

Abstract
Genomic selection (GS) is of interest in breeding because of its potential for predicting the genetic value of individuals and increasing genetic gains per unit of time. To date, very few studies have reported empirical results of GS potential in the context of large population sizes and long breeding cycles such as for boreal trees. In this study, we assessed the effectiveness of marker-aided selection in an undomesticated white spruce (Picea glauca (Moench) Voss) population of large effective size using a GS approach. A discovery population of 1694 trees representative of 214 open-pollinated families from 43 natural populations was phenotyped for 12 wood and growth traits and genotyped for 6385 single-nucleotide polymorphisms (SNPs) mined in 2660 gene sequences. GS models were built to predict estimated breeding values using all the available SNPs or SNP subsets of the largest absolute effects, and they were validated using various cross-validation schemes. The accuracy of genomic estimated breeding values (GEBVs) varied from 0.327 to 0.435 when the training and the validation data sets shared half-sibs that were on average 90% of the accuracies achieved through traditionally estimated breeding values. The trend was also the same for validation across sites. As expected, the accuracy of GEBVs obtained after cross-validation with individuals of unknown relatedness was lower with about half of the accuracy achieved when half-sibs were present. We showed that with the marker densities used in the current study, predictions with low to moderate accuracy could be obtained within a large undomesticated population of related individuals, potentially resulting in larger gains per unit of time with GS than with the traditional approach.
Introduction
Marker-assisted selection for individuals with superior phenotypes has been suggested for quite some time, particularly to increase selection efficiency for low-heritability traits and for improving gains per time unit for traits requiring long testing periods (Lande and Thompson, 1990; Strauss et al., 1992). However, despite some promising results from experimental studies at the genome scale in forest trees (Resende et al., 2012a, 2012b), further investigation is still needed to bring marker-assisted selection to the operational level. This is especially true in forestry, where tree breeding programmes are frequently dealing with species with long breeding cycles involving essentially undomesticated populations and where large population sizes are often considered for the maintenance of high genetic diversity in reforestation stocks (Mullin et al., 2011; Namroud et al., 2012).
Various approaches have been taken over the past 15 years to identify genetic markers that could explain a significant part of the variation observed in quantitative traits of interest. These approaches evolved from detecting quantitative trait loci (QTLs) in single families, to candidate gene approaches in larger populations and, finally, to genome-wide association studies or genomic selection (GS) using either first-generation or advanced breeding populations (Burdon and Wilcox, 2011).
Association studies have been carried out with some success for major crops (Buckler et al., 2009) and tree geneticists have also focussed their more recent efforts on association studies in large populations with low linkage disequilibrium (LD), aiming to overcome weaknesses in QTL analyses (Burdon and Wilcox, 2011). Promising results have been reported for various traits in several species (see, for example, González-Martínez et al., 2007; Beaulieu et al., 2011). However, as for QTL mapping (Pelgas et al., 2011), the variation in quantitative traits explained by individual markers is generally low, rarely reaching 5%.
Meuwissen et al. (2001) proposed an alternative approach relying on genome-wide distributed markers to model the entire complement of QTL effects across the genome, whether these effects are significant or not, and to estimate genomic estimated breeding values (GEBVs). This method, called ‘genomic selection', overcomes the problem of the limited proportion of total genetic variation captured by each significant individual marker such as in association genetics with properly designed training populations. GS is particularly well adapted for marker-assisted selection as it makes it possible to predict the genetic value of an individual using all its known genotypic information. The utility of GS models has already been shown for the selection of complex traits in numerous species, such as plants (Heffner et al., 2010) and animals (Habier et al., 2010).
The potential usefulness of GS for marker-assisted selection in forest trees was first shown by deterministic and simulation models (Grattapaglia and Resende, 2011). These simulations indicated that high accuracy of GEBVs could be obtained with populations of small effective size and high LD with a reasonably limited number of markers, but these represented idealized conditions. Recent results from empirical studies in small breeding populations confirmed that quite a high accuracy of GEBVs could be obtained (Resende et al., 2012a, 2012b; Zapata-Valenzuela et al., 2012). They also showed that this approach could help speed up breeding cycles and significantly increase gains per time unit.
However, no attempt has been made to estimate the value of GS for populations with large effective size, such as for undomesticated tree populations or for commonly used first-generation breeding populations of boreal conifer species (Mullin et al., 2011). Conifer breeders generally assemble large breeding populations of several hundred individuals in order to maintain a high level of genetic diversity for long-term adaptive capacity. The objectives of the present study in that context were to: (1) compare the potential of GS with that of pedigree-based models, that is, based on phenotypic resemblance between relatives using pedigree information, to predict the genetic merit of individual trees for wood and growth traits in a large white spruce (Picea glauca (Moench) Voss) population; (2) evaluate the accuracy of GS predictions and the impacts of marker density as well as the design of the training and the validation data sets on these accuracies; (3) compare the efficiency of GS models relative to the pedigree-based ones; and (4) evaluate empirical genetic gains expected when selecting the top 5% trees based on GEBVs and compare gains per unit of time with the conventional approach.
Materials and methods
Plant material and tissue sampling
A white spruce provenance–progeny test was established by the Canadian Forest Service in Québec, Canada, on three sites (Mastigouche Arboretum: latitude (Lat.) 46°38′ N, longitude (Long.) 73°13′ W, elevation (Elev.) 230 m; Dablon Arboretum: Lat. 48°21′ N, Long. 72°13′ W, Elev. 323 m; and LaPatrie Arboretum: Lat. 45°20′ N, Long. 71°15′ W, Elev. 457 m) in May 1979, with 4-year-old seedlings that had been raised in a greenhouse at the Canadian Forest Service, Laurentian Forestry Centre (Québec City, January to May 1976), and then transplanted into a nursery bed at the Valcartier Forest Experiment Station (Lat. 46°52′ N, Long. 71°32′ W, Elev. 152 m). The test consisted of 214 open-pollinated families, that is, 5 families from each of 43 provenances (except one) sampled in Québec. Seedlings were planted following a randomized complete block design with six blocks, five-tree row plots, and a spacing of 2.4 m between row plots and 1.2 m between trees within plots. At 27 years after planting, three trees from each family covering the range of diameters at breast height were sampled in the Mastigouche Arboretum test. After 4 years, five additional trees per family were selected with the same diameters at breast height constraint to complete the discovery population used in the present study. Needle tissue taken in the upper part of the crown for DNA extractions and a 12-mm increment core taken at 1.3 m from the ground were collected on each tree for wood character assessment, kept on ice and transported to the Canadian Forest Service facilities where they were stored at −10 °C until further treatment.
Two validation populations were also assembled. The first population was sampled in the Dablon Arboretum test. It comprised 125 trees from a random sample of 100 of the 214 open-pollinated families. The second validation population was sampled in a range-wide provenance–progeny test established at Mirabel (Lat. 45°37′ N, Long. 74°05′ W, Elev. 61 m). It comprised 219 trees representing 100 of the 214 open-pollinated families present in the previous tests established in arboreta (Mastigouche, Dablon and LaPatrie). This test was set up in 1984 also using 4-year-old seedlings raised in the nursery of the Valcartier Forest Experiment Station. Foliage and wood core samples were collected 27 years after planting in both tests and the same collection procedure as for the discovery population was followed. Family-wise, there was 47% overlap between the two validation populations. DNA extraction for all the samples collected was conducted as reported in Pavy et al. (2013).
Phenotypic data
Pith to bark profiles of 11 different wood physical characters were obtained for 1694 trees (the discovery population), as in Beaulieu et al. (2011), using the SilviScan technology at FPInnovations facilities (Vancouver, BC, Canada) (Table 1). Measurements were taken from the radial surface of 2 × 7 mm wood flitch samples cut with a twin blade pneumatic saw from the thawed 12-mm wood cores. Wood density was measured at a resolution of 25 μm by X-ray densitometry that corresponds to density at 8% moisture content. Microfibril angle was measured in 1-mm steps using X-ray diffractometry, and wood modulus of elasticity was calculated using both the density and microfibril angle data. Cell wall thickness and cell dimensions were obtained using optical microscopy and image analysis. Trait averages of each trait for each tree were weighted by the annual ring area.
