

Abstract
The application of immunotherapy in combination with chemotherapy is considered an effective treatment strategy against persistent Mycobacterium tuberculosis (Mtb) infection. In this study, we constructed a novel recombinant Mycobacterium smegmatis (rMS) strain that expresses Ag85B and ESAT6 fusion protein (AE–rMS). Immunization of C57BL/6 mice with AE–rMS generated mainly Th1-type immune responses by strongly stimulating IFN-γ- and IL-2-producing splenocytes and increasing antigen-specific cytotoxic T lymphocyte (CTL) activity. To test the immunotherapeutic efficacy of AE–rMS, a persistent tuberculosis infection (PTBI) model was established via tail-vein injection of C57BL/6 mice with 1 × 104 colony forming units (CFU) of Mtb strain H37Rv in combination with concurrent chemotherapy drugs isoniazid (INH) and pyrazinamide (PZA). PTBI mice immunized with AE–rMS showed high levels of IFN-γ secreted by splenocytes and decreased bacteria loads in lung. Treatment with only the anti-tuberculosis (anti-TB) drugs RFP and INH (RI), decreased bacteria loads to low levels, with the Th1-type immune response further attenuated. Moreover, AE–rMS, when combined with RI treatment, further reduced the bacteria load as well as the pathological tissue damage in lung. Together, these results demonstrated the essential roles of AE–rMS-induced Th1-type responses, providing an effective treatment strategy by combining AE–rMS and RI for persistent TB.
Keywords: Ag85B, ESAT6, immunotherapy, Mycobacterium tuberculosis, recombinant Mycobacterium smegmatis
Introduction
Tuberculosis (TB) is a global health issue that causes high mortality rates. Currently, Mycobacterium bovis Bacille Calmette and Guerin (BCG) is the only available vaccine against TB but has limited protective effects ranged from 0% to 80% across different regions as screened.1 Moreover, the emergence of resistant strains, particularly the multidrug-resistant (MDR) Mycobacterium tuberculosis (Mtb) strains, has further worsened the disease control.2 Currently available anti-TB chemotherapy drugs have shown high efficacy for killing most of the actively replicating bacteria within the first few days of a course of treatment, but long-term therapy is still required for a persistent population of slowly replicating or dormant bacilli.3 However, long-term use of anti-TB drugs has shown significantly induced lung inflammation and pathogenesis of lung diseases. Besides the potential side effects and the associated financial burden, long-course therapy often results in some unwanted symptoms, such as relapse and drug resistance. Host immunity holds the greatest importance for effectively inhibiting the growth of intracellular pathogen, such as Mtb.4 Indeed, immunotherapy has shown the potential to improve the control of TB by enhancing host immune responses to eliminate the bacteria, such as persistent bacteria, and to shorten the protracted period of chemotherapy required for TB patients.5 Therefore, development of new anti-microbial agents and optimization of the host responses with adjuvant immunotherapy hold promising opportunities for improved TB cure rates, particularly for active TB or latent TB infection. Different attempts have been made to develop an effective adjunct to chemotherapy,6 including the modulation of cytokine levels,7 administration of environmental mycobacteria,8 and antibody therapy.9 It has been previously reported that a plasmid DNA encoding M. leprae HSP65, as a valuable adjunct to antibacterial chemotherapy, could shorten the duration of therapy, improve the treatment of latent TB infection and decrease MDR-Mtb.10 However, the single use of the Hsp65-DNA vaccine may increase the risk of immunopathological damage.11 M. vaccae is a relatively low virulence species with strong capacity to shift Th2 to Th1 responses in the host, which also can act as an immunotherapeutic agent, particularly when drug treatment failed.12
Mycobacterium smegmatis (MS) is a rapidly growing non-pathogenic environmental species that can function as a strong cellular immune adjuvant,13 with deficiency in arresting phagolysosomal maturation or evading intracellular killing in macrophages.14,15 Recombinant MS (rMS) has been demonstrated to stimulate T-lymphocyte proliferation, initiate Th1-type immune responses, promote the secretion of various cytokines, such as IFN-γ, IL-2, and IL-12, and enhance the phagocytosis and killing of invading pathogens.16 The application of rMS has been further advanced by the construction of a rapidly growing and efficient MS vector, which can stably express secreted recombinant proteins under the promoter of select gene clone(s), with 5- to 10-fold higher in gene expression levels compared with BCG.17
We previously evaluated a recombinant MS vaccine expressing the ESAT6–CFP10 fusion protein. Immunized mice with this rMS showed induced protection against Mtb challenge and dramatic decrease of bacteria loads in lung.18 We also demonstrated that the rMS strain expressing the fusion protein of heparin-binding hemagglutinin and human IL-12 (HBHA-IL-12) enhanced immunogenicity and improved Th1-type responses against TB, with the protective effects equivalent to that of the conventional BCG vaccine in mice.19 Moreover, this rMS also decreased the bacterial load and pathological damage in lung of Mtb-infected mice. These results in aggregate suggest that rMS may exert potent immunological effects and enhance host immune responses against Mtb infections.
It has been shown that immunizing mice with the fusion protein Ag85B–ESAT6 achieved stronger protective effects than the use of either individual protein.20 In addition, the Ag85B–ESAT6 fusion protein was found to induce long-term anti-Mtb immune memory in mice.21 In this study, we generated a novel live recombinant MS strain that expresses the Ag85B–ESAT6 fusion protein and also evaluated its immunotherapeutic efficacy in the persistent tuberculosis infection (PTBI) mouse models.
