

Abstract
India lacks a national policy on the prevention and control of genetic disorders. Although the haemoglobinopathies have received some attention, there are scarce data on the epidemiology of other genetic disorders in India. Haemophilia, an inherited single gene disorder with an incidence of 1 per 10,000 births, manifests as spontaneous or trauma-induced haemorrhagic episodes in patients, progressing to chronic disability and premature mortality in untreated patients or patients with sub-optimal treatment. Although the genetic basis of this disorder has been well studied in India, data on the number of patients, trends of the disorder in India, social costs of the condition and opportunities and competencies for offering genetic counselling through a public health programme have not been reported. This review article summarizes the available Indian data, which show that the country harbours the second highest number of global patients with haemophilia A. The reported number of patients with haemophilia A is 11,586 while the estimated prevalence could be around 50,000 patients. This review also identifies the need to immediately initiate a national programme for haemophilia, with components of prevention, care for patients, surveillance and education and support for families.
Keywords: Bleeding disorders, epidemiology, genetic disorders, haemophilia, India, prevention
Introduction
Genetic disorders are rare conditions that are accorded a low public health priority in India, even though these account for significant suffering of patients and their family members1,2. Subsidized treatment services are of limited availability causing significant out of pocket (OOP) expenditure for parents in India. Genetic disorders are known to impact health services through repeated hospital admissions3 (such as routine transfusion for patients with thalassaemia). Sickle cell anaemia is prevalent in isolated, primarily inbreeding tribal groups in the country4,5,6,7,8,9. Recent studies suggest a significant burden of thalassaemia in India10,11. For other genetic disorders (such as haemophilia, cystic fibrosis, spinomuscular atrophy, myotonic dystrophy, fragile X syndrome), there are limited systematic epidemiological data. This evidence gap enhances the perception that genetic disorders other than the haemoglobinopathies are rare, self-limiting conditions, for which prevention programmes are not required on a priority basis12,13. Due to its demographic characteristics, in absolute numbers, genetic disorders may affect significant numbers of individuals in India. India has the largest birth cohort globally, with 27 million new births in 201014. The country has the second largest global population of 1.21 billion15 and a high birth rate of 21.816. As genetic disorders arise in the population through spontaneous mutations and through affected births in families with known risk of a genetic disorder, in absolute numbers, the birth prevalence of genetic disorders is likely to be high in India. Genetic counselling is the main tool for the prevention and control of genetic disorders, targeted at families with an affected birth or with a family history of the disorder. Although genetic services are available through the private health sector, a public health programme is needed to ensure that prevention and control of genetic disorders can be implemented on a nation-wide basis.
Due to the methodological challenges of studying rare disorders, it is difficult to approach genetic disorders from a public health/epidemiology perspective and explore prevention, that is delivery of genetic counselling from a health systems standpoint. This review uses haemophilia, a rare bleeding disorder to determine the available information on the reported and estimated number of patients in India, the trends of the disorder in the country, the social costs of this disorder, in terms of morbidity, mortality and access to care, and the evidence on opportunities for prevention. It also discusses a framework for a national haemophilia programme, identifying the existing opportunities and challenges in delivering this programme. Some States in India have started providing treatment for patients with haemophilia, and the Government of India is piloting a haematology programme through the National Rural Health Mission, Maharashtra17.
Haemophilia
Bleeding disorders reportedly affect 1 in 1,000 men and women globally18. Haemophilia A and B19,20,21 (Table I) and von Willebrand disease32,33, make up the most prevalent types of bleeding disorders. Other conditions include the rare factor deficiencies (Table II). Haemophilia A and B are single gene disorders, occurring due to a mutation in either the coagulation factor VIII gene (haemophilia A) or the coagulation factor IX gene (haemophilia B), resulting in deficient synthesis of coagulation factor VIII and IX, presenting as haemorrhagic tendencies in the patient35,36. Patients with haemophilia suffer from repeated haemorrhagic episodes in joints and soft tissues (Fig. 1). The frequency of bleeding depends on the residual coagulation factor level and the genotype of the patient20. Patients are classified as having severe, moderately severe or mild haemophilia based on <1, 1-5 and 5-40 per cent of normal clotting factor levels23,37. Patients with severe haemophilia A constitute nearly 70 per cent of patients with bleeding disorders. The haemorrhagic episodes of haemophilia are treated with replacement therapy, with either clotting factor VIII for haemophilia A and clotting factor IX for haemophilia B, or with cryoprecipitate or fresh frozen plasma23,37,38,39,40,41. Clotting factor concentrate is the preferred treatment option. Clotting factor concentrate is an orphan drug42, and its high cost places the drug out of reach of the majority of Indian patients. The lack of sufficient treatment product for pain relief, for life threatening haemorrhagic episodes and for the prevention of disability results in orthropaedic morbidity, premature mortality and extensive OOP expenditure26. The quality of life and wellbeing of patients and family members are significantly affected43.
Table I.
Clinico-epidemiological characteristics of haemophilia

Table II.
Rare clotting factor deficiencies
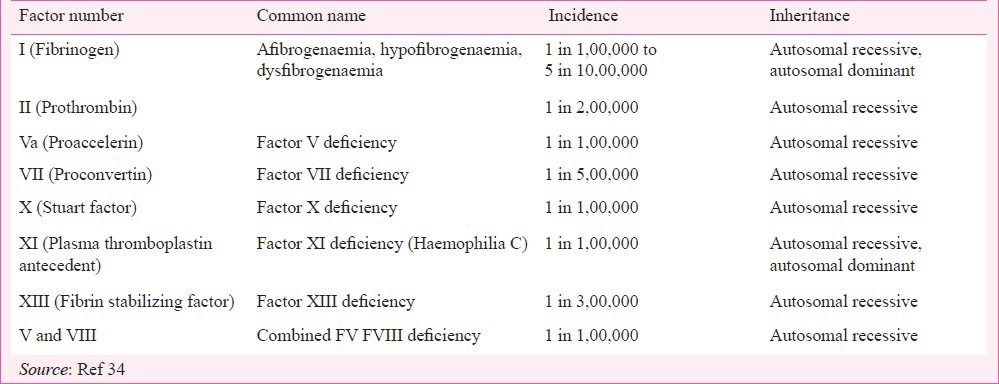
Fig. 1.