Table 1. Variance components of wood and growth characters estimated in genomic selection analyses with three different models using all 6385 single-nucleotide polymorphisms (SNPs) and the full data set of 1694 trees.
| Traita | Modelb | σ2ac | V′A | σ2u | σ2e | hi2 |
|---|---|---|---|---|---|---|
| Cell population (number per mm2) | Polygenic | — | — | 6423.27 | 19 929.96 | 0.24 |
| Markers | 2.89 | 5631.45 | — | 20 568.07 | 0.22 | |
| Combined | 2.63 | 5117.93 | 2107.22 | 190 60.96 | 0.27 | |
| Fibre coarseness (μg m−1) | ||||||
| Polygenic | — | — | 417.67 | 860.10 | 0.33 | |
| Markers | 0.17 | 330.40 | — | 940.78 | 0.26 | |
| Combined | 0.15 | 290.85 | 162.35 | 825.04 | 0.35 | |
| Crystallite width (nm) | ||||||
| Polygenic | — | — | 1.68E−3 | 1.28E−3 | 0.57 | |
| Markers | 3.26E−7 | 6.34E−4 | — | 2.26E−3 | 0.22 | |
| Combined | 1.65E−7 | 3.21E−4 | 1.11E−3 | 1.49E−3 | 0.49 | |
| Wood density (kg m−3) | ||||||
| Polygenic | — | — | 461.52 | 713.88 | 0.39 | |
| Markers | 0.15 | 283.49 | — | 888.90 | 0.24 | |
| Combined | 0.11 | 203.88 | 252.43 | 721.23 | 0.39 | |
| Microfibril angle (degrees) | ||||||
| Polygenic | — | — | 6.60 | 10.76 | 0.38 | |
| Markers | 2.2E−3 | 4.22 | — | 13.10 | 0.24 | |
| Combined | 1.7E−3 | 3.25 | 3.01 | 11.08 | 0.37 | |
| Wood stiffness (GPa) | ||||||
| Polygenic | — | — | 1.08 | 2.40 | 0.31 | |
| Markers | 4.07E−4 | 0.79 | — | 2.67 | 0.23 | |
| Combined | 3.49E−4 | 0.68 | 0.35 | 2.45 | 0.31 | |
| Ring width (mm) | ||||||
| Polygenic | — | — | 1.54E−2 | 0.39 | 0.04 | |
| Markers | 1.73E−5 | 3.36E−2 | — | 0.37 | 0.08 | |
| Combined | 1.44E−5 | 2.81E−2 | 1.18E−2 | 0.36 | 0.10 | |
| Specific fibre surface (m2 kg−1) | ||||||
| Polygenic | — | — | 413.99 | 451.77 | 0.48 | |
| Markers | 0.14 | 272.81 | — | 589.97 | 0.32 | |
| Combined | 0.11 | 217.89 | 178.36 | 467.19 | 0.46 | |
| Cell radial diameter (μm) | ||||||
| Polygenic | — | — | 1.60 | 2.07 | 0.44 | |
| Markers | 5.27E−4 | 1.03 | — | 2.58 | 0.29 | |
| Combined | 4.69E−4 | 0.91 | 0.49 | 2.22 | 0.39 | |
| Cell tangential diameter (μm) | ||||||
| Polygenic | — | — | 0.76 | 2.13 | 0.26 | |
| Markers | 3.10E−4 | 0.60 | — | 2.29 | 0.21 | |
| Combined | 2.65E−4 | 0.52 | 0.34 | 2.04 | 0.30 | |
| Cell wall thickness (μm) | ||||||
| Polygenic | — | — | 1.31E−2 | 2.08E−2 | 0.39 | |
| Markers | 4.72E−6 | 9.19E−3 | — | 2.46E−2 | 0.27 | |
| Combined | 3.86E−6 | 7.53E−3 | 6.11E−3 | 2.03E−2 | 0.40 | |
| 22-Year height (cm) | ||||||
| Polygenic | — | — | 3015.16 | 8818.00 | 0.25 | |
| Markers | 0.91 | 1873.04 | — | 9903.84 | 0.16 | |
| Combined | 0.83 | 1621.10 | 1483.05 | 8711.36 | 0.26 |
Except for 22-year height, all other traits were assessed from wood cores collected when trees were either 31 or 35 years old.
Polygenic indicates pedigree information; markers indicates SNP information only; and combined indicates pedigree and SNP information.
Here, σ2a is additive
genetic variance explained by marker loci;
V′A= is total additive genetic
variance estimated as 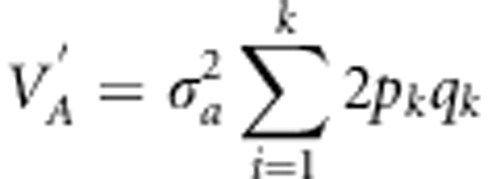 ;
σ2u is polygenic
additive genetic variance;
σ2e is residual
variance; hi2 is individual
heritability.
;
σ2u is polygenic
additive genetic variance;
σ2e is residual
variance; hi2 is individual
heritability.
SNP genotyping
For this experiment, a total of 7338 valid single-nucleotide polymorphisms (SNPs) from 2814 genes were successfully genotyped using the Illumina Infinium HD iSelect bead chip PgAS1 (Illumina, San Diego, CA, USA) as described in Pavy et al. (2013). The genes were from the white spruce gene catalogue GCAT (Rigault et al., 2011) and represented a large variety of functions (Pavy et al., 2013). The SNPs were separated by a minimum distance of 200 bp within genes, given that generally weak LD is observed within gene limits in white spruce genes (Pavy et al., 2012a). The final SNP data set was filtered to retain segregating SNPs that had a GenTrain quality score ⩾0.25, a fixation coefficient |Fe| <0.50, a minor allele frequency ⩾0.003 given the large size of the discovery population, corresponding to a minimum number of heterozygotes of 10, and a call rate at each SNP locus of ⩾50%. A total of 6385 SNPs harboured by 2660 distinct gene loci met these criteria and were used for the present study. Despite the liberal call rate used, >96% of these SNPs had a call rate above 95%.
Population structure
A population structure analysis was used to assess whether there were differences in allele frequencies among unobserved ancestral populations that could bias prediction accuracy estimates. Given the results obtained in previous studies conducted in the same area (Jaramillo-Correa et al., 2001; Namroud et al., 2008, 2010; Beaulieu et al., 2011), we were expecting a weak or no population differentiation from SNPs. We used all available SNPs (m=6385) in multidimensional scaling and principal component analysis (Price et al., 2006) to estimate population covariates for each individual. The results of the two analyses were similar, and two covariates obtained with multidimensional scaling were used in further analyses.
Estimated breeding values
As a control representative of the conventional pedigree-based approach, the EBVs of each tree for each character were obtained using the polygenic model in GS3 (Legarra et al., 2013, http://snp.toulouse.inra.fr/~alegarra/). This mixed linear model is as follows:
where β is a vector of fixed effects (including an overall mean and population structure), u is a vector of random additive genetic polygenic effects with a distribution ∼N(0, Aσ2u), X and T are the incidence matrices and A is the additive genetic (or numerator) relationship matrix (Lynch and Walsh, 1998).
GS analyses
Genomic estimated breeding values
The GEBV of every single tree was estimated for each wood and growth trait using all of the 6385 SNPs available. The effect of each marker was estimated with the mixed linear model 2 in GS3 (Legarra et al., 2013). This model is:
where β is a vector of fixed effects (including an overall mean and population structure), a and e are vectors of random marker and random error effects, respectively, and X and Z are the incidence matrices. The Z matrix was built from the number of alleles observed at each individual and marker pair (0, 1 and 2), the coding representing the number of copies of the minor allele. Missing genotypes (total of 1.21%) at any given marker were replaced by the mean of the genotype coding rounded up to the nearest genotype coding for that marker (average of the population for additive effect).