Results
Characterization of AE–rMS
As shown in Figure 1A, we clearly detected a 40-kDa band that is equivalent to the sum of the molecular weight of Ag85B and ESAT6 by western blotting analysis. This suggests that the fusion protein was successfully expressed in the AE–rMS strain. Moreover, similar growth patterns between the parental and the AE–rMS strains were observed. Both strains entered the plateau phase growth at the same time and did not show significant differences in proliferation rates. Humoral immune responses were determined by measuring total IgG in sera collected from the immunized mice. The specific IgG levels in the sera of immunized mice were increased continuously along with the immune duration and reached the highest levels at week 8 after first immunization (Fig. 1B). The antibody titers of AE–rMS in AE group were significantly higher than MS group at both week 6 and 8 (P < 0.05).

Figure 1. Schedule of PTBI model establishment and treatments. After infection with Mtb, mice were treated with INH and PZA for 4 weeks to establish the PTBI model. AE–rMS was given for treatment group at week 10 and 14. In the RI/AE–rMS group, mice were first treated with RI for 4 weeks from week 10 to 14, and AE–rMS immunotherapy was given at week 14 and 18. The chemotherapy group was treated with RI from week 10 to 20. Dexamethasone was injected at week 22. Four mice from each group were sacrificed at week 8, 10, 14, 18, 22, and 26. (A) MC, model control group without therapy. (B) rMS, AE–rMS immunotherapy group. (C) RI+rMS, AE–rMS plus RI therapy group. (D) RI, RI chemotherapy group. (E) NC, normal control group.
Detection of cytokine-secreting lymphocytes and CTL activity in immunized mice
The frequency of lymphocyte-producing Th1 cytokines were shown in Figure 2A. AE–rMS strongly stimulated mouse lymphocytes to produce higher levels of both IFN-γ and IL-2 (P < 0.05) in comparison with the NC group. AE–rMS immunization also significantly increased CTL activity (P < 0.05) with the effector/target ratio of 25:1 at week 6 since the initial immunization (Fig. 2B). BCG immunization also induced comparable cytolytic activity against target cells when compared with AE or AE–rMS vaccinated mice even though BCG does not express ESAT6. This indicates an essential role of Ag85B in determining the cytolytic activities.
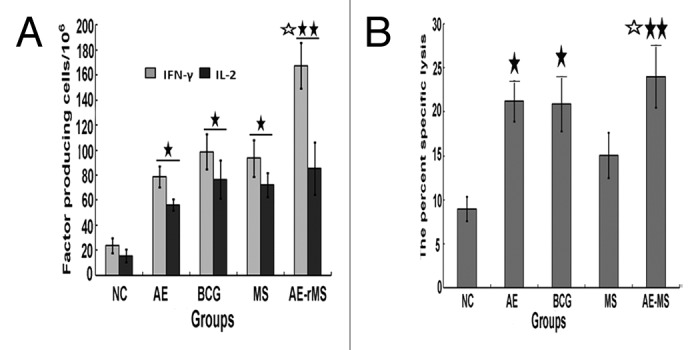
Figure 2. Fusion protein expression and specific antibody detection. (A) Ag85B–ESAT6 fusion protein was detected by western blot. M, protein marker; 1, negative control, supernatant of MS culture; 2, supernatant of AE–rMS culture. Proteins were incubated with monoclonal antibody against ESAT6 or polyclonal antibody against Ag85B. (B) The specific IgG titers in immunized mouse serum (★P < 0.05 vs. MS group).
Evaluation of PTBI mouse-model
To establish the PTBI mouse model, mice were infected with 1 × 104 CFU Mtb via the intravenous route. Four weeks later, the mice were treated with a 4-week course of isoniazed (INH) and pyrazinamide (PZA) at the doses of 0.1 g/L and 15 g/L, respectively (Fig. 3). Bacterial loads (lg CFU) in one lung and spleen were 4.06 ± 0.40 and 3.11 ± 0.78, respectively, at week 8, and 4.02 ± 0.55 and 3.19 ± 0.47, respectively, at week 10. Mice did not show severe signs of infection, such as decreased body weight or scruffy hair during week 8 to 10. In line with these observations, we conclude the successful establishment of PTBI mice model.

Figure 3. Frequency of IFN-γ- and IL-2-producing splenocytes and induction of antigen-specific CTL activity (effector/target ratio, 25:1) (n = 10). (A) Frequency of IFN-γ and IL-2. IFN-γ- and IL-2-producing splenocytes (SFCs) in AE, BCG, MS, and AE–rMS groups were significantly higher than those in the saline group (★P < 0.05, ★★P < 0.01). The frequency of cells to produce IFN-γ in the AE–rMS group was significantly higher than that of MS group (☆P < 0.05). (B) CTL activities. CTL activities of the BCG group, AE group, and AE–rMS group were higher than that of the saline group (★P < 0.05, ★★P < 0.01). In addition, CTL activity of AE–rMS group was significantly higher than that of the MS group (☆P < 0.05).
Therapeutic effects of AE–rMS
To examine the therapeutic effects of AE–rMS in PTBI mice, we determined bacteria loads per lung in mice at different weeks. As shown in Figure 4A, RI alone and RI plus AE–rMS demonstrated distinct effects on the CFU of bacteria. At week 22, the bacterial load in lung of PTBI decreased to undetectable levels in both groups of RI and RI/AE–rMS. Notably, any CFU values below 100, the minimum detection limit in the present CFU assay system, turns to be undetectable levels (i.e., 0) in data presentation. At week 26, the bacterial load in lung increased quickly in response to dexamethasone, and reached 3.98 ± 0.89 and 2.31 ± 0.77 for RI and RI/AE–rMS groups, respectively. Despite the same relapse rate, the RI and AE–rMS combinational treatment inhibited bacterial growth more effectively than RI alone in PTBI mice (P < 0.05).

