethylene glycol 6000, 2.5 M NaCl.
Purified fd-phage antibody library.
Centrifuge tube (Nalgene, 3119-0050).
Methods
Culture the target tumor cells in T75 flasks to 80–90% confluence, which normally takes about 3 days.
Change media 1 hour before selection.
Remove the culture media and add 1 mL of 1012 phage antibodies diluted in 3 mL cold culture media to the target tumor cells and incubate for 2 hours at 4°C with occasional rocking. After incubating at 4°C, the cells are incubated for 30 minutes at 37°C in a 5% CO2 gassed incubator to allow internalization to occur. Rock the flasks several times during incubation to keep the phage evenly distributed.
After the 37°C incubation, aspirate the supernatant and wash cells 3 times in 10 mL of cold PBS.
Wash three times to remove uninternalized phage by adding 4 mL of cell stripping buffer to the cells for 5 minutes per wash. After the last wash, neutralize any remaining stripping buffer by adding 1 mL of 1M Tris-HCl, pH 7.4. Save the stripping and neutralization buffers for phage titration.
Wash the cells in 10 mL of complete culture media twice at RT for 1 minute per wash.
Add 5 mL of trypsin/EDTA solution to the cells and incubate at 37°C until the cells start detaching from the flask. Add 10 mL of PBS to the culture plate triturate and transfer cells to a 15 mL centrifuge tube. Pellet cells by centrifugation at 300g for 5 minutes. Discard the supernatant.
Wash cells twice by resuspending in 10 mL of complete culture media and then pelleting the cells by centrifugation at 300g for 5 minutes. Discard the supernatant.
Lyse the cell pellet by resuspending in 1 mL of fresh 100 mM TEA solution to the cell pellet, pipetting to mix and incubating for 2 minutes at room temperature or 10 minutes on ice.
Neutralize the cell lysate by adding 0.5 mL of 1M Tris-HCl, pH 7.4 and mixing.
Add half of the neutralized cell lysate (0.75 mL) to 10 mL of exponentially growing E. coli TG1 (OD600~0.5) and incubate the bacterial culture without shaking at 37°C for 30 minutes.
Titer the phage in the cell lysate and in the stripping buffer washes by plating 10 mL, 1 mL, and 0.1 mL of each sample, in a total volume of 10 mL, on 100 mm TYE/tet plates.
Centrifuge the remainder of the bacterial culture at 3000g for 15 minutes, resuspend in 0.5 mL of 2 × TY, plate on two 150mm TYE/tet plates, and incubate overnight at 30°C.
The next day, add 5 mL of 2 × TY/tet media to each plate and scrape to harvest the bacteria. Make glycerol stocks by mixing 1.4 ml of bacteria and 0.6 ml of 50 % glycerol. Store glycerol stocks at −80°C. Prepare phage particles for the next round of selection by inoculating the glycerol stocks in 500 mL of 2 × TY/tet media, to an initial OD600nm of 0.01–0.05. Grow the culture at 30°C with shaking (300 rpm) for 12–18 hours.
Centrifuge the bacteria at 6000g in 500 mL centrifuge tubes in a GS3 rotor for 30 minutes.
Transfer the supernatant to new 500 mL centrifugre tubes and precipitate the phage by adding 1/10–1/5 volume of PEG/NaCl solution and leave on ice for 1 hour. Phage should be visible as a clouding of the supernatant.
Pellet the phage in 500 mL bottles by centrifuging at 3000g for 15 minutes at 4°C. Discard the supernatant. Centrifuge the ‘dry’ pellet again for 30 seconds to bring down the last drops of supernatant, and remove the liquid. Resuspend the pellet in 1/10 volume of PBS and transfer the phage to high-speed centrifuge tubes (Nalgene, 3119-0050).
Centrifuge the tubes at 15000g in a SA-600 rotor for 15 min to pellet bacterial debris and transfer the supernatant to a new tube.
Repeat steps 16–18 to further purify the phage, resuspending in a final volume 1/50 of the original culture volume.
Titer the purified phage by following steps 11 and 12 (above).
Repeat the selection process, but this time incorporate a ‘depletion’ step prior to the selection step (step 3, above). For depletion, incubate 1012 phage antibodies diluted in 3 mL of cold culture media with an appropriate cell line grown to 90 % confluence in T75 flask or use 107 cells in suspension. After incubation, aspirate the culture media and the depleted phage antibodies to a culture of the target cells as described in step 3 to initiate the next round of positive selection. Typically only two to three rounds of selection are required.
Profiling the specificity of phage antibodies on tumor cells using flow cytometry
This method is used to identify monoclonal phage antibodies with the desired binding specificity after two to three rounds of selection. For initial screening of phage antibodies, binding is measured to the cells used for positive selection and the cells used for depletion. After identification of phage that preferentially bind the selecting cell line, positive phage can be further screened for binding to a larger panel of cell lines.
Materials
Tumor cell lines used for depletion and selection, as well as additional relevant tumor cell lines. These are typically available from the American Type Culture Collection (ATCC) and should be grown and maintained as described in their accompanying literature.
Cell culture media.
Fetal bovine serum (FBS) (Equitech) for growing cell lines.
Sterile 96-well round bottom microtiter plates made for bacterial culture, e.g. Nunc 62162 (VWR Scientific).
2 x TY media containing 50 mg/mL tetracycline.
96-pin transfer device (Nunc)
0.05% Trypsin-EDTA (Gibco).