In model 2, only biallelic markers are considered and the value +½aj is arbitrarily assigned to the first allele, whereas −½aj is assigned to the second allele. Hence, the effects of homozygotes for the first allele are +aj, whereas those of the alternate homozygotes are −aj, and 0 for the heterozygotes. It is also assumed that m follows a normal distribution (∼N(0, Iσ2a)), and I is an identity matrix. Such a model with a normal distribution of marker effects is often called ridge regression best linear unbiased prediction (Meuwissen et al., 2001; VanRaden, 2008). The total additive genetic variance estimated with the markers was estimated as:
 |
where pk and qk are respectively the frequencies of alleles 1 and 2 of the locus k in the population, and the predicted individual GEBVs were calculated as:
 |
where âj is the estimated effect of the
jth SNP locus, and 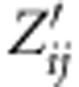 is an indicator covariate (−1, 0 or 1) for the
ith tree and the jth SNP locus.
is an indicator covariate (−1, 0 or 1) for the
ith tree and the jth SNP locus.
Breeding values estimated from combining marker and polygenic effect estimates
Marker and additive polygenic (pedigree-based) effects were jointly estimated using model 3 in GS3 (Legarra et al., 2013), and the following model:
With the GS3 software, best linear unbiased estimation and best linear unbiased prediction of fixed and random variables, respectively, are obtained using the Gauss–Seidel algorithm with residual update (Legarra and Misztal, 2008) that was extended to estimate variance components by Markov chain Monte Carlo (Gibbs sampling). The Gibbs sampler was run for 100 000 iterations with a burn-in of 20 000 iterations, and priors drawn from an inverted χ2 distribution with two degrees of freedom. Convergence of the posterior distribution (800 samples retained) was checked visually using trace plots.
Cross-validation (CV)
To validate the models built to predict breeding values based on the complete set of 6385 SNPs, and to determine the limits of their applicability, we first carried out three CV schemes. For the first one (CV1), one individual per half-sib family was randomly assigned to each of 10 iteration sets, for a total of 214 individuals in each training set. With this validation scheme, half-sib relationships occurred between training and validation sets. The second CV scheme (CV2) was for between-family CV, where entire families were assigned to folds of the training and validation sets. In each of the 10 CV iterations, 27 randomly selected families, corresponding to 12.5% of the 214 available families, composed the validation data set, all the remaining trees made up the training data set. Although all known maternal (seed parent) relatedness was eliminated between training and validation sets in CV2, the unknown paternal (pollen parent) contribution to relatedness within provenance is unaccounted for, and could be traced by markers if present (Doerksen et al., 2014). Note also that spruces have a monoecious mating system allowing individuals to act as both seed and pollen parents. A third CV scheme (CV3) was designed to control for any possible contribution of the pollen parent to relatedness within provenance between CV sets. The scheme was conducted by assigning entire families of geographically distinct provenances to four validation sets, thus eliminating as much as possible any possibility of paternal (and maternal) coancestry between sets. Consequently, if accuracy estimates obtained were different from zero, results from CV3 could likely help determine if historical LD with some traits is still present in extant white spruce populations.
Models were then developed to estimate the marker effects using each of the
training sets per CV scheme, and genomic breeding values of individuals making
up the corresponding validation set were predicted using these models. The
accuracy of prediction was estimated using the correlation r(g,
ĝ) between the breeding value of an individual (g) as
obtained with the conventional approach (that is, (û) from
polygenic model 1), and its estimated value using markers 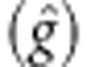 . The predictive ability was
estimated as the correlation between the observed and the estimated phenotype
. The predictive ability was
estimated as the correlation between the observed and the estimated phenotype
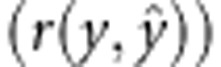 . Both GEBV
accuracy and predictive ability were reported as the average of the correlation
coefficients of the CV scheme used. An accuracy estimate was also estimated for
the pedigree method using a 10-fold CV scheme. Relative efficiency of GS models
was then estimated as the ratio of the accuracy of GS models to that of
pedigree-based ones.
. Both GEBV
accuracy and predictive ability were reported as the average of the correlation
coefficients of the CV scheme used. An accuracy estimate was also estimated for
the pedigree method using a 10-fold CV scheme. Relative efficiency of GS models
was then estimated as the ratio of the accuracy of GS models to that of
pedigree-based ones.
Validation across sites
The three previous CV schemes were conducted on a single (Mastigouche) site, and hence there is an additional need to assess the impact of possible genotype-by-interaction (G × E) on prediction accuracy. Because of financial constraints, a full across-site validation using all the SNPs available as well as all the traits assessed in populations assembled on different testing sites was not possible. We thus limited the validation to two phenotypic traits, that is, wood density and microfibril angle. Moreover, only ∼350 SNPs per trait that were found to be most significantly associated (P<0.05) with these two traits in the discovery population (Mastigouche Arboretum), after an association study carried out using a mixed linear model implemented in Tassel (http://www.maizegenetics.net/tassel), were retained to genotype the two validation populations established at the Dablon Arboretum and Mirabel site. Thus, the training data sets consisted of all individuals from the Mastigouche Arboretum, whereas the individuals sampled at the Dablon Arboretum and Mirabel site were used as testing data sets. Genotyping was conducted at the Genome Quebec Innovation Centre (McGill University, Montreal, QC, Canada) using the highly multiplexed Illumina GoldenGate assay following procedures described in Beaulieu et al. (2011).
Results
Population structure
A weak population structure with no geographical pattern was detected with both principal component analysis and multidimensional scaling analysis, with 1.3% of variance explained by the first two principal component analysis eigenvectors. Weak population structure in white spruce from Québec has been reported in several studies using neutral genetic markers (Jaramillo-Correa et al., 2001; Namroud et al., 2008, 2010) and was expected based on our previous analysis of a subset of the present discovery population (Beaulieu et al., 2011). As a cautionary measure, we used the multidimensional scaling coefficients as population covariates for the GS analyses to control for any potential bias in prediction accuracy that could be brought about by such a population structure.
Variance component estimates with the full data set
Estimates of variance components with the full data set and the 6385 markers for the three models, that is, polygenic additive genetic effects (Equation 1), marker effects (Equation 2) and combined marker and polygenic effects (Equation 5), are presented for each wood and growth trait in Table 1. When comparing the first two models, a slight increase was observed in the estimates of error variance (σ2e) for the marker-effect model. In addition, the estimates of additive genetic variance explained by marker loci (V′A) were lower than those estimated using pedigree information (σ2u). However, the marker-effect model could nevertheless capture at least 60% of the additive genetic variance (V′A/σ2u) for most of the traits. Models combining both marker and pedigree information generally lead to the highest estimates of individual heritability. This is likely because of the fact that pedigree information makes it possible to capture unmarked loci that are also involved in the genetic control of the traits.
Goodness of fit of the models built with the full data set
The goodness of fit of the various models was first estimated using the full data set (Table 2). For all the traits, except for average ring width, the correlation between observed and estimated phenotypes was the highest with the polygenic model, followed by the combined marker-pedigree model and the marker-only model. The correlation between the individual breeding values estimated with the polygenic model and those estimated with the other two models ranged from 0.788 to 0.985 (Table 2). The lowest value was obtained for ring width.