Figure 4. Bacterial loads in lung and IFN-γ frequencies after immunotherapy (n = 4). (A) Bacterial loads per lung. At week 14 and 18, bacterial loads in lung of AE–rMS or RI/AE–rMS group were significantly lower than control group (P < 0.05). At week 22, the bacterial loads in lung of PTBI mice decreased to undetectable levels in both groups of RI and RI/AE–rMS. Since the sensitivity of the CFU assay in mouse was ~100, which didn’t indicate the zero CFUs for the RI and RI+rMS groups. After dexamethasone injection, CFU counts of all groups increased, with CFU counts in RI/rMSgroup lower than RI group at week 26. (B) IFN-γ frequencies. The frequency of IFN-γ-secreting cells in both AE–rMS and RI/AE–rMS groups were significantly higher than RI group (★P < 0.05). Purified AE fusion proteins were used to stimulate splenocytes. MC, model control group receiving no therapy; rMS, AE–rMS immunotherapy group; RI+rMS, AE–rMS plus RI combination therapy group; RI, RI chemotherapy group; NC: normal control group.
Antigen-stimulated secretion of IFN-γ from mouse splenocytes
Since the initiation of immunotherapy with AE–rMS in PTBI mice, the peak values of the observable number of spot-forming cells values (SFC) were captured at week 18 (Fig. 4B). Moreover, the detectable frequency of IFN-γ-secreting cells in both AE–rMS and RI/AE–rMS groups was significantly higher than that of RI group (P < 0.05). Hence, immunization of PTBI mice with AE–rMS strongly stimulated Th1-type immune responses by increasing the number of IFN-γ-secreting cells compared with the anti-TB drug treatment alone.
Lung histology
Immunized mice were sacrificed at different time points, and the pathological changes in lung were examined by H&E staining. The histological morphology, the size of lesions and the degree of inflammation were generally uniform in lung sections among different groups of mice. All groups of mice except NC group showed inflammatory cell infiltration that was mainly composed of lymphocytes and monocytes. After 4-week immunotherapy (Fig. 5A), partial alveolar rupturing was observed in both MC and AE–rMS groups, with additional alveolar septal widening, inflammation and edema detected in AE–rMS group. In contrast, fewer and smaller lesions in lung tissues were shown in RI group, which was capable of maintaining structural integrity with negligible damages in limited number of alveolar cells. However, more serious lesions in lung tissue were found in either RI/AE–rMS or AE–rMS group. Eight weeks after immunotherapy (Fig. 5B), inflammatory terminal bronchi tubes was observable in lung and the alveolar septum was increased by width, resulting in interstitial pneumonia in different treatment groups; whereas a lower degree of inflammation was demonstrated in the RI group. At week 12 post-immunotherapy (Fig. 5C), inflammatory exudate and local tissue destruction were clearly present in the RI group, while this inflammatory responses tended to decline by RI and AE–rMS combinational therapy. At week 16 post-immunotherapy (Fig. 5D), inflammatory fibrosis was detected in all groups. Notably, the inflammation extent was exacerbated in RI group in sharp contrast to that under combination treatment with RI and AE–rMS, which was reduced further with fewer lesions in comparison to the AE–rMS group. We further used different semi-quantitative scoring methods to analyze the pathological changes in lung. As shown in Figure 5E, the scores of lung pathology in the RI/AE–rMS group remained comparable to those in AE–rMS or RI group at either week 22 or 26 (P < 0.05). Acid-fast bacilli could also be detected in lung of the PTBI mice at week 26 (Fig. 6). In addition, bacterial counts in AE–rMS or RI group were less than MC group, which was further reduced to undetectable levels in response to both treatment of AE–rMS and RI.

Figure 5. Histopathology of PTBI mouse lung after immunotherapy (H&E, 20×) (A) Histopathology of PTBI mouse lung at 4 weeks after immunotherapy (week 14 post-infection). The widened alveolar septal, inflammation, and edema were detected in the AE–rMS group. In contrast, fewer and smaller lesions in lung tissues were shown in the RI group, which was capable maintaining structural integrity. However, more serious lesions in lung tissue were found in either the RI/AE–rMS or the AE–rMS group. (B) Histopathology of PTBI mouse lung at 8 weeks after immunotherapy (week 18 post-infection). Inflammatory terminal bronchi tubes were observable in lung, and the alveolar septum was increased by width in different treatment groups. A lower degree of inflammation was demonstrated in the RI group. (C) Histopathology of PTBI mouse lung at 12 weeks after immunotherapy (week 22 post-infection). Inflammatory exudate and local tissue destruction were clearly present in the RI group, while these inflammatory responses tended to decline by the combination therapy of RI plus AE–rMS. (D) Histopathology of PTBI mouse lung at 16 weeks after immunotherapy (week 26 post-infection). Inflammation extent was exacerbated in the RI group in sharp contrast to the combination treatment of RI and AE–rMS, which was reduced further. (E) Results of the semi-quantitative analysis of histopathological changes. Each histogram represents a set of data for four mice. (★P < 0.05, vs. RI group). MC, model control group receiving no therapy; rMS, AE–rMS immunotherapy group; RI+rMS, AE–rMS plus RI therapy group; RI, RI chemotherapy group; NC, normal control group.
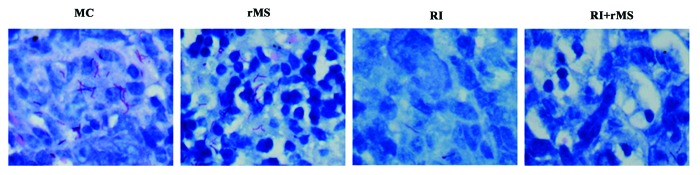
Figure 6. Acid-fast staining of PTBI mouse lung at week 26. Bacterial counts in the AE–rMS or RI group were less than the MC group, which was further reduced to undetectable levels for RI /AE–rMS group. MC, model control group receiving no therapy; rMS, AE–rMS immunotherapy group; RI+rMS, RI plus AE–rMS therapy group; RI, RI chemotherapy group; NC, normal control group.