Flow cytometry buffer (PBS supplemented with 1 mM MgCl2, 0.1 mM CaCl2 and 1% FBS.
Sterile 96 well V-bottom plates (Becton-Dickinson).
Paraformaldehyde.
Biotinylated rabbit anti-fd with biotin conjugates (Sigma-Aldrich).
Streptavidin-Phycoerythrin (PE) (Invitrogen-Biosources).
FACS LSRII (BD Biosciences)
Methods
Preparation of fd scFv phage antibodies in 96-well microtiter plates
Pick individual bacterial colonies using sterile toothpicks, dip into 96-well microtiter plates containing 100 μL of 2 × TY/tet media/well, and grow at 30°C overnight with shaking (300 rpm).
Add 100 μL of 2 × TY/tet containing 30% glycerol to each well and store the plate at −80°C. This is designated as the master plate.
Inoculate about 2 μL of the bacterial culture from the master plate into a new 96-well plate containing 150 μL of 2 × TY/tet/well using a 96-well sterile transfer device (96-pin duplicator), and grow the culture with shaking (300 rpm) at 30°C for 12–18 hours.
Centrifuge the plate at 2,000g for 10 minutes and use 50 μL of supernatant for flow cytometric analysis.
Quantitating cell binding by flow cytometry in 96-well microtiter plates
Grow depleting and selecting tumor cell lines to 80–90% confluence.
Detach the adherent cells from the flask by trypsinizing the cells in 3 mL of 0.05% trypsin.
Transfer the cells to a 50 mL Falcon tube and centrifuge at 300g for 5 minutes. Discard the supernatant.
Wash cells once by resuspending in 10 mL of flow cytometry buffer, followed by centrifugation, resuspend in 2 mL of flow cytometry buffer.
Count the cells using a hematocytometer.
Aliquot 50 μL containing 5 × 104 cells into 96-well V-bottom plates.
Add 50 μL of phage supernatant from sub-section 3.2.1 into each well.
Incubate overnight at 4°C with rocking.
Wash cells twice with 200 μL of FACS buffer.
Resuspend cells in 100 μL of flow cytometry buffer containing 1 μg/mL of biotinylated anti-M13 mouse mAb. Incubate for 1 hour at 4°C.
Wash cells once with 200 μL of flow cytometry buffer.
Resuspend cells in 100 μL of flow cytometry buffer containing 1 μg/mL of streptavdin-PE. Incubate for 30 minutes at 4°C.
Wash cells twice with 200 μL of flow cytometry buffer and resuspended in PBS containing 1% paraformaldehyde.
Measure cell fluorescence in a FACS LSRII flow cytometer or other comparable cytometer using the PE channel.
Identification of the cognate antigen recognized by tumor specific scFv by using mass spectrometry
This method utilizes purified native scFv expressed in E. coli to immunoprecipitate antigen from cell lysates followed by mass spectrometry to identify the antigen.
To immunoprecipitate antigen from cell lysates, Protein-A agarose is used to capture scFvs that bind protein A (the majority of scFv that have a VH gene from the human VH3 family) while Ni-NTA agarose used for non-Protein-A binding scFvs. Protein-A agarose gives less immunoprecipitation of unrelated proteins than Ni-NTA agarose. Regardless, the same type of agarose bead (Protein A or Ni-NTA) should be used for the pre-clearing step and the immunoprecipiation step.
Materials
Tumor cells and culture media.
Sulfo-NHS-LC-biotin (Pierce).
Phosphate buffered saline (PBS).
Cell lysis buffer: 10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 % NP 40, 1 mM EDTA, 1 mM Vanadate, 1 % protease inhibitor cocktail for mammalian cells (Sigma).
Immunoprecipitation (IP) buffer: 10mM Tris-HCl (pH 8.0), 150mM NaCl, 1% NP40.
Nco I (New England Biolabs)
Not I (New England Biolabs)
GeneClean Turbo (MP Biomedicals, LLC)
T4 DNA Ligase (New England Biolabs)
Plasmid vector pSYN1 as described [38]
100 mm TYE plates containing 100 mg/mL Ampicillin and 1 % glucose (TYE/Amp/Glu)
E. coli TG1 chemically competent cells (Zymo Research)
2 × TY media containing 100 μg/mL Ampicillin and 2 % glucose
2 × TY media containing 100 μg/mL Ampicillin and 0.1 % glucose
96-pin transfer device (Nunc)
1 M IPTG
PPB buffer: 200g/L Sucrose, 1mM EDTA, 30mM Tris-HCl, pH8.0
5 mM MgSO4
DNase I (Boerhinger Mannheim)
Protease inhibitor cocktail for bacterial (Sigma)
Centrifuge tube (Nalgene, 3119-0050).
Dialysis tubing, MWCO: 6–8 kDa (Spectrum Laboratories, Inc.)
10 mM Immidazole in PBS, pH 7.4
250 mM Immidazole in PBS, pH 7.4
Poly-Prep chromatography column (Bio-Rad)
Protein A-agarose (Sigma-Aldrich).
Ni-NTA-agarose (Quiagen).
PD 10 column (Pharmacia)
Pre-poured SDS-PAGE gels (8–16%) (Invitrogen).
HRP-conjugated streptavidin (Pierce).
Siliconized tube (PGC Scientific)
NH4HCO3, HPLC grade (Fisher)
Acetonitrile, HPLC grade (Fisher)
Dithiothreitol (DTT) (Sigma-Aldrich)
Iodoacetamide (Sigma-Aldrich).
