

Abstract
Background & objectives:
Multiple suphphatase deficiency (MSD) is an autosomal recessive disorder affecting the post translational activation of all enzymes of the sulphatase family. To date, approximately 30 different mutations have been identified in the causative gene, sulfatase modifying factor 1 (SUMF1). We describe here the mutation analysis of a case of MSD.
Methods:
The proband was a four year old boy with developmental delay followed by neuroregression. He had coarse facies, appendicular hypertonia, truncal ataxia and ichthyosis limited to both lower limbs. Radiographs showed dysostosis multiplex. Clinical suspicion of MSD was confirmed by enzyme analysis of four enzymes of the sulphatase group.
Results:
The patient was compound heterozygote for a c.451A>G (p.K151E) substitution in exon 3 and a single base insertion mutation (c.690_691 InsT) in exon 5 in the SUMF1 gene. The bioinformatic analysis of the missense mutation revealed no apparent effect on the overall structure. However, the mutated 151-amino acid residue was found to be adjacent to the substrate binding and the active site residues, thereby affecting the substrate binding and/or catalytic activity, resulting in almost complete loss of enzyme function.
Conclusions:
The two mutations identified in the present case were novel. This is perhaps the first report of an insertion mutation in SUMF1 causing premature truncation of the protein.
Keywords: Dysostosis, icthysosis, multiple sulphatase, neuroregression
Multiple sulphatase deficiency (MSD) is an autosomal recessive disorder with an estimated prevalence of 1 in 1.4 million live births1. There is a deficient activity of multiple enzymes of the sulphatase family in MSD patients. The lysosomal sulphatases are involved in the hydrolysis of sulphate esters present in glycosaminoglycans, glycolipids, glycopeptides and hydroxysteroids2. Grouped together due to structural similarities, substrate specificity is highlighted by the different disease phenotypes observed in individual enzyme deficiency. Interestingly, the activity of all the sulphatases’ requires a common post-translational modification involving conversion of a conserved cysteine residue at position 69 to alpha-formylglycine3. The formyl glycine generating enzyme (FGE) is encoded by sulphatase modifying factor 1 (SUMF1) gene that has been highly conserved during evolution, from bacteria to humans4. The gene is localized on chromosome 3 and comprises nine coding exons5. Patients with MSD carry mutations in this gene5,6. The clinical features in MSD, partly intersect with as well as extend beyond those observed in mucopolysaccharidosis2. There are less than 100 cases published in the literature accounting for approximately 30 different SUMF1mutations7. We describe here a case of MSD where mutation analysis revealed the proband to be a compound heterozygote for two novel mutations.
Material & Methods
A 4-year old boy, born to non-consanguineous parents hailing from north India was referred to the Genetic clinic of Sir Gangaram Hospital, New Delhi, India, in September 2011 with a history of developmental delay and loss of attained milestones. Born at term, he weighed 2275 g. The neonatal period was uneventful. Parents recalled that he was cruising with support by 15 months of age, though independent ambulation was never attained. At 24 months of age the boy had frequent falls and truncal instability followed by gradual loss of attained milestones. At the time of examination at 4 yr, he was non ambulatory and could sit only with support. Speech was limited to vocalizations only. There was no history of seizures. The family history was non-contributory.
On examination, his weight was 12 kg (3rd -5th centile), length – 97cm (3rd-5th centile) and occipito frontal circumference was 47.5 cm (< 3rd centile). He had coarse facies with depressed nasal bridge, short neck, low posterior hairline and a small mouth. The eyebrows were thick and bushy with medial flaring. Both great toes were broad and medially deviated while the rest of the fingers and thumbs were normal. He had ichthyosis, mainly in both lower limbs (Fig. 1). Bilateral limitation of ankle extension was present, while movements at the other joints were normal. He had appendicular hypertonia in all four limbs with brisk reflexes and truncal ataxia. There was no organomegaly, cardiac involvement, gum hypertrophy or corneal clouding. Detailed opthalmological examination was normal. The radiographs of the proband showed narrowing of the base of metacarpals 2-5, bullet shaped proximal phalanges, beaking of the lumbar vertebrae, and thickened calvaria with a J shaped sella turcica. A brain MRI at 2 yr of age showed cerebral atrophy with diffuse involvement of the cerebral white matter. Central structures including the corpus callosum, posterior limb of internal capsule and the brain stem were affected while the subcortical U fibres were spared. A radiating stripe like pattern of low signal intensity was seen on T2 weighted images. The cerebellar white matter also depicted an abnormal signal intensity.