Discussion
Recent attention has been drawn to immunotherapy against drug-resistant TB, as this type of therapy has shown the potential for improved treatment for MDR and latent TB which received relatively low cure rates by conventional therapies.22 RUTI as therapeutic vaccines reduced significantly the bacterial loads and showed effectiveness for the persistent MTB with slow replication rates and drug sensitivity.23 However, immunotherapy also brought side effects for patients who lack intact immune system, such as tissue damages and certain harmful responses involving exacerbated release of TNF-α and the activation of downstream pro-inflammatory cytokines.24 Moreover, previous studies have also indicated limited effectiveness of therapeutic adjuncts for PTBI.25,26 Recent therapeutic development has brought into focus an attractive strategy by screening and validating specific target antigens to boost the existed Th1-type immune responses in persistent Mtb-infected individuals, which minimizes pathological damages induced by granuloma formation and tissue inflammation.27,28 Both Ag85B and ESAT6 can induce strong immune responses in animal models of tuberculosis.29 ESAT6 is a dominant inducer of immune responses during primary or latent infection, and this antigen shows strong association with latent infection.30 It has been reported that immunotherapy constituting of DNA vaccines with three antigens (Ag85B, MPT-83, and MPT-64), when in combination with chemotherapy, could be effective in preventing TB from entering reactivation, and thus could be a promising strategy for controlling Mtb infection.31 The Salmonella-based live vector vaccine that expresses Ag85B–ESAT6 fusion protein could deliver the foreign antigens directly into the antigen-presenting cells (APCs), providing a strong Th1-cytokine milieu and other immunomodulatory signals to clear tuberculosis.32 Consistently, our results demonstrate that MS can be used as an auxiliary anti-TB treatment supplement to currently available anti-TB drugs,33-35 and the universal components within MS, such as lipopolysaccharide (LPS), may serve as an adjuvant for a potential TB immunotherapy.36 Therefore, AE–rMS-induced immune responses were not only targeting the population of actively replicating bacilli that express high levels of Ag85B and ESAT6, but also capable of killing effectively the persistent population.37,38 Immunization with rMS that expresses Ag85B–ESAT6 fusion protein could increase CTL activity and stimulate high-level production of antigen-specific IFN-γ- and IL-2-secreting splenocytes significantly in mice.
Currently, there are two established persistent tuberculosis models by mimicking human PTBI. In the lose-dose persistent model induced by host immunity, immunocompetent mice are infected with low doses of Mtb, and the acquired immunity inhibits the bacterial growth but fails to clear the bacteria, leading to a persistent infection with stable bacterial loads in organs. The other model is the conventional Cornell model with persistence induced by chemotherapy. However, the bacteria in response to prolonged anti-biotic treatment in these mouse models may be fundamentally different from those causing PTBI in humans. In order to determine the immunotherapeutic efficacy of AE–rMS against Mtb persistent infection, we have established the PTBI mouse model by the same methodology similar to that used for generating the low-dose and Cornell models.39 In our model, only 4-week treatment of INH and PAZ allowed the bacilli to rapidly enter an altered physiological state characterized by stationary-state CFU counts. Following the termination of treatment, the bacterial counts in lung were reduced to about 104 CFU that was stably maintained for the next 12 weeks as shown in Figure 4A (MC group). In contrary to the previous studies that reported undetectable levels of bacillus but a subsequent reactivation since the treatment termination,39,40 we could still detect 104 bacilli in lung after 4-week treatment in our model. This difference may result from the different sensitivities of the CFU assay when applied in mouse lung.
The immunotherapeutic effect of AE–rMS alone in PTBI mice were accompanied by an increase in inflammatory responses in lung at the early stage, which could be due to the induction of strong immune-mediated inflammation by the Ag85B–ESAT6 fusion protein41 and thus interfere with the therapeutic effect of AE–rMS. Early inflammatory responses were also observed in the combination therapy with AE-rMS and RI. The bacterial load in lung was lower in the RI group compared with the RI/AE–rMS group, and the differences in the lesions of lung tissues were also noted at week 14 between these two groups. Although the treatment with RI alone attenuated the inflammation in lung tissues at the early stage, the lesions were augmented after long-term treatment. Because it takes a long period for AE–rMS to stimulate effective cellular immune response to eliminate bacteria, the CFU remained unaffected at the initial stage of infection. However, when combined with chemotherapy drugs, the bacteria loads were decreased dramatically. At week 22, the combination therapy reduced the bacteria burden to undetectable levels comparable to the effects achieved by chemotherapy alone, though neither could completely eliminate the bacterial infection in lung. When the reactivation was induced, the relapse rates of both groups reached 100%, but the bacterial burden was lower in the RI/AE–rMS group than that in the RI group (P < 0.05) at week 26. This suggests that the administration of either single or combinational use of AE–rMS is capable of effectively inhibiting persistent bacterial counts in organs, but AE–rMS in the presence of chemotherapy could further reduce the bacterial counts in TB relapse. At week 22, congestion and edema were found in lung in the RI/AE–rMS group. After the withdrawal of chemotherapy drugs, inflammatory fibrosis was also observed in the RI/AE–rMS group at week 26, with alleviation in the extent of inflammatory responses in lung. These findings collectively indicate that early pathological lesions could be improved by long-term AE–rMS immunotherapy, but a combinational immunochemotherapy may extend the protection against persistent TB with sustainably reduced bacterial loads. In addition, given the strong Th1-type immune responses induced by AE–rMS, prolonged immunochemotherapy could be more beneficial for the ultimate cure of PTBI.