Trypsin, Mass Spectrometry grade (Promega).
Trifluoroacetic Acid (Fisher).
Speed Vac (Savant)
Methods
Preparation of biotinylated cell lysates
Use the tumor cell line that gives the highest mean fluorescent intensity when phage antibody binding is measured by flow cytometry. Grow these cells to 90% confluence in 150 mm culture dish.
Wash cell twice with PBS, then add 5 mL of sulfo-NHS-LC-biotin (0.1 mg/mL in PBS) to each dish and incubate for 20 minutes at 4°C with rocking.
Remove biotinylation solution by aspiration and wash the cells twice with 20 mL of cold PBS containing 50 mM glycine.
Add 3 mL of lysis buffer to each dish and incubate for 1 hour at 4° C with rocking.
Harvest the cell lysates by scraping the dish with a plastic scraper.
Pellet any remaining cells in the cell lysate by centrifugation for 10 minutes at 4°C.
The cleared cell lysates can be used for immunoprecipiation right away or stored at −80°C.
Preparation of hexahistidine-tagged single chain Fv (scFv) antibodies
Digest the scFv-fd DNA with Nco I and Not I restriction enzymes in the manufacturer’s NEB 3 buffer and 1 × BSA solution, under conditions recommended by the manufacturer for double digests. Note 8.
Purify the digested scFv DNA on a 0.8% agarose gel. Extract the scFv fragment from the gel using GeneClean Turbo Kit.
Digest the pSYN1 vector DNA [38] with the Nco I and Not I restriction enzymes in the manufacturer’s NEB 3 buffer and 1 × BSA solution, under conditions recommended by the manufacturer.
Purify the digested vector DNA on a 0.8% agarose gel. Extract the vector fragment from the gel using GeneClean Turbo Kit.
Ligate the digested scFv DNA and the digested vector DNA in a 20 μL reaction volume of T4 DNA reaction buffer under conditions recommended by the manufacturer.
Transform the ligated DNA into chemically competent E. coli TG1 cells under conditions recommended by the manufacturer. After transformation, dilute the cells in 2 x TY media, plate different amounts of cells on 100 mm TYE/Amp/Glu plates, and incubate overnight at 30°C.
A single colony of E. coli TG1 bearing pSYN1-scFv is inoculated into 5 mL of 2 × TY media with 100 μg/mL ampicillin and 2 % glucose and grown overnight at 30 °C with shaking at 250 rpm.
Inoculate 500 mL of 2 × TY media containing 100 μg/mL ampicillin and 0.1 % glucose in a 2 L flask with a 5 mL of the overnight culture and grow at 37 °C to an OD600 of 0.9.
Induce scFv expression by adding isopropyl-b-D-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM.
Grow the culture for four hours at 30 °C with shaking at 250 rpm.
Harvest the culture by centrifugation for 20 minutes at 4000g. Pour off the supernatant and resuspend the pellets in 12.5 mL of periplasmic extraction buffer (PPB) containing protease inhibitor cocktail and DNase I (100 μg/mL) and incubate on ice for 30 minutes.
Centrifuge the PPB extracted cell at 5000g for 20 minutes and transfer the supernatant to a high-speed centrifuge tube. Resuspend the pelletes in 12.5 mL of osmotic shock buffer (5 mM MgSO4) and incubate for 20 minutes on ice.
Centrifuge the osmotic shock fraction at 5000g for 20 minutes and combine the supernatant with the PPB extract supernatant.
Centrifuge the combined PPB and osmotic shock fractions in high-speed centrifuge tubes (Nalgene, 3119-0050) at 32000g for 30 minutes at 4 °C to remove any remaining cellular debris.
Load the cleared periplasmic preparation from step 14 into dialysis tubing (6–8 kDa MWCO) and dialyze in 4 L PBS for 3 hours with two changes at 4°C.
Purify the hexahistidine tagged scFv by immobilized metal affinity chromatography (IMAC) under conditions recommended by the manufacturer.
Change the buffer after IMAC purification by desalting on a PD 10 column equilibrated with PBS and following the protocol recommended by the manufacturer.
Determine the scFv concentration spectrophotometrically at A280 using an extinction coefficient ε = 1.4.
Immunoprecipitation with scFv antibodies
Express and purify hexahistidine tagged native scFv by following the protocol above.
Set up a reaction to deplete the cell lysates of any proteins binding nonspecifically to scFv. Incubate 10 mL of the biotinylated cell lysates and 300 μg of an irrelevant scFv antibody together for 2 hours at 4°C with rocking.
Wash 200 μL of either Ni-NTA-agarose or protein A-agarose three times in IP buffer, re-suspend in an equal volume of IP buffer, and add to the depleting reaction (1 above) and incubate for 1 hour at 4°C with rocking.
Set an unpacked Bio-Rad column in a rack, pour the pre-clearing immunoprecipitation reaction into the column and let it run by gravity while collecting the pre-cleared cell lysates flowing through.
Add 200 μg of the relevant scFv antibody to the pre-cleared cell lysates and incubate for 2 hours at 4°C with rocking followed by incubating with 200 μL of washed Ni-NTA-agarose or protein A-agarose for 1 hour at 4°C with rocking.
Set an unpacked Biorad column in a rack, pour the immunoprecipitation reaction into the column and let it run by gravity while collecting the cell lysate flowing through which can be used for immunoprecipitation with other scFv antibodies.
Wash the column with 5 mL of IP buffer five times.