Table 2. Goodness of fit of the models based on pedigree information, on marker-locus information or on combined marker-locus and pedigree information and correlations of breeding values estimated with the pedigree-based and the two other models using all 6385 SNPs.
| Traita |
Goodness of
fitb |
Correlation of
EBVs |
|||
|---|---|---|---|---|---|
| Pedigreec | Markers | Combined | Markers | Combined | |
| Cell population | 0.852 | 0.749 | 0.833 | 0.850 | 0.985 |
| Fibre coarseness | 0.942 | 0.817 | 0.907 | 0.866 | 0.977 |
| Crystallite width | 0.977 | 0.763 | 0.959 | 0.838 | 0.892 |
| Wood density | 0.918 | 0.760 | 0.903 | 0.871 | 0.950 |
| Microfibril angle | 0.951 | 0.810 | 0.921 | 0.872 | 0.962 |
| Wood stiffness | 0.921 | 0.783 | 0.870 | 0.870 | 0.982 |
| Ring width | 0.371 | 0.515 | 0.566 | 0.788 | 0.978 |
| Specific fibre surface | 0.961 | 0.834 | 0.936 | 0.891 | 0.967 |
| Cell radial diameter | 0.942 | 0.799 | 0.891 | 0.872 | 0.978 |
| Cell tangential diameter | 0.906 | 0.775 | 0.882 | 0.851 | 0.972 |
| Cell wall thickness | 0.939 | 0.810 | 0.922 | 0.878 | 0.965 |
| 22-Year height | 0.914 | 0.745 | 0.879 | 0.832 | 0.958 |
Abbreviations: EBV, estimated breeding value; SNP, single-nucleotide polymorphism.
For units, see Table 1.
Correlation between the observed and the estimated phenotypic values.
Pedigree indicates pedigree information only; markers indicates SNP information only; and combined indicates pedigree and SNP information.
Predictive ability of the models
The predictive ability of the models based on markers was always slightly better than those based on pedigree information alone when the validations were made using the CV1 scheme (within-family CV; Table 3, upper part). Combining information from both the pedigree and the markers in the GS models did not markedly increase the predictive ability (Table 3). When CV was conducted by controlling for known relatedness (half-sibs) between training and testing/validation sets (CV2 scheme), the predictive ability of the models was substantially lower for most of the traits (Table 3, lower part). As expected, models based on pedigree information had lower predictive abilities ranging from −0.074 for fibre coarseness to 0.257 for wood density.
Table 3. Predictive ability of wood and growth traits of the different models for genomic selection using all 6385 SNPs.
| Traita | Pedigreeb | Markers | Combined |
|---|---|---|---|
| Within families (CV1)c | |||
| Cell population | 0.205 (0.015) | 0.265 (0.019) | 0.265 (0.019) |
| Fibre coarseness | 0.174 (0.014) | 0.235 (0.011) | 0.237 (0.011) |
| Crystallite width | 0.271 (0.013) | 0.206 (0.013) | 0.250 (0.012) |
| Wood density | 0.330 (0.017) | 0.325 (0.018) | 0.339 (0.017) |
| Microfibril angle | 0.222 (0.012) | 0.221 (0.023) | 0.236 (0.021) |
| Wood stiffness | 0.177 (0.011) | 0.209 (0.016) | 0.210 (0.016) |
| Ring width | 0.183 (0.015) | 0.208 (0.014) | 0.200 (0.015) |
| Specific fibre surface | 0.256 (0.015) | 0.293 (0.019) | 0.296 (0.018) |
| Cell radial diameter | 0.296 (0.015) | 0.351 (0.016) | 0.352 (0.018) |
| Cell tangential diameter | 0.157 (0.019) | 0.184 (0.028) | 0.191 (0.027) |
| Cell wall thickness | 0.262 (0.014) | 0.293 (0.015) | 0.303 (0.014) |
| 22-Year height | 0.122 (0.010) | 0.166 (0.012) | 0.166 (0.012) |
| Between families (CV2)d | |||
| Cell population | 0.184 (0.049) | 0.250 (0.032) | 0.246 (0.033) |
| Fibre coarseness | −0.074 (0.027) | 0.152 (0.011) | 0.147 (0.013) |
| Crystallite width | 0.105 (0.013) | 0.151 (0.023) | 0.142 (0.023) |
| Wood density | 0.257 (0.052) | 0.268 (0.047) | 0.270 (0.049) |
| Microfibril angle | −0.007 (0.015) | 0.117 (0.024) | 0.109 (0.024) |
| Wood stiffness | 0.113 (0.026) | 0.169 (0.026) | 0.168 (0.026) |
| Ring width | 0.239 (0.041) | 0.245 (0.039) | 0.245 (0.039) |
| Specific fibre surface | 0.137 (0.034) | 0.197 (0.030) | 0.194 (0.025) |
| Cell radial diameter | 0.211 (0.044) | 0.311 (0.025) | 0.311 (0.030) |
| Cell tangential diameter | 0.075 (0.038) | 0.158 (0.020) | 0.152 (0.019) |
| Cell wall thickness | 0.143 (0.038) | 0.176 (0.025) | 0.177 (0.027) |
| 22-Year height | 0.073 (0.028) | 0.142 (0.019) | 0.138 (0.020) |
Abbreviations: CV, cross-validation; SNP, single-nucleotide polymorphism.
For units, see Table 1.
Pedigree indicates pedigree information only; markers indicates SNP information only; and combined indicates pedigree and SNP information.
Both training and testing data sets share individuals of the same families.
Training and testing data sets are made up of individuals of different families.
Accuracy of EBVs and GEBVs
The accuracy of predicted breeding values obtained with the different prediction models varied greatly depending on the CV scheme (Table 4). When model validations were conducted using the CV1 scheme, accuracies varied from 0.457 (crystallite width) to 0.517 (cell wall thickness) in models relying on pedigree information alone. They were slightly lower (0.327–0.435) when estimated with the 6385 SNPs. Nevertheless, the relative efficiency of GS models reached 76% of that of pedigree-based ones on average, varying from 68 to 89%, depending on the trait. When validations were carried out using the CV2 scheme, the accuracy of breeding values fell to ∼0 when predicted from pedigree information only and decreased by ∼50% when predicted with all SNPs (Table 4, CV2 scheme). We tested whether population structure might have an effect on the accuracy estimates obtained by running analyses without population structure covariates and we found only a marginal effect. When the choice of families to constitute the training and the validation data sets was constrained by the origin of the families (belonging to different provenances; CV3), the accuracy estimates decreased but were still different from 0 for 7 out of the 12 traits studied (Table 4), and went up to 0.132±0.039 for fibre coarseness.
Table 4. Accuracy of prediction of the genetic value for wood and growth traits using models based on pedigrees or genotypes using all 6385 SNPs.
| Traita |
Within-family
(CV1)b |
Between-family
(CV2)c |
Between-family (CV3)d | ||
|---|---|---|---|---|---|
| Pedigree | Markers | Pedigree | Markers | Markers | |
| Cell population | 0.514 (0.010) | 0.393 (0.015) | 0.035 (0.031) | 0.198 (0.022) | 0.091 (0.027) |
| Fibre coarseness | 0.494 (0.010) | 0.388 (0.012) | −0.015 (0.038) | 0.175 (0.011) | 0.132 (0.039) |
| Crystallite width | 0.457 (0.010) | 0.327 (0.015) | 0.063 (0.027) | 0.173 (0.026) | −0.046 (0.024) |
| Wood density | 0.513 (0.010) | 0.370 (0.012) | 0.012 (0.032) | 0.132 (0.024) | 0.072 (0.033) |
| Microfibril angle | 0.505 (0.010) | 0.378 (0.019) | 0.022 (0.026) | 0.164 (0.029) | −0.026 (0.092) |
| Wood stiffness | 0.505 (0.012) | 0.383 (0.018) | 0.003 (0.039) | 0.184 (0.031) | 0.026 (0.070) |
| Ring width | 0.490 (0.015) | 0.333 (0.012) | −0.045 (0.041) | 0.127 (0.038) | −0.023 (0.047) |
| Specific fibre surface | 0.466 (0.014) | 0.388 (0.023) | 0.000 (0.036) | 0.158 (0.018) | 0.124 (0.011) |
| Cell radial diameter | 0.490 (0.017) | 0.435 (0.018) | 0.038 (0.024) | 0.277 (0.022) | 0.129 (0.048) |
| Cell tangential diameter | 0.500 (0.013) | 0.352 (0.020) | −0.067 (0.034) | 0.150 (0.037) | 0.113 (0.037) |
| Cell wall thickness | 0.517 (0.011) | 0.411 (0.013) | −0.010 (0.040) | 0.129 (0.019) | 0.127 (0.014) |
| 22-Year height | 0.498 (0.010) | 0.360 (0.013) | −0.034 (0.023) | 0.178 (0.029) | 0.090 (0.047) |
Abbreviations: CV, cross-validation; SNP, single-nucleotide polymorphism.