In summary, this study demonstrates that a combinational use of chemotherapy and AE–rMs treatment induced relevant Th1-type immune responses, providing an effective strategy against PTBI. However, considering the transient bacteria-growth inhibition effects compromised by this strategy, future studies are warranted to either improve the current treatment or develop alternative ones aiming for longer therapeutic efficacy.
Materials and Methods
Bacteria, plasmids, and reagents
The MS strain MC2 155 was purchased from the American Type Culture Collection. E. coli DH5α, Mtb strains H37Rv and BCG (Denmark strain), and the shuttle expression vector pDE22 were maintained in our laboratory.42 Middlebrook 7H9 liquid medium, 7H10 solid medium, and oleic acid-albumin-dextrose-catalase enrichment medium were obtained from BD Biosciences. Both rifampin (RFP) and isoniazid (INH) were purchased from Sigma-Aldrich. Reagents and kits for the lymphocyte proliferation assay, cytotoxicity assay, and cytokine detection of IL-2 and IFN-γ were purchased from Mabtech. The EL4 cell line stably expressing the Ag85B–ESAT6 fusion protein (EL4–Ag85B–ESAT6) was established in our laboratory.
Construction of AE–rMS
Primers were designed based on the nucleotide sequences of the ag85b and esat6 genes of the Mtb H37Rv strain. The ag85b gene was amplified using primers 5′-TAGGATCCAT GACCGCGGGC GCGTTCTC-3′ (forward) and 5′-GCATCGATTC ATGCGAACAT CCCAGTGA-3′ (reverse), and the esat6 gene was amplified using primers: 5′-GCATCGATGG TGGCTCAGGT GGCTCCGGTG GAGGCGGAAG CGGCGGTGGA GGATCAACAG AGCAGCAGTG GAATTT-3′ (forward) and 5′-GCAAGCTTTC ATGCGAACAT CCCAGTGA-3′ (reverse). A 48-bp sequence encoding a hydrophobic linker was added between the ag85b 3′ end and the esat6 5′ end to ensure proper folding of the encoded Ag85B–ESAT6 fusion protein. The PCR conditions were 30 cycles of 94 °C for 45 s, 65 °C for 45 s, 72 °C for 50 s, followed by a final extension step at 72 °C for 5 min. After validating the sequences (AuGCT Biotechnology), the ag85b and esat6 PCR products were inserted into corresponding sites of the multiple cloning site region of the shuttle expression vector pDE22. The recombinant plasmid was transformed into MS by electroporation, and the transformed recombinant MS bacteria were selected on solid 7H10 agar containing hygromycin (50 μg/ml, Sigma-Aldrich) for 3 d.18 After selection, the bacteria were harvested, boiled in loading buffer and analyzed by western blotting using monoclonal antibody against ESAT6 and polyclonal antibody against Ag85B (Santa Cruz Biotechnology). Positive rMS strain after screening was designated AE–rMS.
Determination of growth rates of MS and AE–rMS
Wild-type MS and AE–rMS were cultured in Middlebrook 7H9 medium supplemented with 0.5% glycerol and 10% albumin-dextrose-catalase. For determining growth rates, 200 µl of each starting culture was added to a conical tube containing 100 ml of culture medium and incubated at 37 °C with shaking at 200 rpm. Two-milliliter aliquot of each culture was sampled after 24, 30, 36, 48, 54, 60, 72, 78, 84, 96, and 108 h of incubation for optical density measurement at the wavelength of 600 nm.
Mouse immunizations
Six-week-old specific pathogen-free C57BL/6 female mice provided by the Animal Center of the Fourth Military Medical University were randomly divided into five groups (n = 20 for each group) to receive respective subcutaneous injections as follows: Normal control group (NC) receiving 0.2 mL saline/mouse, BCG group receiving 5 × 106 CFU/mouse, MS group receiving 1 × 106 CFU/mouse, Ag85B-ESAT6 protein (AE) group receiving 50 µg AE protein with 0.2 ml adjuvant mixture of DDA-MPL and AE–rMS group receiving 1 × 106 CFU/mouse. Mice in the AE protein group received three injections every two weeks, and all the other groups were given another injection one month after the first shot. All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the Fourth Military Medical University (ID11014).
Detection of serum specific antibodies
Mice were bled at the week 2, 4, 6, and 8 since the first immunization to obtain sera. Antibody responses were measured by an enzyme-linked immunosorbent assay (ELISA) using microplates. The plates were coated with the purified Ag85B-ESAT6 fusion protein (50 μg/ml, produced in E.coli) at 4 °C and blocked with 1% bovine serum albumin at 37 °C for 1 h. Serum samples were diluted appropriately. The titers of specific antibodies were determined using a spectrophotometer at the wavelength of 490 nm.
Cytokine enzyme-linked immunosorbent spot (ELISPOT) assays
The spleen was removed aseptically from each group and placed in RPMI-1640 medium containing 10% fetal calf serum. After gently grinding the spleens through a 200-gauge stainless steel mesh, single-cell suspensions were prepared by density gradient centrifugation using Lymphocyte-M. Purified cells were counted and plated at 2 × 105 cells per well. Purified AE fusion protein was added (5 µg/well) as the stimulator. The IFN-γ and IL-2 ELISPOT assays were conducted using a kit according to the manufacturer’s instructions.
Cytotoxicity assays
Mouse lymphocytes were prepared as described above and added to 96-well plates in serial 2-fold dilutions with 2 × 106, 1 × 106, 5 × 105, and 2.5 × 105 cells per well in triplicate. The EL40–Ag85B–ESAT6 target cells were co-cultured with effector cells at various ratios (10:1, 25:1, 50:1, and 100:1) at 37 °C under 5% CO2 for 5 h. Subsequently, supernatants were collected and assayed for lactate dehydrogenase (LDH) activity using a kit (Promega) with a spectrophotometer at the wavelength of 490 nm. Cytolytic activity was calculated as follows: cytotoxicity rate (%) = (experimental release − effector spontaneous release − target spontaneous release) / (target maximum release − target spontaneous release).