Frequency and sites of bleeding in patients with severe and moderate haemophilia A. Data from 480 patient weeks of follow up (Source: Ref. 30). The numbers indicate the total number of bleeding episodes at that site (denominator) and the number of haemmorrhagic episodes treated with clotting factor concentrate (numerator).
Prevention of haemophilia
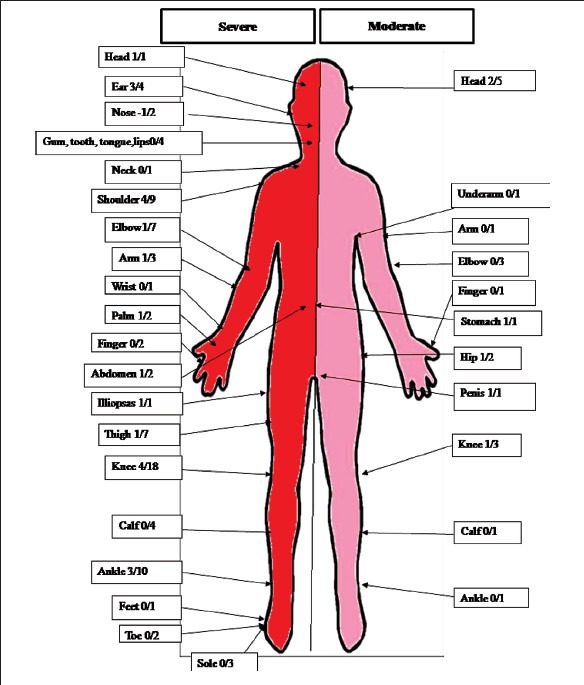
Haemophilia A and B are sex-linked inherited disorders, with the conditions being typically transmitted through asymptomatic, heterozygous females to the male child (Fig. 2). New cases appear in the population through birth of offspring with haemophilia to carrier females, and through new mutations. About 30 per cent of haemophilia A cases arise through spontaneous mutation, presenting as an affected male child in a family without a history of haemophilia27. Prenatal diagnosis (Fig. 2a) followed by the option of medical termination of pregnancy is the method of prevention and control of the disorder. Detection of women who are carriers of the mutant gene (Fig. 2b) is another method of prevention, as carrier detection empowers parents to make informed reproductive choices.
Fig. 2.
Prevention of haemophilia. (a) Prenatal diagnosis is offered to women who are obligate carriers (arrow), having one or more sons with haemophila, or whose father has haemophilia, or who has one or more relative with haemophilia. (b) Carrier detection is offered to possible carriers (arrow), who are daughters of carriers, mothers with one son with haemophilia but no other affected relative, or female relatives of carriers.
Epidemiology of haemophilia in India
(i) Sources of data: Data on the number of patients is of critical importance for any public health intervention. A few hospital based studies have estimated the burden of haemophilia44. As standard epidemiological methods such as surveys are not cost-effective for measuring rare disorders, registries remain the best source of obtaining epidemiological data on these conditions45. In India, a disease registry for haemophilia is available with the Hemophilia Federation of India (HFI)31. This non-governmental organization (NGO) collates data on the number of patients through 76 centers located across India. The organization is responsible for importation and distribution of treatment product, patient care, education and counselling and advocating for the rights of patients with bleeding disorders and their families. The availability of this registry makes haemophilia a model for the study of the epidemiology and for surveillance of trends of this condition in India. The quality of the registry data has been checked in a haemophilia surveillance study in Maharashtra46. Of the 2,219 patients registered during the period 1989 to 2005, the data had 8.3 per cent duplication, and lacked information on nearly 32 per cent patients (273 deceased patients and 882 relatives). Thus this data set may have issues of accuracy, but still remains the best available source of data on this rare condition in the country.
(ii) Incidence: The most frequently cited incidence estimate for haemophilia is from Haldane27, who estimated an incidence of 1 per 5,000 male births. Soucie et al47 estimated a similar incidence of 1 per 5,032 male births (that is around 20 per 1,00,000 population) from a haemophilia surveillance established in the United State of America (USA). Between 2009 and 2010, 260 new patients were reported from India48,49, while there were 1,38,41,667 male births15. This implies that the incidence of haemophilia in India in 2010 was around 2 per 1,00,000 male births, or around 277 new patients being registered each year in the country. If the incidence of 20 per 1,00,000 is used, the number of affected new births in India in 2010 would be 2,768, that is approximately 0.01 per cent of total annual births in India. Although apparently insignificant as compared to infectious disease burden, in the case of haemophilia, where patients survive to adulthood, patient accrual over time results in increasing population prevalence of a disorder whose management is extremely resource intensive.
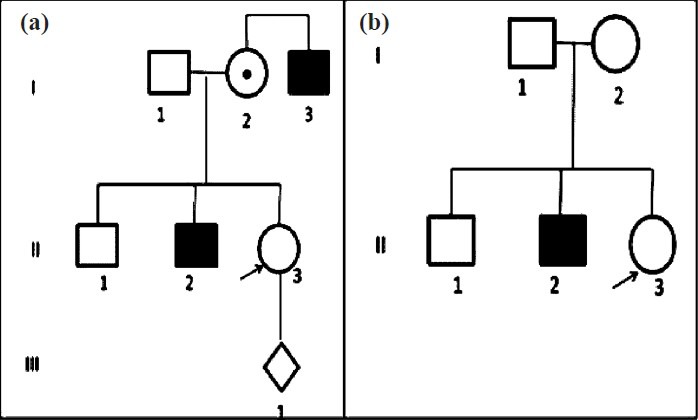
(iii) Prevalence and global rank: The rarity of haemophilia challenges estimates of the disorder50. The best available prevalence data are from a global survey on haemophilia and bleeding disorders, conducted by the World Federation of Haemophilia (WFH), a global organization involved in promoting the care of people with haemophilia and bleeding disorders51. This survey is conducted globally, and in 2011 included 108 countries and covered 90.6 per cent of the global population29. Indian data are reported by the HFI. The data showed that in 2011, India reported 14,718 patients with bleeding disorders and 11,586 patients with haemophilia A29. Fig. 3a and c show the distribution of patients with bleeding disorders and with haemophilia A in the five countries reporting the highest number of patients. India, the only developing country in this list, reports the third largest number of patients with bleeding disorders, and the second highest number of patients with haemophilia A. In terms of proportionate burden of patients, data indicated that India harbours 5 and 9 per cent of the global number of patients with bleeding disorders and with haemophilia A respectively (Fig. 3b and d) (unpublished observation).