For units, see Table 1.
Both training and testing data sets share individuals of the same families.
Training and testing data sets are made up of individuals of different families.
Families making up the validation data sets are from populations that are not represented in the training data sets.
Genetic gains
Expected genetic gains from 5% selection intensity were estimated empirically under both within- (CV1) and between-family (CV2) validation schemes (Tables 5 and 6). For the CV1 scheme, 30–60% of the maximum genetic gains could be captured by superior trees identified by markers only (Table 5, see ratio GEBVCV1/EBV). However, compared with gains predicted using pedigree information (EBVCV1, EBV obtained after CV), the relative efficiency of the markers varied between 65% (ring width) and 111% (height) (ratio GEBVCV1/EBVCV1). Moreover, with the assumption that a breeding cycle could be completed in 10 years with GS instead of 30 years using the conventional approach, gain per time unit in favour of markers would be two- to threefold (Table 5). When using markers to predict breeding values of trees from families that were different from those used in the training data set (CV2), the predicted gains dropped considerably but were still positive. But in contrast to CV1, no genetic gain was expected when selection was based on pedigree information alone for this validation scheme (Table 6).
Table 5. Empirical genetic gains from a 5% selection intensity made within families using EBVs (model based on pedigree information) and GEBV (model based on all 6385 SNPs) and gain per time unit under the assumption that with GS, a breeding cycle can be completed within a period of 10 years whereas it can be completed in 30 years with the conventional approach.
| Traita |
Empirical genetic
gainb |
|||||
|---|---|---|---|---|---|---|
| EBV (full data set) | GEBVCV1c | Ratio GEBVCV1/EBV (%) | EBVCV1d | Ratio GEBVCV1/EBVCV1 (%) | Difference in gain per time unit (%)e | |
| Cell population | 99.79 | 44.72 | 44.8 | 46.99 | 95.2 | 286 |
| Fibre coarseness | 26.86 | 10.79 | 40.2 | 13.89 | 77.7 | 233 |
| Crystallite width | 0.06 | 0.02 | 36.3 | 0.03 | 75.3 | 226 |
| Wood density | 32.89 | 9.63 | 29.3 | 14.02 | 68.7 | 206 |
| Microfibril angle | −2.73 | −1.02 | 37.3 | −1.05 | 96.9 | 291 |
| Wood stiffness | 1.31 | 0.60 | 45.7 | 0.71 | 84.5 | 253 |
| Ring width | 0.06 | 0.02 | 38.5 | 0.03 | 65.2 | 196 |
| Specific fibre surface | 32.70 | 19.60 | 59.9 | 18.50 | 105.9 | 318 |
| Cell radial diameter | 1.81 | 0.71 | 39.3 | 0.77 | 92.5 | 277 |
| Cell tangential diameter | 1.02 | 0.39 | 38.8 | 0.49 | 80.7 | 242 |
| Cell wall thickness | 0.18 | 0.05 | 30.0 | 0.07 | 75.4 | 226 |
| 22-Year height | 60.60 | 27.94 | 46.1 | 25.19 | 110.9 | 333 |
Abbreviations: CV, cross-validation; EBV, estimated breeding value; GEBV, genomic estimated breeding value; GS, genomic selection; SNP, single-nucleotide polymorphism.
For units, see Table 1.
Average of the 10 best trees out of the 214 making up each of the 10 validation data sets.
Both training and testing data sets share individuals of the same families.
Gain estimated after CV using pedigree information.
(GEBVCV1/TE)/( EBVCV1/TC); TC=30 and TE=10, where TC is the breeding cycle in years with conventional breeding and TE is the number of years needed to complete a breeding cycle with GS.
Table 6. Empirical genetic gains from a 5% selection intensity made between families using EBVs (model based on pedigree information) and GEBV (model based on all 6385 SNPs).
| Traita |
Empirical genetic
gainb |
|||
|---|---|---|---|---|
| EBV (full data set) | GEBVCV2c | Ratio GEBVCV2/EBV (%) | EBVCV2d | |
| Cell population | 91.82 | 22.27 | 24.25 | −0.42 |
| Fibre coarseness | 24.89 | 4.20 | 16.89 | −0.71 |
| Crystallite width | 0.06 | 0.01 | 15.93 | 0.003 |
| Wood density | 30.52 | 3.32 | 10.86 | −2.74 |
| Microfibril angle | −2.70 | −0.88 | 32.77 | 0.09 |
| Wood stiffness | 1.18 | 0.27 | 20.52 | −0.06 |
| Ring width | 0.06 | 0.01 | 9.48 | −0.01 |
| Specific fibre surface | 32.14 | 3.94 | 12.27 | 1.48 |
| Cell radial diameter | 1.89 | 0.59 | 31.36 | 0.17 |
| Cell tangential diameter | 0.96 | 0.19 | 19.62 | −0.02 |
| Cell wall thickness | 0.16 | 0.02 | 12.32 | −0.01 |
| 22-Year height | 59.32 | 17.26 | 29.09 | 1.72 |
Abbreviations: CV, cross-validation; EBV, estimated breeding value; GEBV, genomic estimated breeding value; GS, genomic selection; SNP, single-nucleotide polymorphism.
For units, see Table 1.
Average of the 10 best trees out of ∼220 making up each of the 10 validation data sets and representing families that were not involved in the training data set (CV2).
Training and testing data sets are made up of individuals of different families.
Gain estimated after CV using pedigree information.
GEBV estimates for trees sampled at the Dablon Arboretum and Mirabel site were obtained using the GS models built with all 1694 trees belonging to the 214 families sampled at the Mastigouche Arboretum and genotyped using the subsets of significant SNPs (P<0.05). The accuracy of their wood density and microfibril angle GEBVs was estimated as the ratio of the correlation between the phenotype and the GEBVs divided by the square root of trait heritability (Dekkers, 2007). At the Dablon Arboretum, the accuracy estimates obtained for wood density and microfibril angle were 0.25 and 0.14, respectively, whereas they were 0.29 and 0.27 at the Mirabel site.
Discussion
Predictive ability and accuracy of GS models
To our knowledge, this is the first experimental study presenting estimates of predictive ability and accuracy of GS models for wood and growth traits in spruces for a population of large effective size representative of first-generation breeding populations, as often used in boreal conifer improvement programmes (Mullin et al., 2011). The number of parents that contributed to generating the 1694 white spruce trees of our study was estimated to be 620, using the status effective number (Ns) defined as Ns=0.5/f (Lindgren and Mullin, 1998) where the group's coancestry coefficient (f) is half of the relatedness coefficient derived from numerator the relationship matrix among all 1694 offspring.
Relatedness between training and testing data sets was either known (within-family CV) or unknown (between-family CV). Estimates of predictive ability were reduced by ∼20% when the coefficient of relatedness between CV2 sets was presumably much smaller than 0.25 (Table 3, CV2). Accuracy estimates were slightly higher than predictive ability for most of the traits with within-family CV and they could capture, depending on the trait, between 68 and 89% of the accuracy obtained with pedigree information. For the between-family validation (CV2), the prediction accuracy of the marker-based models was much better than that of the pedigree-based models (Table 4).