Fig. 1.
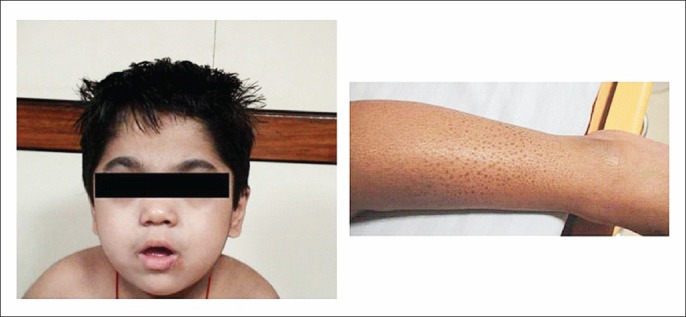
Coarse facial features and icthyosis in the lower limbs.
Urine examination for glycosaminoglycans (GAGs) was positive, with chromatography revealing mainly chondrotin and heparin sulphate excretion. The combination of a storage phenotype with ichthyosis and neuroregression led us to consider the possibility of MSD.
Biochemical analysis: Leukocytes were separated from heparinized whole blood samples of the patient and normal subject. Cells lysate was prepared by sonicating leukocytes in water for enzyme assays. Protein was measured in mg/ml. Four sulfate containing enzymes (aryl sulphatase A, aryl sulphatase B, heparan sulphamidase and iduronate 2 sulphatase) assays were analyzed using spectrophotometric and fluorometric methods for diagnosis of MSD. The substrates 4-nitrocatechol sulphate and 4- methylumbelliferyl sulphate were obtained from Sigma, USA, for the estimation of aryl sulphatase A and B enzymes, respectively8,9. The fluorometric substrates 4 methylumbelliferyl α- L-iduronide-2-sulphate and 4-methylumbelliferyl α-D- N-sulphoglucosaminide (MU-alpha-GlcNS). Na were obtained from Moscerdam, The Netherland, for the analysis of iduronate 2 sulphatase and heparan sulphamidase enzymes, respectively10,11. All four sulphatases levels demonstrated deficiencies of these enzymes, confirming the diagnosis of MSD.
Molecular analysis: Genomic DNA was extracted from peripheral blood. The nine exons of SUMF1 were amplified with primers designed using Primer3 software (University of Massachusetts, Medical School, USA) spanning the exon intron boundries. PCR was performed in Sir Gangaram Hospital, New Delhi using 10pmol of each primer, 1U of Taq DNA polymerase, 1.5mM MgCl2 and 0.1mM dNTPs in the recommended buffer with the initial denaturation at 94°C for 5 min, 35 cycles of denaturation at 94°C for 30 sec, annealing at 58°C for 30 sec and extension at 72°C for 45 sec. Complete SUMF1 gene sequencing was carried out on ABI 3500 sequencer (Applied Biosystems, USA) covering all the nine coding exons encompassing exon intron boundaries.
Results & Discussion
The patient was compound heterozygote for a c.451A>G (p.K151E) substitution in exon 3 and a single base insertion mutation (c.690_691 InsT) in exon 5 on sequencing of SUMF1 gene, establishing the diagnosis of MSD (Fig. 2a). The first variation was a missense mutation causing replacement of lysine with glutamic acid at 151 amino acid position. The second insertion mutation changed the sequence after glutamic acid 230 leading to stop codon downstream of 32 bases (after the residue 240) in the tryptophan rich domain.
Fig. 2.