Fig. 3.
Global distribution of total reported cases of bleeding disorders (a) and haemophilia A (c) in five countries reporting the highest number of patients. (b) and (d) show that nearly 5 and 9% of global patients with bleeding disorders and haemophilia A are from India. (Source: Authors’ calculation based on data from Ref. 29).
(iv) Estimated number of patients: Table III shows the number of cases reported (prevalence) in five high burden countries29. The USA and Brazil report the highest prevalence of over 4 per 1,00,000 population. In comparison, the number of prevalent cases in India is 0.9 per 1,00,000 population, suggesting that underdiagnosis, mortality and incomplete case recording could be a reason for lower case reporting. The very high prevalence of 8.7 reported by the United Kingdom is a phenomenon that has been observed in countries where the small population magnifies the prevalence estimates29. China reports the third highest global number of haemophilia patients, but like India has a low prevalence of 0.7 per 1,00,000 population. As compared to the USA, which has an excellent haemophilia surveillance system52, the current case detection in India is 4.7 times lower. Adjusting the underdiagnosis of patients with haemophilia A in India to the prevalence rate of haemophilia A in the USA, the estimated number of patients in India would be around 54,454. In case of haemophilia B, the prevalence rate of 0.1 per 1,00,000 population in India is 13 times lower to the prevalence rate of the USA (1.3 per 1,00,000). Adjusting for this difference in prevalence, there would be an estimated 21,931 patients with haemophilia B in the country (unpublished observation).
Table III.
Case detection rate (prevalence) of haemophilia A (HA) and haemophilia B (HB) in five countries reporting the highest global number of patients
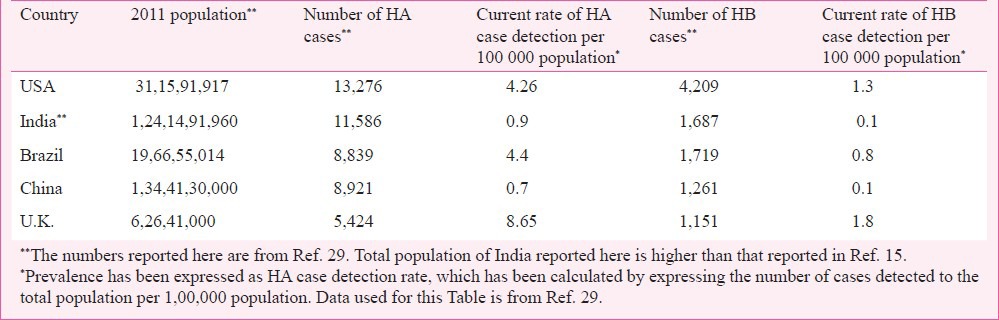
Using the population data for 2011 from the Census of India15 and a prevalence of haemophilia A of 4 per 1,00,000, the estimated number of haemophilia patients in India would be around 48,407. Thus, India may have over 70,000 patients with haemophilia A and B. Fig. 4 shows the observed number of cases (calculated at a prevalence of 0.9 per 1,00,000 population) and estimated number of patients (calculated at 4 per 1,00,000 population) with haemophilia A in States/UT of the country. The large patient burden indicates the need for a public health intervention for prevention and care.
Fig. 4.
Observed (calculated at 0.9 per 1,00,000) and estimated (calculated at 4 per 1,00,000 population) prevalence of haemophilia A for states and UT of India. (Source: Authors’ calculation based on data from Ref. 15).
(v) Trends in case detection: For public health services, data on time trends of the disorder are needed for decision making on implementation of services. While studies predicting the trends of haemophilia in India are lacking, gross deductions are possible from the data available from the Annual Global Surveys29,48,49,53,54,55. Fig. 5a shows the trends in the diagnosis of new patients in the four countries reporting the highest numbers of haemophilia patients. Over the last five years, India (1,474) and Brazil (1,952) have reported more newly diagnosed patients than the USA (1,376) or UK (713), reiterating the need for a prevention programme.
Fig. 5.
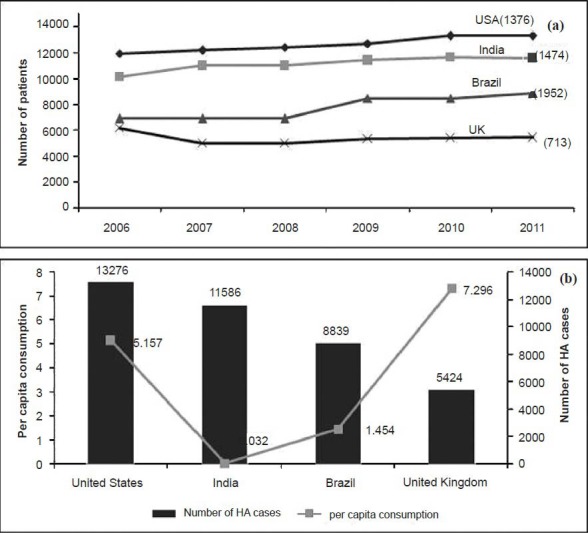
(a) Haemophilia A trends in four high burden countries. Numbers in parenthesis show the number of patients reported in the last five years by each country (Source: Refs 29, 48, 49, 53-55. (b) Per capita clotting factor VIII consumption (Yaxis) and number of haemophilia patients reported in 2011 for the four high burden countries (Source: Ref. 29).
Social costs of haemophilia: One of the major roles of public health agencies is to link people to needed services. Lack of such services contributes to suffering of patients and families. The social costs of haemophilia occur as a consequence of lack of access to treatment and impact patients in terms of morbidity and mortality, schooling and employment. Parents are financially affected due to the OOP expenditure26, and experience significant guilt and distress at being unable to offer appropriate treatment to the child.