After the final wash, add 1 mL of IP buffer to the column and transfer the slurry into a 1.5 mL eppendorf tube.
Spin down the slurry at top speed for 1 minute and remove the supernatant completely.
Add 50 uL of 2.5-fold SDS loading buffer (non-reducing) and boil the beads for 8 minutes at 94°C.
Centrifuge the tube containing the boiled beads to pellet the beads at top speed for 5 minutes, transfer the supernatant to a new microtube and and store the supernatant at −20°C.
Resolve the immunoprecipitates on an 8–16% SDS-PAGE (Invitrogen) in duplicate. One gel should be transferred to a PVDF membrane and proteins stanied with HRP-conjugated streptavidin (Pierce). This will stain only proteins that were on the cell surface and labeled with the sulfo-NHS-LC-biotin. The other gel should be stained with Coomassie R250. A dominant band should be observed on the HRP-streptavidin stained gel that is the antigen recognized by the scFv. The two gels should be aligned and the corresponding band on the Coomassie R250 gel excised for mass spectrometry evaluation.
In-gel digestion and mass spectrometry analysis
The excised protein gel slice is diced into small pieces (~ 1 mm2), placed in 0.65 mL siliconized tubes (PGC Scientific), and destained in 150 μL–250 μL of 25 mM NH4HCO3/50% ACN vortexing for 10 minutes, which can be repeated several times until gels have been stripped of stain.
After removing the supernatant, add 100% acetonitrile to cover the gel pieces, and wait until the gel pieces shrink and turn white. Dry the gel pieces in a Speed Vac with no heating.
Add 40 μL of 10 mM DTT solution to the dried gels, vortex, spin briefly, and incubate 45 minutes at 56°C to reduce the target protein.
After removing the supernatant, add 40 μL of 55 mM iodoacetamide solution to the gel pieces, vortex, spin briefly, and incubate in the dark for 30 minutes at room temperature to alkylate the reduced –SH group.
Remove the supernatant, wash the gel pieces with 100 μL of 25 mM NH4CO3, vortex for 15 minutes, and spin briefly. Repeat step 2 to dry the gel pieces.
Estimate the volume of the dried gel pieces, add about 3 × volume of 12.5 ng/μL trypsin solution to cover the gel pieces, re-hydrate the gel pieces on ice for 15–30 minutes until the trypsin solution has been absorbed, add 25 mM NH4CO3 as needed to cover the gel pieces, spin briefly and incubate for 4 hours to overnight at 37°C to digest the target protein.
Remove samples from heat, spin briefly on a microcentrifuge, and transfer the solution into a clean 1.5 mL siliconized tube. Add 30 μL of 50% ACN/5% TFA to the gel pieces, vortex for 20–30 minutes, and spin briefly. Transfer supernatant to the previously extracted solution, and repeat this step once more. Concentrate the extractions in a Speed Vac to approximately 10 μL. The peptide extracts can be stored at −80°C or −20°C until mass spectrometric analysis.
Analyze the tryptic peptide digests by LC MS/MS using a qTOP mass spectrometer (QSTAR XL, Applied Biosystems/PE Sciex), as described [39].
Identify the protein by searching protein databases using the Protein Prospector search engine and the peptides identified by mass spectrometry (http://prospector.ucsf.edu).
Selection of antigen-specific phage antibodies by sequential selection tumor cells followed by selection on yeast cells displaying specific tumor antigens
Internalizing phage antibodies to a specific cell surface antigen can be generated by first selecting the phage antibody library for internalization into a mammalian cell line followed by selection of the polyclonal phage output on Saccharomyces cerevisiae (yeast) displaying the relevant antigen on their surface as a C-terminal fusion to AgaII [40–42] (Figure 3). We successfully utilized this approach to generate internalizing antibodies to CD44 and EphA2 [37]. Many proteins can be displayed on the surface of yeast including T-cell receptors, domains of EGFR, NY-Eso-1, CD44, and EphA2 [37, 43–46]. Recently, we cloned 65 different cDNA from 19 different single pass membrane proteins. These included the full length extracellular domains (ECD) for some of the proteins, single ECD domains (e.g. 10 Ig domains, 4 L domains, 5 cysteine rich domains, 3 FnIII domains, 3 SEMA domains, etc) and multiple domains. Eighty six percent (56 of 65) of the proteins had detectable display on the surface of yeast. Since most of the 65 proteins were represented by more than one domain, there was at least one domain from each of the 19 proteins that displayed. Thus it can be expected that most protein targets can be successfully displayed on yeast.
For yeast antigen display, the cDNA encoding the full length antigen or antigen domain is amplified by PCR and cloned into NcoI-NotI-digested pYD2 vector by using gap repair [47, 48]. Gap repair instead of digestion of the antigen cDNA fragment allows cloning of virtually all cDNA fragments including those with internal digestion sites [49]. Since many publications describe the cloning and expression of yeast displayed cDNA, a protocol is not provided here [37, 42, 44]. Once cloned, antigen display is induced and the level of display quantitated (Figure 4). If possible, assaying for binding of the natural ligand or existing antibodies to the yeast displayed protein will help validate the quality of display (Figure 4).
Figure 4. Display of antigen domains on the surface of yeast.