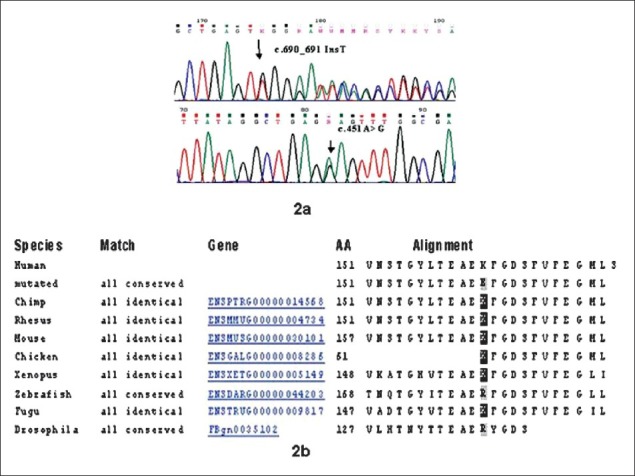
Chromatogram of the novel mutations and sequence conservation at the amino acid position 151. Chromatogram, sequence conservation and surface topography of K151E substitution. 2a: Chromatogram of the novel variants identified in the present case. 2b: Conserved amino acid lysine (K) at 151 position across the seven species.
The K151E mutation lies in the N-terminal subdomain [residues 91-154 in human formyl glycine generating enzyme (FGE)] which has a sequence identity of 39 per cent and a similarity of 85 per cent within the six known full-length eukaryotic FGE orthologs5. In human FGE, this domain carries the N-glycosylation site at Asn-141, which is conserved in the other orthologs. The amino acid lysine at the 151 position of the FGE protein is conserved across the species with exception of Zebrafish and Drosophila (Fig. 2b). We performed a computational comparative analysis of the sequences through Ensembl variant effect predictor (http://asia.ensembl.org/tools.html) which employs Polyphen II (Polymorphism Phenotyping) and SIFT (Sorting Intolerant From Tolerant) for determining whether the novel missense variant identified in the present case was pathogenic or not. The missense mutation c.451A>G was found to be deleterious with a score of 0.04 by SIFT although Polyphen II showed it to be benign. The prediction of SIFT was corroborated by Mutation Taster (http://www.mutationtaster.org/) which also gave the result as disease causing.
Since the p.K151E was a novel missense mutation, we proceeded to perform bioinformatic analysis. The mutant SUMF1 protein was modelled based on the FGE structure (PDB ID: 1Y1E) using Modeller12,13. The two structures were aligned and visualized using PyMOL Molecular Graphics system (Schrödinger, LLC). Strikingly, there was no apparent effect of the mutation on the overall structure as indicated by the Root mean square deviation (RMSD) value of only 0.125 Å with the 1Y1E template structure of FGE protein (data not shown). However, the amino acid residue-151 (the site of mutation) was found to be adjacent to the active site as well as substrate binding residues. Therefore, the mutation at this site probably changes the surface topography which in turn affects substrate binding and/or catalytic activity, resulting in almost non-functional enzyme. This was substantiated by the finding that the pocket size in the mutated protein was reduced by approximately 20 per cent in comparison to the normal protein (calculated using CASTp with a probe radius of 1.0 Å)14.
Eight known disorders associated with individual sulphatase enzyme deficiencies include metachromatic leukodystrophy (MLD, arylsulphatase A), Hunter disease (MPS II, iduronate - 2- sulphatase), Sanfillipo A (MPS IIIA, sulphamidase), Sanfillipo D (MPS IIID, Glucosamine -6-sulphatase), Morquio A (MPS IVA, N-acetylgalactosamine-6-sulphatase), and Morteaux Lamy (MPS VI, N-acetylygalactosamine-4-sulphatase alias arylsulphatase B), X- linked ichthyosis (arylsulphatase C) and chondrodysplasia punctata (arylsulphatase E)2. The phenotype of MSD combines features of different sulphatase deficiencies. Developmental delay, coarse facies and organomegaly as in mucopolysaccharidosis, neurodegenerative course like metachromatic leucodystrophy, radiographic changes reminiscent of chondrodysplasia punctata and X-linked ichthyosis like skin changes. A clinical clue to suspect the diagnosis was the presence of a combination of “multiple phenotypes” like coarse facies with dysostosis, neuroregression and ichthyosis in our patient.
Based on the age of presentation, three different variants are known: neonatal, late infantile and a juvenile type of MSD15. In the neonatal presentation symptoms appear in the first few months and death usually occurs by one year. The late infantile type usually has an onset after 1 year and can be further divided into mild and severe forms. Our patient was the severe form of the late infantile variety. The juvenile form, though very rare, presents later with mild symptoms. At the molecular level, this heterogeneity is explained by the degree of protein stability and amount of residual enzyme activity7.