(i) Orthopaedic complications and disability: The long-term consequences of repeated joint bleeds with sub-optimal treatment are the development of chronic and progressive joint damage and disability56. Studies in developed countries have primarily focused on the development of musculoskeletal scores which can be used for optimization of dose of clotting factor concentrate for maintaining joint health. In contrast, lack of treatment results in rampant disability in patients with haemophilia in India. Only one study by Kar et al57 has measured the prevalence of disability. This study, conducted at five centres across the country showed that of the 148 patients with severe haemophilia A, only nine were free of disability. The knee joint was affected in all of the 148 patients. Of concern was that in the age group of 5 to 12 years, only 15 per cent of patients were disability-free57. A significant association was found between the socio-economic status of the patient's family and the severity of disability, suggesting that disability was most likely to be prevalent amongst the most vulnerable strata of society. Increased orthopaedic vulnerability of patients was demonstrated in another study that reported that the incidence of osteoporosis (T score: -2.5 or more) and the incidence of fractures in adult life were significantly higher in patients with haemophilia as compared to controls58.
(ii) Prevalence of transfusion transmitted infections (TTI): In the absence of access to treatment, there are anecdotal reports of use of whole blood by patients with haemophilia, especially in remote areas. There is, however, no data on the frequency of whole blood use by patients. Patients with haemophilia, therefore, remain vulnerable to transfusion transmitted infections (TTI). Data from the Annual Global Report (2011) show that currently, 1.12 per cent (159/14,152) patients with haemophilia are reported infected with the human immunodeficiency virus (HIV)29. Only one large study reported the prevalence of HIV, hepatitis B virus surface antigen (HBsAg) and hepatitis C virus (HCV) infection amongst 323 severe and 77 moderately severe haemophilia patients. In the five year long study initiated in 1995, the prevalence of HIV, HBsAg and HCV positivity was found to be 3.8, 6 and 23.9 per cent59. One of the earliest studies conducted in 1991 had detected a 12 per cent HIV seropositivity (n=124)60. A retrospective review of HIV, HBsAg, and HCV status of the 39,060 healthy donors identified the overall prevalence as 0.44, 1.27 and 0.23 per cent, respectively61. The risk of TTIs in patients was also reflected in a study which reported that the prevalence of hepatitis C virus (HCV) infection was 7.1 per cent, (258 out of 3,589) when RT-PCR (reverse transcriptase - polymerase chain reaction) was used. Of concern was that ELISA (enzyme-linked immunosorbent assay) routinely used for the screening at blood banks detected only 6.1 per cent (221/3,589) of the HCV positive samples, suggesting that a substantial number of HCV infected samples in blood banks go undetected62.
(iii) Prevalence of inhibitors: One of the complications of replacement therapy is the development of alloantibodies called inhibitors63. These are high affinity IgG molecules to factor VIII which neutralize clotting factor concentrate, so that the patient becomes non-responsive to treatment. The treatment of inhibitors is done through immune tolerance induction. The latter treatment is however, expensive so that the most patients with inhibitors in India remain untreated. A systematic review reported the prevalence of patients with inhibitors between 5 to 7 per cent, with the risk of inhibitor development being higher in patients with severe rather than mild or moderate haemophilia A64. Studies conducted in India reported the prevalence of inhibitors ranging from 8.2 to 13 per cent65,66. These estimates are likely to be overestimates, as these studies have been conducted at tertiary care centers.
(iv) Access to treatment: In developed countries, use of clotting factor concentrate has resulted in increased longevity, so that the life expectancy of patients with haemophilia is now equal to that of the general population67. Although data on survival of haemophilia patients in India are lacking, access to treatment can be used as a surrogate indicator, as lack of access to treatment reduces longevity. Haemophilia can be treated with replacement therapy on demand, or through prophylactic infusions of clotting factor concentrate68. Prophylaxis reduces the risk of joint damage69, and possibly reduces the number of bleeding episodes and the risk of life threatening bleeds. In India, lack of access to treatment remains a major factor that severely compromises the quality of life of patients. Dharmarajan et al30 measured the bleeding episodes experienced by 24 Indian patients followed up over a 20 week period (Fig. 1). Nearly one-fifth of the injuries were caused by lifestyle-related factors (such as travelling to school by public transportation) whilst the remaining bleeding episodes were spontaneous in nature. The annualized bleeding episodes for patients with severe haemophilia A were 10.8, implying that a patient with severe haemophilia A may bleed as frequently as 11 times in a year. Only one fourth of bleeding episodes were treated with clotting factor concentrate. Data on usage of treatment product in India are available from the global surveys. The data reveal that India has one of the lowest usage of treatment product (Fig. 5b). The per capita use of clotting factor concentrate in India in 2011 was 0.032, as compared to the US (5.157) or even Thailand (0.074)29. The extremely low per capita use of treatment product, despite having the second largest number of global patients with haemophilia A, illustrates the large treatment gap that exists in the country.
(v) Impact on schooling and employment: Without access to treatment, patients with haemophilia experience frequent absenteeism from school and work, as physical activity is limited due to the extreme pain and discomfort associated with the haemorrhagic episode. A cross-sectional study reported that the proportion of school dropouts due to bleeding was 36.5 per cent (n=148)57, while a prospective study estimated that the annual number of school days lost due to haemorrhagic episodes was 19.2 (range 0-50 days)30. The consequence of chronic bleeding episodes and poor education was apparent in the overall proportion of unemployed patients aged above 18 years of age, which was 51 per cent57.
(vi) Emotional costs of haemophilia: Genetic disorders like haemophilia impact parents and families due to the chronic nature of the disorder, the progressive disability arising in the patient due to poor treatment, high treatment costs and the premature mortality25. Parents experience significant emotional distress due to the handicap of the child25. Studies on quality of life (QoL) need assessment and studies on the gender impact of a sex-linked recessive disorder in India have not been reported.
Opportunities for prevention
Apart from clinical reports, the largest thrust of Indian haemophilia research is in the area of mutation analysis of the factor VIII and factor IX genes, and on carrier detection and prenatal diagnosis. The factor VIII gene is 186 kb long and mutations have been identified from the entire length of the gene22. Direct mutation detection as well as indirect methods using linkage analysis have been reported. Studies have estimated the prevalence of inversion 22 (responsible for the aetiology of 50% of severe haemophilia) and inversion 1 rearrangements in patients with severe haemophilia A70,71. Mutation analysis has revealed known and novel mutations72,73,74. Studies have investigated allelic frequency of various intragenic or extragenic markers and their utility in carrier detection and prenatal diagnosis through linkage analysis75,76,77. A diagnostic algorithm for genetic diagnosis has been established78. One of the institutions offering prenatal diagnosis has reported that the quality and easy availability of medical support overwhelmingly determines the attitude and belief towards genetic testing79.