The low to moderate estimates of accuracy (Table 4) obtained with the markers for between-family validation (CV2) may have been because of the presence of LD between markers and QTLs persisting across families in the population. However, because the paternal contribution to within-provenance relatedness was not accounted for, markers may have been capturing this cryptic/unknown relatedness. Indeed, markers can capture additive genetic relationships between individuals (Fernando, 1998) that affect the estimates of accuracy of GEBVs even in the absence of LD between markers and QTLs (Habier et al., 2007, 2013). This observation supports the need to carefully design CV schemes in order to better identify the origin of the accuracies obtained, and CV should account for family structure in the data to allow for long-lasting genomics-based breeding plans (Habier et al., 2010). When the objective is to develop GS models to be used to select recombined individuals from the same population, separating prediction accuracy due to long-range LD and relatedness is less problematic. However, if one would like to apply GS models to select individuals from other populations, a much larger marker density might be required to do so (Meuwissen, 2009).
The white spruce genome is ∼2100 cM (Pavy et al., 2012b); therefore, the full SNP data set analyzed in the present study represented 2660 gene loci with an average rate of 2.4 SNPs per locus that would result in average genome coverage of ∼1.27 marker locus per cM. Although it is true that LD appears to be low in white spruce genes in essentially unrelated trees from natural populations, many exceptions were found where LD was sizeable (Namroud et al., 2010; Pavy et al., 2012a). Therefore, the true coverage would lie between the number of SNPs sampled and the number of gene loci sampled. For most traits in the present study, not all SNPs were likely to be close to a QTL and the assumption that all SNP effects were non-null is likely unrealistic, considering that the genotyping chip was built for several classes of traits and a large gene representation, but nevertheless is only a fraction of the complete white spruce gene space (Rigault et al., 2011). In theory, differential shrinkage methods proposed to estimate GEBVs should ensure that false-positive or uninformative effects are regressed towards zero. But in practice, the false-positive or uninformative effects are not strictly equal to zero, and pre-selecting SNPs could be crucial for improving the quality of genomic predictions (Croiseau et al., 2011). This might also be important for reducing the cost of GS implementation in breeding programmes. However, lower marker densities could negatively affect the capture of LD that would be detrimental to prediction accuracy over the long term.
Grattapaglia and Resende (2011) provided theoretical expectations for up to a maximum effective population size of 100. For training populations of 1000 individuals, effective population sizes of 100 parents, a number of 100 QTLs controlling characters with an heritability varying from 0.2 to 0.4 and a marker density of 1 to 2 SNPs per cM, they reported that prediction accuracy of GS models should vary approximately between 0.3 and 0.4 (see Figures 1 and 2 in Grattapaglia and Resende, 2011). Thus, it appears that our results are quite in line with theoretical expectations when both training and validation data sets came from the same populations. As demonstrated by Grattapaglia and Resende (2011), the number of QTLs controlling the characters might have some impact at low marker densities, but it is difficult to determine at the present time the most probable number of QTLs involved in growth and wood traits in spruces, though their numbers are likely to be large (Pelgas et al., 2011). Similarly, the larger the effective population sizes, the lower the expected accuracy of predictions, especially when the marker density per cM is low. More empirical data are needed to have a better idea of the prediction accuracy that GS models can achieve with forest trees in various population setups.
The predictive ability of models developed for remotely related individuals (CV2) was only slightly lower than that of those built for closely related individuals (CV1). This is likely an indication that some LD might be present but also that relatedness was picked up by the markers. Hayes et al. (2009), using multibreed dairy cattle populations, reported that prediction equations in one breed do not predict accurate GEBVs when applied to other breeds. Meuwissen (2009), using computer simulation of small populations, showed that a substantially higher marker density and number of training records would be needed to obtain accurate predictions of breeding values of unrelated individuals. However, the notion of unrelatedness is tricky because some pairs of individuals are more closely related than others even if they are believed to be unrelated (Powell et al., 2010). When the possibility of paternal (and maternal) coancestry between CV sets was eliminated, and confirmed by an average estimated kinship coefficient of zero between the training and validation sets used (CV3), the prediction accuracy was reduced, but it was still clearly different from zero for several of the traits. This result supports to some extent the presence of historical LD with trait loci.
Such an historical LD pattern may have arisen if all populations considered in the present study were from the same glacial lineage. The main phylogeographic patterns for North American trees detected with DNA markers were recently reviewed (Jaramillo-Correa et al., 2009). The study identified converging patterns, such as the location of glacial refugia and postglacial recolonization routes, and the inference of common causes of vicariance. The glacial and postglacial history of white spruce was inferred based on chloroplast DNA and nuclear microsatellite polymorphisms (de Lafontaine et al., 2010). Trees of an east Appalachian refugium population would have migrated into New England and northwards into the province of Québec and the Maritimes (de Lafontaine et al., 2010) <10 000 years ago, suggesting that all populations considered in the present study are from the same glacial lineage, as supported by our analyses of population structure and previous ones (Jaramillo-Correa et al., 2001; Namroud et al., 2008, 2010). In addition, a molecular footprint of recent expansion following the last glacial maximum was found in the genes of white spruce and other boreal spruces (Namroud et al., 2010). Given these phylogeographical and demographic inferences, and the limited number of generations that occurred since the beginning of recolonization, the presence of shared ancestry and historical LD are likely in the populations sampled (Doerksen et al., 2014).
So far, empirical accuracy estimates in forest trees have been reported only for small breeding populations of Pinus taeda and Eucalyptus (Resende et al., 2012a, 2012b; Zapata-Valenzuela et al., 2012). As expected from deterministic and simulation models (Grattapaglia and Resende, 2011; Iwata et al., 2011), the accuracy of GS models for wood and growth traits in these species was slightly higher than that reported in the present study.
When GS models were built with ∼350 markers found to be associated with wood density and microfibril angle in the discovery population (Mastigouche Arboretum) and validated (VS scheme) using half-sibs sampled on two different sites (Dablon Arboretum and Mirabel), the GEBV accuracy estimates obtained were reasonably high compared with those obtained using the CV1 scheme (up to 70% of accuracy values) in the Mastigouche Arboretum. This is likely because of the fact that G × E interactions in white spruce are generally low for growth and wood traits (Li et al., 1997; Lenz et al., 2011). Thus, when both the training and the validation data sets are related, moderate accuracy estimates could likely be expected in white spruce even when trees for training and for validation are located on different sites. The fact that the GS models were built using a larger genetic base (214 families) than that of the testing sets on these two other sites might also have contributed to better trace relatedness and consequently, to increasing the accuracy of predictions.
Implementing GS models in white spruce breeding programmes
Our results suggest that between 65 and 110% of the gain predicted with pedigree information alone (Table 5) could be obtained with GS models when selecting the top 5% of individuals closely related to those used in the training data set. However, when the degree of relatedness between training and testing data sets was lower, the gains predicted were lower, as expected (Table 6).
The major advantage of GS over conventional phenotypic pedigree-based selection is the possibility to shorten breeding cycles and increase gains per time unit in long-lived species. One generation of genetic improvement for white spruce, from the selection of parent trees to the production of genetically improved seed, can last two to three decades. It generally takes 10–15 years, sometimes even more, for a white spruce tree to reach sexual maturity and produce seeds (Nienstaedt and Zasada, 1990), although this period can be made shorter with the use of flower induction techniques (Beaulieu et al., 1998). Testing is also time consuming, especially for characters such as wood traits; 15–25 years of growth may be needed before trees become large enough for the reliable assessment of wood and fibre properties. GS could eliminate the need for tree testing at least for one or two generations, after which retraining the models would be needed to maintain or increase GEBV accuracy (Iwata et al., 2011; Resende et al., 2012a). New markers could also be added to future models to maintain a high level of GEBV accuracy (Goddard, 2009).
Several approaches can be envisioned for implementing GS in white spruce, but a forward selection scenario using somatic embryogenesis to produce seedlings in selected somatic embryogenesis lines would likely be the most fruitful. Somatic embryogenesis is an intensive vegetative propagation technology that is well developed for white spruce (Wahid et al., 2012; Weng et al., 2012), and somatic embryogenesis varieties are already used in reforestation programmes in eastern Canada. Our results show that in the present unfavourable context of large population size, over twofold gains could be obtained per year using GS if the breeding cycle was reduced to 10 years compared with 30 years with conventional pedigree-based methods (Table 5). However, if GS is implemented following a clonal strategy, additional studies would be required to also model nonlinear genetic effects in order to predict more accurately the total genetic merit of clones.