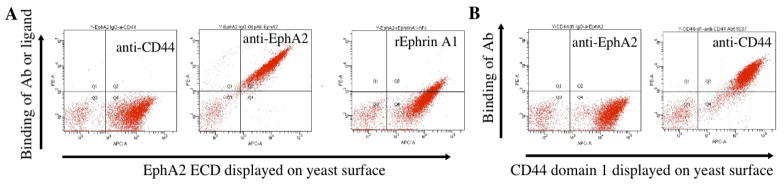
(A) The extracellular domain (ECD) of receptor EphA2 was displayed on the yeast surface and recognized by anti-EphA2 antibody and recombinant mouse Ephrin A1 (R&D) as determined by flow cytometry analysis. (B) The link domain of CD44 (domain 1, or D1) was displayed on the yeast surface and recognized by anti-CD44 rabbit monoclonal antibody as determined by flow cytometry analysis. Both anti-EphA2 and anti-CD44 antibodies did not recognize an irrelevant protein displayed on the yeast surface. The unstained yeast in quadrant 1 represent the parental yeasts that did not induce.
Materials
Yeast for depletion. For example, we used yeast cells displaying an irrelevant protein on the surface, such as an anti-botulinum neurotoxin scFv (labeled as Y-CON in Figure 3) to absorb phage antibodies recognizing yeast cell surface proteins prior to the selection on yeast displaying the target antigen. Yeast cells were cultured and induced following standard protocols [49, 50].
Yeast displaying the target antigen. Yeast cells were cultured and induced following standard protocols [49, 50].
SD-CAA medium (recipe: to 900 mL deionized H2O add: 7 g Yeast Nitrogen base w/o amino acid, 10.19 g Na2HPO4·7H2O or 5.4 g Na2HPO4, 8.56 g NaH2PO4· H2O or 7.4 g NaH2PO4, and 5 g CAA (DIFCO) w/o Tryptophan or Ura. After all components dissolve, add 100 ml of 20% dextrose and 10 ml of 0.6% (100X) Leucine. Sterilize by filtering through 0.22 μm filter.
SG-CAA media (recipe: to 900 mL deionized H2O add: 7 g Yeast Nitrogen base w/o amino acid, 10.19 g Na2HPO4·7H2O or 5.4 g Na2HPO4, 8.56 g NaH2PO4· H2O or 7.4 g NaH2PO4, and 5 g CAA (DIFCO) w/o Tryptophan or Ura. After all components dissolve, add 100 ml of 20% galactose and 10 ml of 0.6% (100X) Leucine. Sterilize by filtering through 0.22 μm filter.
Phosphate buffered saline (PBS)
FACS buffer (PBS supplemented with 1 mM MgCl2, 0.1 mM CaCl2 and 0.3 % BSA)
100 mM triethylamine (TEA)
1 M Tris-HCl, pH 7.4
Exponentially growing E. coli TG1 (OD600 nm approximately 0.8)
2 x TY media
2 x TY media containing 50 mg/mL tetracycline
100 and 150 mm TYE plates containing 50 mg/mL tetracycline (TYE/tet)
50% glycerol
PEG/NaCl solution: 20 % (w/v) polyethylene glycol 6000, 2.5 M NaCl
Purified polyclonal phage antibody output from the second round of selection for internalization on target cancer cells
Biotinylated rabbit anti-fd with biotin conjugates (Sigma-Aldrich)
Streptavidin-Phycoerythrin (PE) (Invitrogen-Biosources)
SV5 Antibody (Invitrogen)
Alexa-647 labeling kit (Molecular Probes)
Methods
Display of tumor antigen domains on yeast surface
Single colonies of transformed EBY100 are inoculated into 5 mL of SD-CAA medium and grown overnight at 30°C with shaking at 250 rpm.
Measure the OD600 of the overnight cultures, transfer 108 cells into a centrifuge tube, pellet the cells, resuspend in 5 mL of SG-CAA medium and grow for 24–48 hrs at 18°C with shaking at 250 rpm.
Transfer 106 cells to a 1.5 mL microfuge tube, and pellet cells by centrifugation at top speed for 15 seconds. Discard the supernatant.
Wash cells once by resuspending in 1mL of FACS buffer, followed by centrifugation, aspiration of the supernatant and resuspension in 1 mL of FACS buffer.
Aliquot 50 μl of the cell suspension containing 5 × 105 cells into 96-well V-bottom plates.
Add 50 μl of 1 μg/mL Alexa-647 labeled anti-SV5 IgG diluted in FACS buffer.
Incubate for 1h at 4°C with rocking. Alexa-647 labeled anti-SV5 IgG should be previously prepared using SV5 antibody and the Alexa-647 labeling kit following the instructions provided by the manufacturer.
Wash cells twice with 200 μl of FACS buffer and resuspend in 150 μL of FACS buffer.
Measure cell fluorescence in a FACS LSRII flow cytometer or other comparable cytometer using the APC channel.
Evaluation of whether an antibody recognizes the yeast displayed antigen or antigen domain
Transfer 107 yeast cells to a 1.5 mL microfuge tube, and pellet cells by centrifugation at top speed for 15 seconds. Discard the supernatant.
Wash cells once by resuspending in 1mL of FACS buffer, followed by centrifugation, resuspend in 1 mL of flow cytometry buffer.
Aliquot 50 μl of the cell suspension containing 5 × 105 cells into 96-well V-bottom plates.
Add 50 μl of antibody known (or hypothesized) to bind the displayed antigen, and diluted in FACS buffer. In this example, the antibody is a human IgG antibody. Antibodies of other species and isotype would require use of a different secondary antibody (below). Incubate for 1h at 4°C with rocking. Note that single chain Fv (scFv) and phage antibodies can also be used to stain yeast displayed antigens. In these instances, binding is detected using secondary antibodies to C-terminal epitope tags for scFv and anti-phage antibody for phage antibodies.