Discussion & future planning
Comparison of prevalence data from developed nations with established haemophilia surveillance shows the possible magnitude of cases in the country. Using the prevalence data of the USA, we estimate that the number of patients with haemophilia could be over 70,000 in India. As more State governments initiate the provision of free treatment, the gap between the observed and estimated number of patients is likely to reduce, increasing the demand on public health services. The large patient load of haemophilia is not unexpected, as haemophilia prevalence is known to increase with improving medical services and increased survival of patients. Like prevalence estimates, the incidence of haemophilia in India is underreported. Considering haemophilia A, and using an estimate of 20,000 IU of clotting factor VIII requirement per patient per year76,80, India would need to acquire 23,17,20,000 IU of clotting factor concentrate for the currently reported number of patients (11,586 patients with haemophilia A). For the estimated number of patients (48,407), an estimated 96,81,40,000 IU of clotting factor VIII concentrate would be needed. This cost suggests that the development of indigenous capacity for the production of clotting factor concentrate may have to be considered in the near future.
The large existing or anticipated burden of patients also suggests that a national programme for haemophilia needs to be initiated. An outline of the key components of a national haemophilia programme is shown in Table IV. Treatment has to be the most important component of a haemophilia service. Apart from issues of training and competency development of health service staff for the management of this disorder, issues of equity will have to be kept in mind, as patients from rural and remote areas are less likely to have access to the service as compared to patients from urban areas or from more socio-economically privileged groups. A method of auditing clotting factor concentrate to ensure equitable use will be needed. Compulsory hepatitis B vaccination of all patients, and routine screening of patients for HIV could be an essential component of the programme directed towards prevention and screening for TTIs. Education of patients, and psychosocial support, especially after diagnosis (which has a devastating impact on parents)25 have to be the major components of haemophilia services.
Table IV.
Components of a programme for the prevention and control of genetic disorders

The prevention of haemophilia needs to be addressed on a priority basis. In contrast to the paucity of data on the epidemiology and social costs of haemophilia, there are numerous studies on the genetic basis of this condition, including reports of prenatal diagnosis being conducted routinely at premier institutions of the country. This wealth of information indicates the biotechnological competencies of the country, but this potential remains untranslated to communities, being limited to referred patients only. Prenatal diagnostic services have to initiate with the identification of facilities for amniocentesis at the district level. Genetic counselling services can be delivered through designated referral centres and national laboratories. Unlike other developing countries, the technological competencies will not be the limiting factor for a haemophilia programme in India. Rather, capacity building for launching genetic services, including building an understanding of genetics in public health will have to be initiated. Critical to this will be the need to develop genetic counselling material which is contextual to the cultural milieu of patients.
Surveillance for haemophilia has to be an integral component of haemophilia services. Maintenance of the existing haemophilia registry is vital to providing surveillance data, that is information on the burden and trends of the disorder, data for estimating treatment needs and monitoring the impact of services. Carefully selected variables can yield significant indicators for tracking the long term trends of haemophilia in the country81,82. The registry may be appropriately used by public health services for protecting the general population. For example, the haemophilia surveillance system in the USA, is used for monitoring the prevalence of TTIs and for monitoring the emergence of new blood-borne infections52. In conclusion, the complacency about the low prevalence of genetic disorders in India may have to be revisited. Even at the current rates of case detection, India appears to harbour a large patient burden. The high cost of providing treatment for patients is likely to overwhelm the public health services, suggesting the need for launching a national programme for haemophilia with components of prevention, care and support.
References
- 1.Kar A. Preventing birth defects in India. Economic Political Weekly. 2011;46:21–2. [Google Scholar]
- 2.March of Dimes Foundation. March of Dimes Global Report on Birth Defects: The hidden toll of dying and disabled children. 2006. [accessed on November 22, 2012]. Available from: http://www.marchofdimes.com/mission/globalprograms_birthdefectsreport. html .
- 3.Dye DE, Brameld KJ, Maxwell S, Goldblatt J, O’ Leary P. The impact of single gene and chromosomal disorders on hospital admissions in an adult population. J Community Genet. 2011;2:81–90. doi: 10.1007/s12687-011-0043-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shrikhande AV, Dani AA, Tijare JR, Agrawal AK. Hematological profile of sickle cell disease in central India. Indian J Hematol Blood Transfus. 2007;23:92–8. doi: 10.1007/s12288-008-0005-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rao SS, Goyal JP, Raghunath SV, Shah VB. Hematological profile of sickle cell disease from South Gujarat, India. Hematol Rep. 2012;4:e8. doi: 10.4081/hr.2012.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jain D, Italia K, Sarathi V, Ghosh K, Colah R. Sickle cell anemia from Central India: a retrospective analysis. Indian Pediatr. 2012;49:911–3. doi: 10.1007/s13312-012-0217-z. [DOI] [PubMed] [Google Scholar]
- 7.Balgir RS. Community expansion and gene geography of sickle cell trait and G6PD deficiency, and natural selection against malaria: experience from tribal land of India. Cardiovasc Hematol Agents Med Chem. 2012;10:3–13. doi: 10.2174/187152512799201190. [DOI] [PubMed] [Google Scholar]
- 8.Nikhar HS, Meshram SU, Shinde GB. A comprehensive review of diverse issues related to sickle cell disease. East Afr J Public Health. 2011;8:164–9. [PubMed] [Google Scholar]
- 9.Balgir RS. Spectrum of hemoglobinopathies in the state of Orissa, India: a ten years cohort study. J Assoc Physicians India. 2005;53:1021–6. [PubMed] [Google Scholar]
- 10.Mohanty D, Colah RB, Gorakshakar AC, Patel RZ, Master DC, Mahanta J, et al. Prevalence of β-thalassemia and other haemoglobinopathies in six cities in India: a multicentre study. J Community Genet. 2013;4:33–42. doi: 10.1007/s12687-012-0114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chatterjee N, Mishra A, Soni R, Kulkarni H, Mamtani M, Shrivasatava M. Bayesian estimates of the prevalence of β-thalassemia trait in voluntary blood donors of central India: a survey. Hemoglobin. 2010;34:548–60. doi: 10.3109/03630269.2010.526488. [DOI] [PubMed] [Google Scholar]
- 12.Christianson A, Modell B. Medical genetics in developing countries. Annu Rev Genomics Hum Genet. 2004;5:219–65. doi: 10.1146/annurev.genom.5.061903.175935. [DOI] [PubMed] [Google Scholar]
- 13.Bellagio, Italy: 2005. Apr 14-20, [accessed on November 22, 2012]. Genome-based Research and Population Health. Report of an expert workshop held at the Rockefeller Foundation Study and Conference Centre. Available from: http://www.phgfoundation.org/file/2205 . [Google Scholar]
- 14.UNICEF India statistics. [accessed on October 15, 2012]. Available from: http://www.unicef.org/infobycountry/india_statistics.html .