Immunotherapy in the PTBI mouse model
The low-dose PTBI mouse model referred to the modified Cornell model39 was established by intravenous inoculation of 6-week-old female C57BL/6 mice with 1 × 104 CFU of Mtb. After 4 weeks, the mice were treated with a 4-week course of the anti-mycobacteria drugs, INH (0.1 g/L), and PZA (15 g/L), delivered in drinking water. Detailed schedule of the generation of PTBI model is provided in Figure 3. At week 8 and 10, viable bacteria loads were determined by plating homogenates of spleen and right lung to confirm the successful establishment of the PTBI animal model.
Two-hundred and forty PTBI mice were randomly divided into four groups with 60 mice each and administered with various therapeutic protocols as outlined in Figure 3. The PTBI mice received the following different treatment: saline only (model control group, MC), AE–rMS (1 × 106 CFU/mouse), Rifampin (105 mg/L) and Isoniazid (108.5 mg/L) (RI) chemotherapy, and combination of RI chemotherapy with AE–rMS. In AE–rMS treatment group, AE–rMS was given at week 10 and 14. In the comibnation group receiving both RI and AE–rMS, mice were first treated with RI for 4 weeks from week 10 to 14, and then AE–rMS immunotherapy was given at week 14 and 18. The chemotherapy group was treated with RI from week 10 to 20 post-infection. At week 22, re-infection was induced in all groups of mice by intramuscular injection with dexamethasone (120 μg/0.1 ml/mouse) given twice with three days apart. At week 26, mice were sacrificed. The right lung of four mice from each group was removed for determination of bacteria CFU. The left lung was stained with H&E for histological observation as described above, In each lung section, the extent of inflammatory was scored semi-quantitatively (0, 1, 2, 3, 4, and 5 representing absent, minimal, slight, moderate, marked, and strong staining, respectively) by 3 pathologists independently. In this scoring system, both the frequency and severity of lesion were incorporated. The morphology of MTB in lung at week 26 was observed by modified acid-fast staining. Normal C57BL/6 mice were used as controls.
Four mice from each group were sacrificed at week 10, 14, 18, 22, and 26. Splenocytes were separated by the same method as described above. The expression level of IFN-γ in the single cell of spleen was determined by ELISPOT kit (Mabtech).
Statistical analysis
Statistical analyses were performed using SPSS software. When analyzing parametric or log transformed data, The Student t test was performed to compare 2 groups of mice, while an ANOVA with a Dunnett test was used to compare more than 2 groups of mice. P values less than 0.05 were considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr Zengshan Li, Dr Peizhen Hu, and Prof Zhe Wang for the pathology analysis, and Yilin Zhao for technical assistance. This work was supported by National major infectious disease prevention and control project (2012ZX10003008-007 and 2012FWPT-03).
References
- 1.Anderson EJ, Webb EL, Mawa PA, Kizza M, Lyadda N, Nampijja M, Elliott AM. The influence of BCG vaccine strain on mycobacteria-specific and non-specific immune responses in a prospective cohort of infants in Uganda. Vaccine. 2012;30:2083–9. doi: 10.1016/j.vaccine.2012.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shao Y, Yang D, Xu W, Lu W, Song H, Dai Y, Shen H, Wang J. Epidemiology of anti-tuberculosis drug resistance in a Chinese population: current situation and challenges ahead. BMC Public Health. 2011;11:110. doi: 10.1186/1471-2458-11-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomioka H. Current status of some antituberculosis drugs and the development of new antituberculous agents with special reference to their in vitro and in vivo antimicrobial activities. Curr Pharm Des. 2006;12:4047–70. doi: 10.2174/138161206778743646. [DOI] [PubMed] [Google Scholar]
- 4.Cooper AM, Khader SA. The role of cytokines in the initiation, expansion, and control of cellular immunity to tuberculosis. Immunol Rev. 2008;226:191–204. doi: 10.1111/j.1600-065X.2008.00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dlugovitzky D, Stanford C, Stanford J. Immunological basis for the introduction of immunotherapy with Mycobacterium vaccae into the routine treatment of TB. Immunotherapy. 2011;3:557–68. doi: 10.2217/imt.11.6. [DOI] [PubMed] [Google Scholar]
- 6.Roy E, Lowrie DB, Jolles SR. Current strategies in TB immunotherapy. Curr Mol Med. 2007;7:373–86. doi: 10.2174/156652407780831557. [DOI] [PubMed] [Google Scholar]