For its practical implementation, the use of GS in white spruce breeding programmes or any other species also depends on the results of a cost–benefit analysis. Testing a very large number of markers on a large number of trees may not be cost effective, although genotyping costs are decreasing very rapidly and may represent a marginal cost when considering the value of genetic gains obtained from genetically improved plantations. But Habier et al. (2009) noted that using smaller SNP sets may require distinct arrays of SNPs for each trait. In the present study and in previous ones (Beaulieu et al., 2011), we found that the list of most significant SNPs varied considerably from one character to the next, even when considering only wood traits. For instance, the average overlap between SNP subsets for wood density and microfibril angle was only 11%. Thus, in the case where improvement for multiple traits is sought, a large number of SNPs would still need to be genotyped. Moreover, a larger number of markers would likely make it possible to develop GS models for unforeseen traits without additional genotyping.
On the other hand, Weigel et al. (2009) showed that a relatively small number of SNPs with large effects on lifetime net merit in Holstein bulls could capture a significant fraction of the gain that was achieved with a large number of SNPs. Resende et al. (2012) also obtained good accuracy estimates with a limited number of SNPs. We tested a posteriori, for both wood density and microfibril angle, whether retaining only the 10% SNPs with the largest effects could make it possible to capture a large fraction of the accuracy obtained with all 6385 SNPs. We identified for each of 10 training data sets the 700 SNPs with the largest absolute effects. New GS models were built for each training data set using only these groups of 700 SNPs and were validated in corresponding validation data sets. Estimates of GEBV accuracies were obtained as previously done for the other CVs. The estimate of accuracy obtained for wood density GEBV was 0.335 (±0.013) for the within-family validation (CV1) compared with 0.370 (±0.012) with all SNPs; it was 0.155 (±0.027) for the between-family cross-validation (CV2) compared with 0.132 (±0.024) with all SNPs. Similarly for microfibril angle, GEBV accuracies were, for the CV1 scheme, 0.313 (±0.018) versus 0.378 (±0.019) with all SNPs, and for CV2, 0.104 (±0.019) versus 0.164 (±0.029) with all 6385 SNPs. Thus, it appears that this strategy could make it possible to capture a large fraction of the maximum accuracy achievable with a much larger number of markers or loci. However, it could be at the expense of developing GS models that would hold for a larger number of generations.
Conclusion
For the time being, considering that for most tree species (1) the number of markers currently available is relatively limited, (2) genotyping large numbers of markers is still expensive, (3) only a few genetic tests are generally in place, (4) relatedness among individuals is picked up by the markers and (5) LD decays rapidly, especially in conifers, we recommend the development of GS models within the same population (CV1 scheme) only and that should make it possible to obtain a quite high efficiency of marker models relative to conventional pedigree-based models even with lower marker coverage, and obtain potentially higher gains per time unit.
Data archiving
Data available from the Dryad Digital Repository: 10.5061/dryad.6rd6f.
Acknowledgments
This study is part of the SMarTForests project. We thank the team of A Montpetit (Genome Québec Innovation Centre, McGill University, Montreal) for assistance with the acquisition of SNP data on the Illumina platform. We also thank M Deslauriers, F Gagnon and S Blais for assistance in DNA extractions, the acquisition of SNP data and for preparing and cleaning SNP data files, D Plourde and É Dussault for wood core sampling, the EvalueTree team of FPInnovations in Vancouver for wood core processing and P-L Poulin for phenotypic data handling. We are also grateful to P Lenz for his constructive comments on a preliminary version of this manuscript and to I Lamarre for her editing work. This work was funded through grants from Genome Canada, Genome Québec, the Canadian Wood Fibre Centre and the Genomics R&D Initiative of the Government of Canada to JBe, JM and JBo.
The authors declare no conflict of interest.
References
- Beaulieu J, Deslauriers M, Daoust G. Flower induction treatments have no effects on seed traits and transmission of alleles in Picea glauca. Tree Physiol. 1998;18:817–821. doi: 10.1093/treephys/18.12.817. [DOI] [PubMed] [Google Scholar]
- Beaulieu J, Doerksen T, Boyle B, Clément S, Deslauriers M, Beauseigle S, et al. Association genetics of wood physical traits in the conifer white spruce and relationships with gene expression. Genetics. 2011;188:197–214. doi: 10.1534/genetics.110.125781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C. The genetic architecture of maize flowering time. Science. 2009;325:714–718. doi: 10.1126/science.1174276. [DOI] [PubMed] [Google Scholar]
- Burdon RD, Wilcox PL.2011Integration of molecular markers in breedingIn: Plomion C, Bousquet J, Kole C (eds)Genetics, Genomics and Breeding of Conifers CRC Press and Edenbridge Science Publishers: New York; 276–322. [Google Scholar]
- Croiseau P, Legarra A, Guillaume F, Fritz S, Baur A, Colombani C, et al. Fine tuning genomic evaluations in dairy cattle through SNP pre-selection with the Elastic-Net algorithm. Genet Res. 2011;93:409–417. doi: 10.1017/S0016672311000358. [DOI] [PubMed] [Google Scholar]
- Dekkers JCM. Prediction of response to marker-assisted and genomic selection using selection index theory. J Anim Breed Genet. 2007;124:331–341. doi: 10.1111/j.1439-0388.2007.00701.x. [DOI] [PubMed] [Google Scholar]
- de Lafontaine G, Turgeon J, Payette S. Phylogeography of white spruce (Picea glauca) in eastern North America reveals contrasting ecological trajectories. J Biogeogr. 2010;37:741–751. [Google Scholar]
- Doerksen T, Bousquet J, Beaulieu J. Inbreeding depression in intra-provenance-crosses driven by founder relatedness in white spruce. Tree Genet Genomes. 2014;10:302–212. [Google Scholar]
- Fernando RL.1998Genetic evaluation and selection using genotypic, phenotypic and pedigree information Proceedings of the 6th World Congress on Genetics Applied to Livestock Production11–16 January 1998; Armidale, NSW, Australia,Vol. 26329–336. [Google Scholar]
- Goddard M. Genomic selection: prediction of accuracy and maximisation of long term response. Genetica. 2009;136:245–257. doi: 10.1007/s10709-008-9308-0. [DOI] [PubMed] [Google Scholar]
- González-Martínez SC, Wheeler NC, Ersoz E, Nelson CD, Neale DB. Association genetics in Pinus taeda L. I. Wood property traits. Genetics. 2007;175:399–409. doi: 10.1534/genetics.106.061127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grattapaglia D, Resende MDV. Genomic selection in forest tree breeding. Tree Genet Genomes. 2011;7:241–255. [Google Scholar]
- Habier D, Fernando RL, Dekkers JCM. The impact of genetic relationship information on genome-assisted breeding values. Genetics. 2007;177:2389–2397. doi: 10.1534/genetics.107.081190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habier D, Fernando RL, Dekkers JCM. Genomic selection using low-density marker panels. Genetics. 2009;182:343–353. doi: 10.1534/genetics.108.100289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habier D, Tetens J, Seefried F-R, Lichtner P, Thaller G. The impact of genetic relationship information on genomic breeding values in German Holstein cattle. Genet Sel Evol. 2010;42:5. doi: 10.1186/1297-9686-42-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habier D, Fernando RL, Garrick DJ. Genomic-BLUP decoded: a look into the black box of genomic prediction. Genetics. 2013;194:597–607. doi: 10.1534/genetics.113.152207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes BJ, Bowman PJ, Chamberlain AC, Verbyla K, Goddard ME. Accuracy of genomic breeding values in multi-breed dairy cattle populations. Genet Sel Evol. 2009;41:51. doi: 10.1186/1297-9686-41-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffner EL, Lorenz AJ, Jannink J-L, Sorrels ME. Plant breeding with genomic selection: gain per unit time and cost. Crop Sci. 2010;50:1681–1690. [Google Scholar]
- Iwata H, Hayashi T, Tsumura Y. Prospects for genomic selection in conifer breeding: a simulation study of Cryptomeria japonica. Tree Genet Genomes. 2011;7:747–758. [Google Scholar]
- Jaramillo-Correa JP, Beaulieu J, Bousquet J. Contrasting evolutionary forces driving population structure at ESTPs, allozymes, and quantitative traits in white spruce. Mol Ecol. 2001;10:2729–2740. doi: 10.1046/j.0962-1083.2001.01386.x. [DOI] [PubMed] [Google Scholar]
- Jaramillo-Correa JP, Beaulieu J, Khasa DP, Bousquet J. Inferring the past from the present phylogeographic structure of North American forest trees: seeing the forest for the genes. Can J For Res. 2009;39:286–307. [Google Scholar]
- Lande R, Thompson R. Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics. 1990;124:743–756. doi: 10.1093/genetics/124.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legarra A, Ricard A, Filangi O.2013. GS3 software. Available at: http://snp.toulouse.inra.fr/~alegarra/ . Version 6. INRA, Toulouse..