- 15.New Delhi: [accessed on October 12, 2012]. Census of India 2011, Office of the Registrar General and Census Commissioner, India, Controller of Publications. Available from: http://www.censusindia.gov.in/2011-prov-results/prov_results_paper1_india.html . [Google Scholar]
- 16.New Delhi: Controller of Publications; [accessed on October 12, 2012]. Sample Registration System, Office of the Registrar General and Census Commissioner, India. Available from: http://www.censusindia.gov.in/2011-Common/srs.html . [Google Scholar]
- 17.National Rural Health Mission PIP: 2012-13 Mission Flexipool. Government of Maharashtra. [accessed on May 9, 2013]. Available from: http://www.nrhm.maharashtra.gov.in/partb.pdf .
- 18.Skinner MW. WFH: closing the global gap - achieving optimal care. Haemophilia. 2012;18(Suppl 4):1–12. doi: 10.1111/j.1365-2516.2012.02822.x. [DOI] [PubMed] [Google Scholar]
- 19.Lee CA. Historical introduction. In: Lee C, Berntorp E, Hoots K, editors. Textbook of hemophilia. 2nd ed. West Sussex: Wiley-Blackwell; 2010. pp. 1–6. [Google Scholar]
- 20.van den Berg HM, Fischer K. Phenotypic - genotypic relationship. In: Lee C, Berntorp E, Hoots K, editors. Textbook of hemophilia. 2nd ed. West Sussex: Wiley-Blackwell; 2010. pp. 33–7. [Google Scholar]
- 21.Mannucci PM, Tuddenham EG. The hemophilias - from royal genes to gene therapy. N Engl J Med. 2001;344:1773–9. doi: 10.1056/NEJM200106073442307. [DOI] [PubMed] [Google Scholar]
- 22.Bowen DJ. Haemophilia A and haemophilia B: molecular insights. Mol Pathol. 2002;55:127–44. doi: 10.1136/mp.55.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berntorp E, Shapiro AD. Modern haemophilia care. Lancet. 2012;379:1447–56. doi: 10.1016/S0140-6736(11)61139-2. [DOI] [PubMed] [Google Scholar]
- 24.Philipp C. The aging patient with hemophilia: complications, comorbidities, and management issues. Hematology Am Soc Hematol Educ Program 2010. 2010:191–6. doi: 10.1182/asheducation-2010.1.191. [DOI] [PubMed] [Google Scholar]
- 25.Cohen JS, Biesecker BB. Quality of life in rare genetic conditions: a systematic review of the literature. Am J Med Genet A. 2010;152A:1136–56. doi: 10.1002/ajmg.a.33380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dharmarajan S, Phadnis S, Gund P, Kar A. Out-of-pocket and catastrophic expenditure on treatment of haemophilia by Indian families. Haemophilia. 2014;20:382–7. doi: 10.1111/hae.12324. [DOI] [PubMed] [Google Scholar]
- 27.Haldane JB. The rate of spontaneous mutation of a human gene. J Genet. 1935;31:317–26. doi: 10.1007/BF02717892. [DOI] [PubMed] [Google Scholar]
- 28.Giannelli F, Choo KH, Rees DJG, Boyd Y, Rizza CR, Brownlee GG. Gene deletions in patients with haemophilia B and anti-factor IX antibodies. Nature. 1983;303:181–2. doi: 10.1038/303181a0. [DOI] [PubMed] [Google Scholar]
- 29.World Federation of Hemophilia (WFH) Canada: WFH; 2013. [accessed on May 9, 2013]. Report on the Annual Global Survey 2011. Available from: http://www1.wfh.org/publications/files/pdf-1488.pdf . [Google Scholar]
- 30.Dharmarajan S, Phadnis S, Gund P, Kar A. Treatment decisions and usage of clotting factor concentrate by a cohort of Indian haemophilia patients. Haemophilia. 2012;18:e27–9. doi: 10.1111/j.1365-2516.2011.02668.x. [DOI] [PubMed] [Google Scholar]
- 31.Hemophilia Federation (India) [accessed on October 12, 2012]. Available from: http://www.hemophilia.in/
- 32.Rodeghiero F, Castaman G. von Willebrand disease: epidemiology. In: Lee C, Berntorp E, Hoots K, editors. Textbook of hemophilia. 2nd ed. West Sussex: Wiley-Blackwell; 2010. pp. 286–93. [Google Scholar]
- 33.Srivastava A, Rodeghiero F. Epidemiology of von Willebrand disease in developing countries. Semin Thromb Hemost. 2005;31:569–76. doi: 10.1055/s-2005-922229. [DOI] [PubMed] [Google Scholar]
- 34.Bolton-Maggs PH. The rare coagulation disorders. World Federation of Haemophilia. 2006. [accessed on May 7, 2013]. Available from : http://www1.wfh.org/docs/en/About_Bleeding_Disorders/RBD_Characteristics_Table.pdf .