Wash cells twice with 200 μl of FACS buffer.
Resuspend cells in 100 μl of FACS buffer containing 1 μg/ml of PE-labeled anti-human Fc specific antibody and 1 μg/ml of Alexa-647 labeled anti-SV5 IgG. Incubate for 1 hour at 4°C.
Wash cells twice with 200 μl of FACS buffer and resuspend in 150 μL of FACS buffer.
Measure cell fluorescence in a FACS LSRII flow cytometer or other comparable cytometer using the PE and APC channels. Note that due to displayed protein partition, at best 50% of yeast will display antigen. This population can be gated for APC staining (antigen display) and should also be the population with PE staining (antigen binding by antibody).
Isolation of internalizing phage antibodies binding to a specific tumor antigen displayed on yeast surface
Induce yeast display of the target antigen following standard protocols [50].
Transfer 109 control yeast cells (Y-CON) to a 15 mL Falcon tube, and pellet cells by centrifugation at 1000 g for 3 minutes. Discard the supernatant.
Wash yeast once by resuspending in 10 mL of FACS buffer, followed by centrifugation, resuspend in 4 mL of FACS buffer.
Aliquot 100 μl of the purified polyclonal phage antibody output from the cancer cell selection (1 × 1010 cfu/μL) to the Y-CON yeast, incubate with rotating for 1 hour at room temperature followed by incubation for 1 hour at 4°C.
Pellet the yeast cells by centrifugation for 5 minutes at 3000 rpm. Filter the supernatant through a 0.45 μm filter.
Aliquot 2 × 107 freshly induced yeast cells displaying the target antigen into a 15 ml Falcon tube, wash yeast twice with 10 ml of FACS buffer, and incubate with the depleted phage output (step 5) for 2 hrs at 4°C with rocking.
Pellet the yeast cells and wash with ice-cold PBS 10 times.
After the final wash, elute the yeast bound phages by adding 500 μl of 100 mM TEA solution for 2 min at RT or 10 min on ice.
Neutralize the eluted phage mixture with 250 μl of 1M Tris-HCl, pH 7.4.
Amplify the eluted phage by following steps 11 to 20 in the Method for selection on cancer cells.
Repeat the selection process if the output phages do not bind to the yeast displayed target antigen (see following protocol).
Screening of phage antibodies for binding to antigen displaying yeast cells
Pick individual bacterial colonies and amplify phage antibodies in 96-well microtiter plates following the method for phage antibody profiling, and use 50 μl of supernatant for flow cytometric analysis.
Transfer 107 yeast cells displaying the antigen of interest to a 1.5 mL microfuge tube, and pellet the yeast by centrifugation at top speed for 15 seconds. Wash the yeast cells once by resuspending in 1mL of FACS buffer, followed by centrifugation, resuspend in 1 mL of flow cytometry buffer.
Aliquot 50 μl of the yeast cell suspension containing 5 × 105 cells into 96-well V-bottom plates.
Add 50 μl of phage antibody supernatant from step 1 to the yeast cells, and incubate for 1 hr a 4°C with rocking. Wash cells twice with 200 μl of FACS buffer.
Resuspend cells in 100 μl of FACS buffer containing 1 μg/ml of anti-fd biotin conjugates for 30 min at 4°C followed by two washes with 200 μl of FACS buffer.
Resuspend cells in 100 μl of FACS buffer containing 1 μg/ml of streptavidin-PE and 1 μg/ml of Alexa-647 labeled anti-SV5 IgG. Incubate for 30 min at 4°C.
Wash cells twice with 200 μl of FACS buffer and resuspend in 150 μL of FACS buffer.
Measure cell fluorescence in a FACS LSRII flow cytometer or other comparable cytometer using the PE and APC channels. Note that due to displayed protein partition, at best 50% of yeast will display the antigen. This population can be gated for APC staining (antigen display) and should also be the population with PE staining (antigen binding by antibody).