- 7.Johnson BJ, Estrada I, Shen Z, Ress S, Willcox P, Colston MJ, Kaplan G. Differential gene expression in response to adjunctive recombinant human interleukin-2 immunotherapy in multidrug-resistant tuberculosis patients. Infect Immun. 1998;66:2426–33. doi: 10.1128/iai.66.6.2426-2433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang CC, Rook GA. Inhibition of an established allergic response to ovalbumin in BALB/c mice by killed Mycobacterium vaccae. Immunology. 1998;93:307–13. doi: 10.1046/j.1365-2567.1998.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamasur B, Haile M, Pawlowski A, Schroder U, Kallenius G, Svenson SB. A mycobacterial lipoarabinomannan specific monoclonal antibody and its F(ab’) fragment prolong survival of mice infected with Mycobacterium tuberculosis. Clin Exp Immunol. 2004;138:30–8. doi: 10.1111/j.1365-2249.2004.02593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silva CL, Bonato VL, Coelho-Castelo AA, De Souza AO, Santos SA, Lima KM, Faccioli LH, Rodrigues JM. Immunotherapy with plasmid DNA encoding mycobacterial hsp65 in association with chemotherapy is a more rapid and efficient form of treatment for tuberculosis in mice. Gene Ther. 2005;12:281–7. doi: 10.1038/sj.gt.3302418. [DOI] [PubMed] [Google Scholar]
- 11.Taylor JL, Ordway DJ, Troudt J, Gonzalez-Juarrero M, Basaraba RJ, Orme IM. Factors associated with severe granulomatous pneumonia in Mycobacterium tuberculosis-infected mice vaccinated therapeutically with hsp65 DNA. Infect Immun. 2005;73:5189–93. doi: 10.1128/IAI.73.8.5189-5193.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernández-Pando R, Aguilar D, Orozco H, Cortez Y, Brunet LR, Rook GA. Orally administered Mycobacterium vaccae modulates expression of immunoregulatory molecules in BALB/c mice with pulmonary tuberculosis. Clin Vaccine Immunol. 2008;15:1730–6. doi: 10.1128/CVI.00286-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang C, He YL, Zhang L, Xu L, Yi Z, Wang Y, Li N, Zhu D. GLS/IL-12-modified Mycobacterium smegmatis as a novel anti-tuberculosis immunotherapeutic vaccine. Int J Tuberc Lung Dis. 2009;13:1360–6. [PubMed] [Google Scholar]
- 14.Cayabyab MJ, Hovav AH, Hsu T, Krivulka GR, Lifton MA, Gorgone DA, Fennelly GJ, Haynes BF, Jacobs WR, Jr., Letvin NL. Generation of CD8+ T-cell responses by a recombinant nonpathogenic Mycobacterium smegmatis vaccine vector expressing human immunodeficiency virus type 1 Env. J Virol. 2006;80:1645–52. doi: 10.1128/JVI.80.4.1645-1652.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hovav AH, Cayabyab MJ, Panas MW, Santra S, Greenland J, Geiben R, Haynes BF, Jacobs WR, Jr., Letvin NL. Rapid memory CD8+ T-lymphocyte induction through priming with recombinant Mycobacterium smegmatis. J Virol. 2007;81:74–83. doi: 10.1128/JVI.01269-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yi Z, Fu Y, Yang C, Li J, Luo X, Chen Q, Zeng W, Jiang S, Jiang Y, He Y, et al. Recombinant M. smegmatis vaccine targeted delivering IL-12/GLS into macrophages can induce specific cellular immunity against M. tuberculosis in BALB/c mice. Vaccine. 2007;25:638–48. doi: 10.1016/j.vaccine.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 17.Goldstone RM, Moreland NJ, Bashiri G, Baker EN, Shaun Lott J. A new Gateway vector and expression protocol for fast and efficient recombinant protein expression in Mycobacterium smegmatis. Protein Expr Purif. 2008;57:81–7. doi: 10.1016/j.pep.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 18.Zhang H, Peng P, Miao S, Zhao Y, Mao F, Wang L, Bai Y, Xu Z, Wei S, Shi C. Recombinant Mycobacterium smegmatis expressing an ESAT6-CFP10 fusion protein induces anti-mycobacterial immune responses and protects against Mycobacterium tuberculosis challenge in mice. Scand J Immunol. 2010;72:349–57. doi: 10.1111/j.1365-3083.2010.02448.x. [DOI] [PubMed] [Google Scholar]
- 19.Zhao S, Zhao Y, Mao F, Zhang C, Bai B, Zhang H, Shi C, Xu Z. Protective and therapeutic efficacy of Mycobacterium smegmatis expressing HBHA-hIL12 fusion protein against Mycobacterium tuberculosis in mice. PLoS One. 2012;7:e31908. doi: 10.1371/journal.pone.0031908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weinrich Olsen A, van Pinxteren LA, Meng Okkels L, Birk Rasmussen P, Andersen P. Protection of mice with a tuberculosis subunit vaccine based on a fusion protein of antigen 85b and esat-6. Infect Immun. 2001;69:2773–8. doi: 10.1128/IAI.69.5.2773-2778.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.You Q, Wu Y, Jiang D, Wu Y, Wang C, Wei W, Yu X, Zhang X, Kong W, Jiang C. Immune responses induced by heterologous boosting of recombinant bacillus Calmette-Guerin with Ag85B-ESAT6 fusion protein in levamisole-based adjuvant. Immunol Invest. 2012;41:412–28. doi: 10.3109/08820139.2012.658940. [DOI] [PubMed] [Google Scholar]
- 22.Uhlin M, Andersson J, Zumla A, Maeurer M. Adjunct immunotherapies for tuberculosis. J Infect Dis. 2012;205(Suppl 2):S325–34. doi: 10.1093/infdis/jis197. [DOI] [PubMed] [Google Scholar]
- 23.Prabowo SA, Gröschel MI, Schmidt ED, Skrahina A, Mihaescu T, Hastürk S, Mitrofanov R, Pimkina E, Visontai I, de Jong B, et al. Targeting multidrug-resistant tuberculosis (MDR-TB) by therapeutic vaccines. Med Microbiol Immunol. 2013;202:95–104. doi: 10.1007/s00430-012-0278-6. [DOI] [PubMed] [Google Scholar]