- 35.Kemball-Cook G, Gomez K. Molecular basis of hemophilia A. In: Lee C, Berntorp E, Hoots K, editors. Textbook of hemophilia. 2nd ed. West Sussex: Wiley-Blackwell; 2010. pp. 24–32. [Google Scholar]
- 36.Gomez K. Hemophilia B-molecular basis. In: Lee C, Berntorp E, Hoots K, editors. Textbook of hemophilia. 2nd ed. West Sussex: Wiley-Blackwell; 2010. pp. 88–93. [Google Scholar]
- 37.Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003;361:1801–9. doi: 10.1016/S0140-6736(03)13405-8. [DOI] [PubMed] [Google Scholar]
- 38.Mannucci PM. Back to the future: a recent history of haemophilia treatment. Haemophilia. 2008;14(Suppl 3):10–8. doi: 10.1111/j.1365-2516.2008.01708.x. [DOI] [PubMed] [Google Scholar]
- 39.Franchini M. The modern treatment of haemophilia: a narrative review. Blood Transfus. 2013;11:178–82. doi: 10.2450/2012.0166-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carcao MD. The diagnosis and management of congenital hemophilia. Semin Thromb Hemost. 2012;38:727–34. doi: 10.1055/s-0032-1326786. [DOI] [PubMed] [Google Scholar]
- 41.Keeling D, Tait C, Makris M. Guideline on the selection and use of therapeutic products to treat haemophilia and other hereditary bleeding disorders. A United Kingdom Haemophilia Center Doctors’ Organisation (UKHCDO) guideline approved by the British Committee for Standards in Haematology. Haemophilia. 2008;14:671–84. doi: 10.1111/j.1365-2516.2008.01695.x. [DOI] [PubMed] [Google Scholar]
- 42.EURORDIS Rare Diseases Europe. About orphan drugs. [accessed on October 9, 2012]. Available from: http://www.eurordis.org/about-orphan-drugs .
- 43.Kar A. Making haemophilia a global priority. Lancet. 2012;380:216–7. doi: 10.1016/S0140-6736(12)61214-8. [DOI] [PubMed] [Google Scholar]
- 44.Dalal A, Pradhan M, Agarwal S. Genetics of bleeding disorders. Int J Hum Genet. 2006;6:27–32. [Google Scholar]
- 45.Forrest CB, Bartek RJ, Rubinstein Y, Groft SC. The case for a global rare-diseases registry. Lancet. 2011;377:1057–9. doi: 10.1016/S0140-6736(10)60680-0. [DOI] [PubMed] [Google Scholar]
- 46.Kar A. Factors influencing haemophilia prevalence estimates from the volunteer-supervised Indian registry in Maharashtra. Haemophilia. 2010;16:952–4. doi: 10.1111/j.1365-2516.2010.02270.x. [DOI] [PubMed] [Google Scholar]
- 47.Soucie JM, Evatt B, Jackson D. Occurrence of hemophilia in the United States. The Hemophilia Surveillance System Project Investigators. Am J Hematol. 1998;59:288–94. doi: 10.1002/(sici)1096-8652(199812)59:4<288::aid-ajh4>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 48.Report on the Annual Global Survey 2010. Montreal, QC, Canada: World Federation of Hemophilia; 2011. [accessed on May 9, 2013]. World Federation of Hemophilia. Available from: http://www.wfh.org/en/pageaspx?pid=492 . [Google Scholar]
- 49.Report on the Annual Global Survey 2009. Canada: WFH; 2011. [accessed on May 9, 2013]. World Federation of Hemophilia (WFH) Available from: http://www1.wfh.org/publications/files/pdf-1428.pdf . [Google Scholar]
- 50.Stonebraker JS, Bolton-Maggs PH, Soucie JM, Walker I, Brooker M. A study of variations in the reported haemophilia A prevalence around the world. Haemophilia. 2010;16:20–32. doi: 10.1111/j.1365-2516.2009.02127.x. [DOI] [PubMed] [Google Scholar]
- 51.Report on the Annual Global Survey 2010. Canada: WFH; 2011. [accessed on May 9, 2013]. World Federation of Hemophilia (WFH) Available from http://www.wfh.org/en/page.aspx?pid=492 . [Google Scholar]
- 52.Universal Data Collection (UDC) System. Centers for Disease Control and Prevention. [accessed on May 9, 2013]. Available from: http://www.cdc.gov/ncbddd/blooddisorders/udc/
- 53.Report on the Annual Global Survey 2008. Canada: WFH; 2009. [accessed on May 9, 2013]. World Federation of Hemophilia (WFH) Available from: http://www1.wfh.org/publications/files/pdf-1429.pdf . [Google Scholar]
- 54.Report on the Annual Global Survey 2007. Canada: World Federation of Hemophilia; 2008. [accessed on May 9, 2013]. World Federation of Hemophilia (WFH) Available from: http://www1.wfh.org/publications/files/pdf-1430.pdf . [Google Scholar]
- 55.Report on the Annual Global Survey 2006. Canada: WFH; 2007. [accessed on May 9, 2013]. World Federation of Hemophilia (WFH) Available from: http://www1.wfh.org/publications/files/pdf-1431.pdf . [Google Scholar]
- 56.Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, et al. Treatment Guidelines Working Group on Behalf of the World Federation of Hemophilia. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1–47. doi: 10.1111/j.1365-2516.2012.02909.x. [DOI] [PubMed] [Google Scholar]
- 57.Kar A, Mirkazemi R, Singh P, Potnis-Lele M, Lohade S, Lalwani A, et al. Disability in Indian patients with haemophilia. Haemophilia. 2007;13:398–404. doi: 10.1111/j.1365-2516.2007.01483.x. [DOI] [PubMed] [Google Scholar]
- 58.Nair AP, Jijina F, Ghosh K, Madkaikar M, Shrikhande M, Nema M. Osteoporosis in young haemophiliacs from western India. Am J Hematol. 2007;82:453–7. doi: 10.1002/ajh.20877. [DOI] [PubMed] [Google Scholar]
- 59.Ghosh K, Joshi SH, Shetty S, Pawar A, Chipkar S, Pujari V, et al. Transfusion transmitted diseases in haemophilics from western India. Indian J Med Res. 2000;112:61–4. [PubMed] [Google Scholar]
- 60.Singh YN, Bhargava M, Malaviya AN, Tripathy SP, Kakkar A, Khare SD. HIV infection in Asian Indian patients with haemophilia & those who had multiple transfusions. Indian J Med Res. 1991;93:12–4. [PubMed] [Google Scholar]