References
- 1.Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, Gianni L, Baselga J, Bell R, Jackisch C, Cameron D, Dowsett M, Barrios CH, Steger G, Huang CS, Andersson M, Inbar M, Lichinitser M, Lang I, Nitz U, Iwata H, Thomssen C, Lohrisch C, Suter TM, Ruschoff J, Suto T, Greatorex V, Ward C, Straehle C, McFadden E, Dolci MS, Gelber RD. N Engl J Med. 2005;353:1659–1672. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- 2.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 3.Hainsworth JD, Burris HA, 3rd, Morrissey LH, Litchy S, Scullin DC, Jr, Bearden JD, 3rd, Richards P, Greco FA. Blood. 2000;95:3052–3056. [PubMed] [Google Scholar]
- 4.Wendtner CM, Ritgen M, Schweighofer CD, Fingerle-Rowson G, Campe H, Jager G, Eichhorst B, Busch R, Diem H, Engert A, Stilgenbauer S, Dohner H, Kneba M, Emmerich B, Hallek M. Leukemia. 2004;18:1093–1101. doi: 10.1038/sj.leu.2403354. [DOI] [PubMed] [Google Scholar]
- 5.Kreitman RJ, Pastan I. Curr Drug Targets. 2006;7:1301–1311. doi: 10.2174/138945006778559139. [DOI] [PubMed] [Google Scholar]
- 6.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, Vielmetter J, Carmichael DF, Hayes RJ, Dahiyat BI. Proc Natl Acad Sci U S A. 2006;103:4005–4010. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adams GP, Shaller CC, Dadachova E, Simmons HH, Horak EM, Tesfaye A, Klein-Szanto AJ, Marks JD, Brechbiel MW, Weiner LM. Cancer Res. 2004;64:6200–6206. doi: 10.1158/0008-5472.CAN-03-2382. [DOI] [PubMed] [Google Scholar]
- 8.Burris HA, 3rd, Rugo HS, Vukelja SJ, Vogel CL, Borson RA, Limentani S, Tan-Chiu E, Krop IE, Michaelson RA, Girish S, Amler L, Zheng M, Chu YW, Klencke B, O’Shaughnessy JA. J Clin Oncol. 2011;29:398–405. doi: 10.1200/JCO.2010.29.5865. [DOI] [PubMed] [Google Scholar]
- 9.Noble CO, Kirpotin DB, Hayes ME, Mamot C, Hong K, Park JW, Benz CC, Marks JD, Drummond DC. Expert Opin Ther Targets. 2004;8:335–353. doi: 10.1517/14728222.8.4.335. [DOI] [PubMed] [Google Scholar]
- 10.Hayes ME, Drummond DC, Hong K, Zheng WW, Khorosheva VA, Cohen JA, CONth, Park JW, Marks JD, Benz CC, Kirpotin DB. Mol Pharm. 2006;3:726–736. doi: 10.1021/mp060040v. [DOI] [PubMed] [Google Scholar]
- 11.Hayes ME, Drummond DC, Kirpotin DB, Zheng WW, Noble CO, Park JW, Marks JD, Benz CC, Hong K. Gene Ther. 2006;13:646–651. doi: 10.1038/sj.gt.3302699. [DOI] [PubMed] [Google Scholar]
- 12.Reichert JM, Valge-Archer VE. Nat Rev Drug Discov. 2007;6:349–356. doi: 10.1038/nrd2241. [DOI] [PubMed] [Google Scholar]
- 13.Bradbury AR, Sidhu S, Dubel S, McCafferty J. Nat Biotechnol. 2011;29:245–254. doi: 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. J Mol Biol. 1991;222:581–597. doi: 10.1016/0022-2836(91)90498-u. [DOI] [PubMed] [Google Scholar]
- 15.Sheets MD, Amersdorfer P, Finnern R, Sargent P, Lindquist E, Schier R, Hemingsen G, Wong C, Gerhart JC, Marks JD. Proc Natl Acad Sci U S A. 1998;95:6157–6162. doi: 10.1073/pnas.95.11.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Nature Biotech. 1996;14:309–314. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- 17.Andersen PS, Stryhn A, Hansen BE, Fugger L, Engberg J, Buus S. Proc Natl Acad Sci U S A. 1996;93:1820–1824. doi: 10.1073/pnas.93.5.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barry MA, Dower WJ, Johnston SA. Nat Med. 1996;2:299–305. doi: 10.1038/nm0396-299. [DOI] [PubMed] [Google Scholar]
- 19.Cai X, Garen A. Proc Natl Acad Sci U S A. 1995;92:6537–6541. doi: 10.1073/pnas.92.14.6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Kruif J, Terstappen L, Boel E, Logtenberg T. Proc Natl Acad Sci U S A. 1995;92:3938–3942. doi: 10.1073/pnas.92.9.3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marks JD, Ouwehand WH, Bye JM, Finnern R, Gorick BD, Voak D, Thorpe SJ, Hughes-Jones NC, Winter G. Biotechnology (N Y) 1993;11:1145–1149. doi: 10.1038/nbt1093-1145. [DOI] [PubMed] [Google Scholar]
- 22.Hoogenboom HR, Lutgerink JT, Pelsers MM, Rousch MJ, Coote J, Van Neer N, De Bruine A, Van Nieuwenhoven FA, Glatz JF, Arends JW. Eur J Biochem. 1999;260:774–784. doi: 10.1046/j.1432-1327.1999.00214.x. [DOI] [PubMed] [Google Scholar]
- 23.Hart SL, Knight AM, Harbottle RP, Mistry A, Hunger HD, Cutler DF, Williamson R, Coutelle C. J Biol Chem. 1994;269:12468–12474. [PubMed] [Google Scholar]
- 24.Becerril B, Poul MA, Marks JD. Biochem Biophys Res Commun. 1999;255:386–393. doi: 10.1006/bbrc.1999.0177. [DOI] [PubMed] [Google Scholar]