The majority of the mutations identified in SUMF1 gene so far are missense followed by nonsense, microdeletion and splicing mutations clustering mainly in the C-terminal subdomain of the protein7. Our patient was compound heterozygous for a c.451A>G substitution in exon 3 and a single base insertion mutation (c.690_691 InsT) in exon 5 adding to the spectrum of SUMF1 mutations that cause MSD. Use of bioinformatic tools helped in predicting the possible pathogenicity of the novel mutation in this rare disorder as in vitro studies were not possible. The two mutations identified in our case were novel, reporting insertion mutation in SUMF1 causing premature truncation of the protein.
Identification of novel mutations adds to the database of disease causing changes in the SUMF1 gene. In rare disorders such as this where mutation reports are infrequent and functional studies difficult, bioinformatics plays an important role to confirm the pathogenicity of the muations.
References
- 1.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–54. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 2.Ballabio A, Shapiro LJ. Steroid sulfatase deficiency and X-linked ichthyosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The metabolic and molecular basis of inherited disease. 8th ed. New York: McGraw-Hill; 2001. pp. 4241–62. [Google Scholar]
- 3.Schmidt B, Selmer T, Ingendoh A, von Figura K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell. 1995;82:271–8. doi: 10.1016/0092-8674(95)90314-3. [DOI] [PubMed] [Google Scholar]
- 4.Landgrebe J, Dierks T, Schmidt B, von Figura KV. The human SUMF1 gene, required for posttranslational sulfatase modification defines a new gene family which is conserved from pro- to eukaryotes. Gene. 2003;316:47–56. doi: 10.1016/s0378-1119(03)00746-7. [DOI] [PubMed] [Google Scholar]
- 5.Dierks T, Schmidt B, Borissenko LV, Peng J, Preusser A, Mariappan M, et al. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human Cα-formylglycine generating enzyme. Cell. 2003;113:435–44. doi: 10.1016/s0092-8674(03)00347-7. [DOI] [PubMed] [Google Scholar]
- 6.Cosma MP, Pepe S, Annunziata I, Newbold RF, Grompe M, Parenti G, et al. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 2003;113:445–56. doi: 10.1016/s0092-8674(03)00348-9. [DOI] [PubMed] [Google Scholar]
- 7.Schlotawa L, Ennemann EC, Radhakrishnan K, Schmidt B, Chakrapani A, Christen HJ, et al. SUMF1 mutations affecting stability and activity of formylglycine generating enzyme predict clinical outcome in multiple sulfatase deficiency. Eur J Hum Genet. 2011;19:253–61. doi: 10.1038/ejhg.2010.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Percy AK, Brady RO. Metachromatic leukodystrophy: diagnosis with samples of venous blood. Science. 1968;161:594–5. doi: 10.1126/science.161.3841.594. [DOI] [PubMed] [Google Scholar]
- 9.Harinath BC, Robins E. Arylsulphatases in human brain: assay, some properties and distribution. J Neurochem. 1971;18:237–44. doi: 10.1111/j.1471-4159.1971.tb00562.x. [DOI] [PubMed] [Google Scholar]
- 10.Voznyi YV, Keulemans JL, van Diggelen OP. A fluorimetric enzyme assay for the diagnosis of MPS II (Hunter disease) J Inherit Metab Dis. 2001;24:675–80. doi: 10.1023/a:1012763026526. [DOI] [PubMed] [Google Scholar]
- 11.Karpova EA, Voznyi YV, Keulemans JL, Hoogeveen AT, Winchester B, Tsvetkova I, et al. A fluoroimetric enzyme assay for the diagnosis of Sanfilippo disease type A (MPS IIIA) J Inherit Metab Dis. 1996;19:278–85. doi: 10.1007/BF01799255. [DOI] [PubMed] [Google Scholar]
- 12.Dierks T, Dickmanns A, Preusser-Kunze A, Schmidt B, Mariappan M, von Figura K, et al. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell. 2005;121:541–52. doi: 10.1016/j.cell.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 14.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Web Server issue):W116–8. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eto Y, Gomibuchi I, Umezawa F, Tsuda T. Pathochemistry, pathogenesis and enzyme replacement in multiple-sulfatase deficiency. Enzyme. 1987;38:273–9. doi: 10.1159/000469216. [DOI] [PubMed] [Google Scholar]