- 61.Pallavi P, Ganesh CK, Jayashree K, Manjunath GV. Seroprevalance and trends in transfusion transmitted infections among blood donors in a university hospital blood bank: a 5 year study. Indian J Hematol Blood Transfus. 2011;27:1–6. doi: 10.1007/s12288-010-0047-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khaja MN, Madhavi C, Thippavazzula R, Nafeesa F, Habib AM, Habibullah CM, et al. High prevalence of hepatitis C virus infection and genotype distribution among general population, blood donors and risk groups. Infect Genet Evol. 2006;6:198–204. doi: 10.1016/j.meegid.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 63.Saint-Remy JM, Jacquemin MG. Inhibitors to factor VIII-immunology. In: Lee C, Berntorp E, Hoots K, editors. Textbook of hemophilia. 2nd ed. West Sussex: Wiley-Blackwell; 2010. pp. 52–6. [Google Scholar]
- 64.Wight J, Paisley S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003;9:418–35. doi: 10.1046/j.1365-2516.2003.00780.x. [DOI] [PubMed] [Google Scholar]
- 65.Ghosh K, Shetty S, Kulkarni B, Nair S, Pawar A, Khare A, et al. Development of inhibitors in patients with haemophilia from India. Haemophilia. 2001;7:273–8. doi: 10.1046/j.1365-2516.2001.00505.x. [DOI] [PubMed] [Google Scholar]
- 66.Mathews V, Nair SC, David S, Viswabandya A, Srivastava A. Management of hemophilia in patients with inhibitors: the perspective from developing countries. Semin Thromb Hemost. 2009;35:820–6. doi: 10.1055/s-0029-1245115. [DOI] [PubMed] [Google Scholar]
- 67.Tagliaferri A, Rivolta GF, Iorio A, Oliovecchio E, Mancuso ME, Morfini M, et al. Mortality and causes of death in Italian persons with haemophilia, 1990-2007. Haemophilia. 2010;16:437–46. doi: 10.1111/j.1365-2516.2009.02188.x. [DOI] [PubMed] [Google Scholar]
- 68.Fischer K, van den Berg HM. Prophylaxis. In: Lee C, Berntorp E, Hoots K, editors. Textbook of hemophilia. 2nd ed. West Sussex: Wiley-Blackwell; 2010. pp. 38–43. [Google Scholar]
- 69.Iorio A, Marchesini E, Marcucci M, Stobart K, Chan AK. Clotting factor concentrates given to prevent bleeding and bleeding-related complications in people with hemophilia A or B. Cochrane Database Syst Rev. (9):CD003429. doi: 10.1002/14651858.CD003429.pub2. [DOI] [PubMed] [Google Scholar]
- 70.Shetty S, Pathare A, Ghosh K, Jijina F, Mohanty D. Intron 22 inversions in factor VIII gene in Indian hemophiliacs. Thromb Haemost. 1998;79:881–2. [PubMed] [Google Scholar]
- 71.Ahmed R, Kannan M, Choudhry VP, Saxena R. Mutation reports: intron 1 and 22 inversions in Indian haemophilics. Ann Hematol. 2003;82:546–7. doi: 10.1007/s00277-003-0704-3. [DOI] [PubMed] [Google Scholar]
- 72.Jayandharan G, Shaji RV, Baidya S, Nair SC, Chandy M, Srivastava A. Identification of factor VIII gene mutations in 101 patients with haemophilia A: mutation analysis by inversion screening and multiplex PCR and CSGE and molecular modelling of 10 novel missense substitutions. Haemophilia. 2005;11:481–91. doi: 10.1111/j.1365-2516.2005.01121.x. [DOI] [PubMed] [Google Scholar]
- 73.Jayandharan GR, Shaji RV, Baidya S, Nair SC, Chandy M, Srivastava A. Molecular characterization of factor IX gene mutations in 53 patients with haemophilia B in India. Thromb Haemost. 2005;94:883–6. [PubMed] [Google Scholar]
- 74.Ghosh K, Quadros L, Shetty S. Spectrum of factor IX gene mutations causing haemophilia B from India. Blood Coagul Fibrinolysis. 2009;20:333–6. doi: 10.1097/MBC.0b013e32832b27d1. [DOI] [PubMed] [Google Scholar]
- 75.Shetty S, Ghosh K, Pathare A, Colah R, Badakare S, Mohanty D. Factor VIII and IX gene polymorphisms and carrier analysis in Indian population. Am J Hematol. 1997;54:271–5. doi: 10.1002/(sici)1096-8652(199704)54:4<271::aid-ajh2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 76.Jayandharan G, Shaji RV, George B, Chandy M, Srivastava A. Informativeness of linkage analysis for genetic diagnosis of haemophilia A in India. Haemophilia. 2004;10:553–9. doi: 10.1111/j.1365-2516.2004.00908.x. [DOI] [PubMed] [Google Scholar]
- 77.Saha A, Mukherjee S, Maulik M, Chandak GR, Ray K. Indian Genome Variation Consortium, Evaluation of genetic markers linked to hemophilia A locus: an Indian experience. Haematologica. 2007;92:1725–6. doi: 10.3324/haematol.11545. [DOI] [PubMed] [Google Scholar]
- 78.Jayandharan GR, Srivastava A, Srivastava A. Role of molecular genetics in hemophilia: from diagnosis to therapy. Semin Thromb Hemost. 2012;38:64–78. doi: 10.1055/s-0031-1300953. [DOI] [PubMed] [Google Scholar]
- 79.Ghosh K, Shetty S. Quality and easy availability of medical support overwhelmingly determines the attitude and belief towards genetic testing in haemophilia. Haemophilia. 2008;14:392–3. doi: 10.1111/j.1365-2516.2007.01641.x. [DOI] [PubMed] [Google Scholar]
- 80.Britten A, Etzel F, Ala F, Carman C, Smit Sibinga C. Supply and need of factor VIII concentrates. Haematologia (Budap) 1983;16:109–20. [PubMed] [Google Scholar]
- 81.Kar A, Potnis-Lele M. Descriptive epidemiology of haemophilia in Maharashtra, India. Haemophilia. 2001;7:561–7. doi: 10.1046/j.1365-2516.2001.00570.x. [DOI] [PubMed] [Google Scholar]
- 82.Kar A, Potnis-Lele M. Haemophilia data collection in developing countries: example of the haemophilia database of Maharashtra. Haemophilia. 2004;10:301–4. doi: 10.1111/j.1365-2516.2004.00883.x. [DOI] [PubMed] [Google Scholar]