- Legarra A, Misztal I. Technical note: computing strategies in genome-wide selection. J Dairy Sci. 2008;91:360–366. doi: 10.3168/jds.2007-0403. [DOI] [PubMed] [Google Scholar]
- Lenz P, MacKay J, Rainville A, Cloutier A, Beaulieu J. The influence of cambial age on breeding for wood properties in Picea glauca. Tree Genet Genomes. 2011;7:641–653. [Google Scholar]
- Li P, Beaulieu J, Bousquet J. Genetic structure and patterns of genetic variation among populations in eastern white spruce (Picea glauca) Can J For Res. 1997;27:189–198. [Google Scholar]
- Lindgren D, Mullin TJ. Relatedness and status number in seed orchard crops. Can J For Res. 1998;28:276–283. [Google Scholar]
- Lynch M, Walsh B. Genetics and Analysis of Quantitative Traits. Sinauer Associates: Sunderland; 1998. [Google Scholar]
- Meuwissen THE. Accuracy of breeding values of ‘unrelated' individuals predicted by dense SNP genotyping. Genet Sel Evol. 2009;41:35. doi: 10.1186/1297-9686-41-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen THE, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001;157:1819–1829. doi: 10.1093/genetics/157.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullin TJ, Andersson B, Bastien J-C, Beaulieu J, Burdon RD, Dvorak WS, et al. 2011Economic importance, breeding objectives and achievementsIn: Plomion C, Bousquet J, Kole C (eds)Genetics, Genomics and Breeding of Conifers CRC Press and Edenbridge Science Publishers: New York; 40–127. [Google Scholar]
- Namroud M-C, Beaulieu J, Juge N, Laroche J, Bousquet J. Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce. Mol Ecol. 2008;17:3599–3613. doi: 10.1111/j.1365-294X.2008.03840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namroud M-C, Guillet-Claude C, Mackay J, Isabel N, Bousquet J. Molecular evolution of regulatory genes in spruces from different species and continents: heterogeneous patterns of linkage disequilibrium and selection but correlated recent demographic changes. J Mol Evol. 2010;70:371–386. doi: 10.1007/s00239-010-9335-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namroud M-C, Bousquet J, Doerksen T, Beaulieu J. Scanning SNPs from a large set of expressed genes to assess the impact of artificial selection on the undomesticated genetic diversity of white spruce. Evol Appl. 2012;5:641–656. doi: 10.1111/j.1752-4571.2012.00242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienstaedt H, Zasada JC.1990Picea glauca. White spruceIn: Burns RM, Honkala BH (eds)Silvics of North America: 1. Conifers. Agriculture Handbook 654 USDA Forest Service: Washington, DC; 204–226. [Google Scholar]
- Pavy N, Namroud M-C, Gagnon F, Isabel N, Bousquet J. The heterogeneous levels of linkage disequilibrium in white spruce genes and comparative analysis with other conifers. Heredity. 2012;108:273–284. doi: 10.1038/hdy.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavy N, Pelgas B, Laroche J, Rigault P, Isabel N, Bousquet J. A spruce gene map infers ancient plant genome reshuffling and subsequent slow evolution in the gymnosperm lineage leading to extant conifers. BMC Biol. 2012;10:84. doi: 10.1186/1741-7007-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavy N, Gagnon F, Rigault P, Blais S, Deschênes A, Boyle B, et al. Development of high-density SNP genotyping arrays for white spruce (Picea glauca) and transferability to subtropical and nordic congeners. Mol Ecol Resour. 2013;13:324–336. doi: 10.1111/1755-0998.12062. [DOI] [PubMed] [Google Scholar]
- Pelgas B, Bousquet J, Meirmans PG, Ritland K, Isabel N. QTL mapping in white spruce: gene maps and genomic regions underlying adaptive traits across pedigrees, years and environments. BMC Genomics. 2011;12:145. doi: 10.1186/1471-2164-12-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JE, Visscher PM, Goddard M. Reconciling the analysis of IBD and IBS in complex trait studies. Nat Rev Genet. 2010;11:800–805. doi: 10.1038/nrg2865. [DOI] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Resende MDV, Resende MFR, Jr, Sansaloni CP, Petroli CD, Missiaggia AA, Aguiar AM, et al. Genomic selection for growth and wood quality in Eucalyptus: capturing the missing heritability and accelerating breeding for complex traits in forest trees. New Phytol. 2012;194:16–128. doi: 10.1111/j.1469-8137.2011.04038.x. [DOI] [PubMed] [Google Scholar]
- Resende MFR, Jr, Muñoz P, Acosta JJ, Peter GF, Davis JM, Grattapaglia D, et al. Accelerating the domestication of trees using genomic selection: accuracy of prediction models across ages and environments. New Phytol. 2012;193:617–624. doi: 10.1111/j.1469-8137.2011.03895.x. [DOI] [PubMed] [Google Scholar]
- Resende MFR, Jr, Muñoz P, Resende MDV, Garrick DJ, Fernando RL, Davis JM, et al. Accuracy of genomic selection methods in a standard data set of loblolly pine (Pinus taeda L.) Genetics. 2012;190:1503–1510. doi: 10.1534/genetics.111.137026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigault P, Boyle B, Lepage P, Cooke JEK, Bousquet J, MacKay J. A white spruce gene catalog for conifer genome analyses. Plant Physiol. 2011;157:14–28. doi: 10.1104/pp.111.179663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss SH, Lande R, Namkoong G. Limitations of molecular-marker-aided selection in forest tree breeding. Can J For Res. 1992;22:1050–1061. [Google Scholar]
- VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–4423. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- Wahid N, Rainville A, Lamhamedi MS, Margolis HA, Beaulieu J, Deblois J. Genetic parameters and performance stability of white spruce somatic seedlings in clonal tests. For Ecol Manag. 2012;270:45–53. [Google Scholar]
- Weigel KA, de los Campos G, González-Recio O, Naya H, Wu XL, Long N, et al. Predictive ability of direct genomic values for lifetime net merit of Holstein sires using selected subsets of single nucleotide polymorphism markers. J Dairy Sci. 2009;92:5248–5257. doi: 10.3168/jds.2009-2092. [DOI] [PubMed] [Google Scholar]
- Weng YH, Park Y-S, Lindgren D. Unequal clonal deployment improves genetic gains at constant diversity levels for clonal forestry. Tree Genet Genomes. 2012;8:77–85. [Google Scholar]
- Zapata-Valenzuela J, Isik F, Maltecca C, Wegrzyn J, Neale D, Mekeand S, et al. SNP markers trace familial linkages in a cloned population of Pinus taeda – prospects for genomic selection. Tree Genet Genomes. 2012;8:1307–1318. [Google Scholar]