- 24.Gil O, Guirado E, Gordillo S, Díaz J, Tapia G, Vilaplana C, Ariza A, Ausina V, Cardona PJ. Intragranulomatous necrosis in lungs of mice infected by aerosol with Mycobacterium tuberculosis is related to bacterial load rather than to any one cytokine or T cell type. Microbes Infect. 2006;8:628–36. doi: 10.1016/j.micinf.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 25.Prior JG, Khan AA, Cartwright KA, Jenkins PA, Stanford JL. Immunotherapy with Mycobacterium vaccae combined with second line chemotherapy in drug-resistant abdominal tuberculosis. J Infect. 1995;31:59–61. doi: 10.1016/S0163-4453(95)91488-9. [DOI] [PubMed] [Google Scholar]
- 26.Changhong S, Hai Z, Limei W, Jiaze A, Li X, Tingfen Z, Zhikai X, Yong Z. Therapeutic efficacy of a tuberculosis DNA vaccine encoding heat shock protein 65 of Mycobacterium tuberculosis and the human interleukin 2 fusion gene. Tuberculosis (Edinb) 2009;89:54–61. doi: 10.1016/j.tube.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Frantz FG, Ito T, Cavassani KA, Hogaboam CM, Lopes Silva C, Kunkel SL, Faccioli LH. Therapeutic DNA vaccine reduces schistosoma mansoni-induced tissue damage through cytokine balance and decreased migration of myofibroblasts. Am J Pathol. 2011;179:223–9. doi: 10.1016/j.ajpath.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang XY, Chen QF, Li YP, Wu SM. Mycobacterium vaccae as adjuvant therapy to anti-tuberculosis chemotherapy in never-treated tuberculosis patients: a meta-analysis. PLoS One. 2011;6:e23826. doi: 10.1371/journal.pone.0023826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andersen P. The T cell response to secreted antigens of Mycobacterium tuberculosis. Immunobiology. 1994;191:537–47. doi: 10.1016/S0171-2985(11)80460-2. [DOI] [PubMed] [Google Scholar]
- 30.Shi L, North R, Gennaro ML. Effect of growth state on transcription levels of genes encoding major secreted antigens of Mycobacterium tuberculosis in the mouse lung. Infect Immun. 2004;72:2420–4. doi: 10.1128/IAI.72.4.2420-2424.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu DH, Hu XD, Cai H. Efficient tuberculosis treatment in mice using chemotherapy and immunotherapy with the combined DNA vaccine encoding Ag85B, MPT-64 and MPT-83. Gene Ther. 2008;15:652–9. doi: 10.1038/gt.2008.13. [DOI] [PubMed] [Google Scholar]
- 32.Hall LJ, Clare S, Pickard D, Clark SO, Kelly DL, El Ghany MA, Hale C, Dietrich J, Andersen P, Marsh PD, et al. Characterisation of a live Salmonella vaccine stably expressing the Mycobacterium tuberculosis Ag85B-ESAT6 fusion protein. Vaccine. 2009;27:6894–904. doi: 10.1016/j.vaccine.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altaf M, Miller CH, Bellows DS, O’Toole R. Evaluation of the Mycobacterium smegmatis and BCG models for the discovery of Mycobacterium tuberculosis inhibitors. Tuberculosis (Edinb) 2010;90:333–7. doi: 10.1016/j.tube.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Wang R, Marcotte EM. The proteomic response of Mycobacterium smegmatis to anti-tuberculosis drugs suggests targeted pathways. J Proteome Res. 2008;7:855–65. doi: 10.1021/pr0703066. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez L, Tirado Y, Reyes F, Puig A, Kadir R, Borrero R, Fernandez S, Reyes G, Alvarez N, Garcia MA, et al. Proteoliposomes from Mycobacterium smegmatis induce immune cross-reactivity against Mycobacterium tuberculosis antigens in mice. Vaccine. 2011;29:6236–41. doi: 10.1016/j.vaccine.2011.06.077. [DOI] [PubMed] [Google Scholar]
- 36.Post FA, Manca C, Neyrolles O, Ryffel B, Young DB, Kaplan G. Mycobacterium tuberculosis 19-kilodalton lipoprotein inhibits Mycobacterium smegmatis-induced cytokine production by human macrophages in vitro. Infect Immun. 2001;69:1433–9. doi: 10.1128/IAI.69.3.1433-1439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaufmann SH, Gengenbacher M. Recombinant live vaccine candidates against tuberculosis. Curr Opin Biotechnol. 2012;23:900–7. doi: 10.1016/j.copbio.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 38.Chao MC, Rubin EJ. Letting sleeping dos lie: does dormancy play a role in tuberculosis? Annu Rev Microbiol. 2010;64:293–311. doi: 10.1146/annurev.micro.112408.134043. [DOI] [PubMed] [Google Scholar]
- 39.Scanga CA, Mohan VP, Joseph H, Yu K, Chan J, Flynn JL. Reactivation of latent tuberculosis: variations on the Cornell murine model. Infect Immun. 1999;67:4531–8. doi: 10.1128/iai.67.9.4531-4538.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aagaard C, Hoang T, Dietrich J, Cardona PJ, Izzo A, Dolganov G, Schoolnik GK, Cassidy JP, Billeskov R, Andersen P. A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nat Med. 2011;17:189–94. doi: 10.1038/nm.2285. [DOI] [PubMed] [Google Scholar]
- 41.Yuan W, Dong N, Zhang L, Liu J, Lin S, Xiang Z, Qiao H, Tong W, Qin C. Immunogenicity and protective efficacy of a tuberculosis DNA vaccine expressing a fusion protein of Ag85B-Esat6-HspX in mice. Vaccine. 2012;30:2490–7. doi: 10.1016/j.vaccine.2011.06.029. [DOI] [PubMed] [Google Scholar]
- 42.Chang-hong S, Xiao-wu W, Hai Z, Ting-fen Z, Li-Mei W, Zhi-kai X. Immune responses and protective efficacy of the gene vaccine expressing Ag85B and ESAT6 fusion protein from Mycobacterium tuberculosis. DNA Cell Biol. 2008;27:199–207. doi: 10.1089/dna.2007.0648. [DOI] [PubMed] [Google Scholar]