- 25.Huie MA, Cheung MC, Muench MO, Becerril B, Kan YW, Marks JD. Proc Natl Acad Sci U S A. 2001;98:2682–2687. doi: 10.1073/pnas.051631798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Connell D, Becerril B, Roy-Burman A, Daws M, Marks JD. J Mol Biol. 2002;321:49–56. doi: 10.1016/s0022-2836(02)00561-2. [DOI] [PubMed] [Google Scholar]
- 27.Poul MA, Becerril B, Nielsen UB, Morisson P, Marks JD. J Mol Biol. 2000;301:1149–1161. doi: 10.1006/jmbi.2000.4026. [DOI] [PubMed] [Google Scholar]
- 28.Heitner T, Moor A, Garrison JL, Marks C, Hasan T, Marks JD. J Immunol Methods. 2001;248:17–30. doi: 10.1016/s0022-1759(00)00340-9. [DOI] [PubMed] [Google Scholar]
- 29.Neve RM, Nielsen UB, Kirpotin DB, Poul MA, Marks JD, Benz CC. Biochem Biophys Res Commun. 2001;280:274–279. doi: 10.1006/bbrc.2000.4104. [DOI] [PubMed] [Google Scholar]
- 30.Nielsen UB, Kirpotin DB, Pickering EM, Hong K, Park JW, Refaat Shalaby M, Shao Y, Benz CC, Marks JD. Biochim Biophys Acta. 2002;1591:109–118. doi: 10.1016/s0167-4889(02)00256-2. [DOI] [PubMed] [Google Scholar]
- 31.Park JW, Hong K, Kirpotin DB, Colbern G, Shalaby R, Baselga J, Shao Y, Nielsen UB, Marks JD, Moore D, Papahadjopoulos D, Benz CC. Clin Cancer Res. 2002;8:1172–1181. [PubMed] [Google Scholar]
- 32.Zhou Y, Drummond DC, Zou H, Hayes ME, Adams GP, Kirpotin DB, Marks JD. J Mol Biol. 2007;371:934–947. doi: 10.1016/j.jmb.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu B, Conrad F, Cooperberg MR, Kirpotin DB, Marks JD. Cancer Res. 2004;64:704–710. doi: 10.1158/0008-5472.can-03-2732. [DOI] [PubMed] [Google Scholar]
- 34.Goenaga AL, Zhou Y, Legay C, Bougherara H, Huang L, Liu B, Drummond DC, Kirpotin DB, Auclair C, Marks JD, Poul MA. Mol Immunol. 2007;44:3777–3788. doi: 10.1016/j.molimm.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth A, Drummond DC, Conrad F, Hayes ME, Kirpotin DB, Benz CC, Marks JD, Liu B. Mol Cancer Ther. 2007;6:2737–2746. doi: 10.1158/1535-7163.MCT-07-0140. [DOI] [PubMed] [Google Scholar]
- 36.Liu B, Conrad F, Roth A, Drummond DC, Simko JP, Marks JD. J Mol Med. 2007;85:1113–1123. doi: 10.1007/s00109-007-0208-z. [DOI] [PubMed] [Google Scholar]
- 37.Zhou Y, Zou H, Zhang S, Marks JD. J Mol Biol. 2010;404:88–99. doi: 10.1016/j.jmb.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schier R, Marks JD, Wolf EJ, Apell G, Wong C, McCartney JE, Bookman MA, Huston JS, Houston LL, Weiner LM, et al. Immunotechnology. 1995;1:73–81. doi: 10.1016/1380-2933(95)00007-0. [DOI] [PubMed] [Google Scholar]
- 39.Liu B, Huang L, Sihlbom C, Burlingame A, Marks JD. J Mol Biol. 2002;315:1063–1073. doi: 10.1006/jmbi.2001.5276. [DOI] [PubMed] [Google Scholar]
- 40.Boder ET, Wittrup KD. Nat Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 41.Boder ET, Wittrup KD. Biotechnol Prog. 1998;14:55–62. doi: 10.1021/bp970144q. [DOI] [PubMed] [Google Scholar]
- 42.Levy R, Forsyth CM, LaPorte SL, Geren IN, Smith LA, Marks JD. J Mol Biol. 2007;365:196–210. doi: 10.1016/j.jmb.2006.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aggen DH, Chervin AS, Insaidoo FK, Piepenbrink KH, Baker BM, Kranz DM. Protein Eng Des Sel. 2010 doi: 10.1093/protein/gzq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cochran JR, Kim YS, Olsen MJ, Bhandari R, Wittrup KD. J Immunol Methods. 2004;287:147–158. doi: 10.1016/j.jim.2004.01.024. [DOI] [PubMed] [Google Scholar]
- 45.Johns TG, Adams TE, Cochran JR, Hall NE, Hoyne PA, Olsen MJ, Kim YS, Rothacker J, Nice EC, Walker F, Ritter G, Jungbluth AA, Old LJ, Ward CW, Burgess AW, Wittrup KD, Scott AM. J Biol Chem. 2004;279:30375–30384. doi: 10.1074/jbc.M401218200. [DOI] [PubMed] [Google Scholar]
- 46.Piatesi A, Howland SW, Rakestraw JA, Renner C, Robson N, Cebon J, Maraskovsky E, Ritter G, Old L, Wittrup KD. Protein Expr Purif. 2006;48:232–242. doi: 10.1016/j.pep.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 47.Gietz RD, Schiestl RH. Yeast. 1991;7:253–263. doi: 10.1002/yea.320070307. [DOI] [PubMed] [Google Scholar]
- 48.Orr-Weaver TL, Szostak JW. Proc Natl Acad Sci U S A. 1983;80:4417–4421. doi: 10.1073/pnas.80.14.4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia-Rodriguez C, Geren IN, Lou J, Conrad F, Forsyth C, Wen W, Chakraborti S, Zao H, Manzanarez G, Smith TJ, Brown J, Tepp WH, Liu N, Wijesuriya S, Tomic MT, Johnson EA, Smith LA, Marks JD. Protein Eng Des Sel. 2010 doi: 10.1093/protein/gzq111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boder ET, Midelfort KS, Wittrup KD. Proc Natl Acad Sci U S A. 2000;97:10701–10705. doi: 10.1073/pnas.170297297. [DOI] [PMC free article] [PubMed] [Google Scholar]