

Abstract
O-GlcNAcylation is a modification that alters the function of numerous proteins. We hypothesized that augmented O-GlcNAcylation levels enhance myosin light chain kinase (MLCK) and reduce myosin light chain phosphatase (MLCP) activity, leading to increased vascular contractile responsiveness. The vascular responses were measured by isometric force displacement. Thoracic aorta and vascular smooth muscle cells (VSMCs) from rats were incubated with vehicle or with PugNAc, which increases O-GlcNAcylation. In addition, we determined whether proteins that play an important role in the regulation of MLCK and MLCP activity are directly affected by O-GlcNAcylation. PugNAc enhanced phenylephrine (PE) responses in rat aortas (maximal effect, 14.2±2 vs 7.9±1 mN for vehicle, n=7). Treatment with an MLCP inhibitor (calyculin A) augmented vascular responses to PE (13.4±2 mN) and abolished the differences in PE-response between the groups. The effect of PugNAc was not observed when vessels were preincubated with ML-9, an MLCK inhibitor (7.3±2 vs 7.5±2 mN for vehicle, n=5). Furthermore, our data showed that differences in the PE-induced contractile response between the groups were abolished by the activator of AMP-activated protein kinase (AICAR; 6.1±2 vs 7.4±2 mN for vehicle, n=5). PugNAc increased phosphorylation of myosin phosphatase target subunit 1 (MYPT-1) and protein kinase C-potentiated inhibitor protein of 17 kDa (CPI-17), which are involved in RhoA/Rho-kinase-mediated inhibition of myosin phosphatase activity. PugNAc incubation produced a time-dependent increase in vascular phosphorylation of myosin light chain and decreased phosphorylation levels of AMP-activated protein kinase, which decreased the affinity of MLCK for Ca2+/calmodulin. Our data suggest that proteins that play an important role in the regulation of MLCK and MLCP activity are directly affected by O-GlcNAcylation, favoring vascular contraction.
Keywords: O-GlcNAcylation, Myosin light chain, Vascular reactivity
Introduction
Vascular smooth muscle cells (VSMCs) are highly specialized cells that regulate blood flow to all tissues and organs through contraction and relaxation. A variety of contractile proteins, ion channels and signaling molecules regulate contraction of VSMCs, but the phosphorylation/dephosphorylation of myosin light chain (MLC) is a key event in the regulation of smooth muscle contraction. The binding of calcium (Ca2+) by calmodulin activates smooth muscle myosin light chain kinase (MLCK) by inducing a conformational change that allows MLCK to phosphorylate the 20-kDa regulatory MLC at serine (1,2). Furthermore, Ca2+-dependent phosphorylation of MLC is followed by a Rho-kinase-mediated Ca2+ sensitization process, which maintains the contracted state of VSMCs by inhibiting MLC phosphatase (MLCP) activity via phosphorylation of the MLCP target subunit (MYPT-1) (2-4).
The phosphorylation state of MLC depends on the balance between MLCK, which phosphorylates MLC leading to smooth muscle cell contraction, and MLCP, which dephosphorylates MLC, resulting in smooth muscle cell relaxation. Therefore, a perfect balance between MLCK and MLCP is critical to modulate VSMCs function, and a disruption in these dynamic processes frequently results in vascular dysfunction (2-4).
Horman et al. (1) have shown that phosphorylation of AMP-activated protein kinase (AMPK), an energy sensor at both the cellular and systemic levels in mammals, decreases the affinity of MLCK for Ca2+/calmodulin. They also showed that aortic rings of α1-AMPK knockout mice displayed an increased contractility in response to high potassium (K+) concentration, as well as to phenylephrine (PE), suggesting that AMPK attenuates contraction by phosphorylating and inactivating MLCK. In addition to the Ca2+-dependent activation of MLCK, the state of MLC phosphorylation is further regulated by MLCP, which is also known as myosin phosphatase (5).
Rho kinase has been shown to phosphorylate MYPT-1 at Thr695, which decreases its phosphatase activity (6), leading to increases in MLC phosphorylation and force development (7). Furthermore, protein kinase C-potentiated inhibitor protein of 17 kDa (CPI-17) can be phosphorylated by protein kinase C (PKC) (5,8) and by Rho kinase (9). When phosphorylated, CPI-17 also inhibits MLC phosphatase activity, prolonging or enhancing smooth muscle contraction (2,9).
Numerous proteins, including kinases and phosphatases, have been identified as targets of O-GlcNAc modification - that is, the O-linked attachment of N-acetyl-glucosamine (O-GlcNAc) to serine and threonine residues of nuclear and cytoplasmic proteins (10-12). This highly dynamic post-translational modification plays a key role in signal transduction pathways; and key contractile proteins, such as myosin heavy chain, actin and MLC also have been shown to be modified by O-GlcNAc in slow and fast skeletal muscle, as well as in VSMCs (11,13,14).
Recent evidence from our laboratory (11,12,15) suggests that endothelin-1 (ET-1) increases O-GlcNAcylation levels in VSMCs, resulting in increased RhoA/Rho kinase activation and consequent augmentation of vascular reactivity to constrictor stimuli. However, a specific role of O-GlcNAcylation on vascular MLC, along with the activation of other components of the RhoA/Rho kinase pathway by O-GlcNAcylation, was not assessed.
Considering that phosphorylation of MLC is a key event in VSMC contraction (4,9) and that kinases and phosphatases have been identified as targets of O-GlcNAc modification (11), we hypothesized that augmented O-GlcNAcylation levels enhance MLCK and reduce MLCP activity, leading to increased vascular contractile responsiveness.
To address our hypothesis, we used a pharmacological approach to increase O-GlcNAcylation by exposing vascular smooth muscle tissue to O-(2-acetamido-2-deoxy-D-glucopyranosylidene) amino-N-phenylcarbamate - referred to as PugNAc - which blocks O-GlcNAcase activity by mimicking the enzyme-stabilized transition state (16-18). We also determined whether proteins that play an important role in the regulation of MLCK and MLCP activity are directly affected by O-GlcNAcylation.
Materials and Methods
Animals
Male Wistar rats (8-10 weeks-old, 230-250 g; Harlan Laboratories, USA) were used in this study. All procedures were performed in accordance with the Guiding Principles in the Care and Use of Animals, approved by the Georgia Health Sciences University Committee on the Use of Animals in Research and Education and in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. The animals were maintained on a 12-h light/dark cycle and fed a standard chow diet with water ad libitum.
Vascular functional studies
After euthanasia, the thoracic aorta was removed and cleaned of fat tissue in an ice-cold physiological salt solution, containing 130 mM NaCl, 14.9 mM NaHCO3, 4.7 mM KCl, 1.18 mM KH2PO4, 1.18 mM MgSO4·7H2O, 1.56 mM CaCl2·2H2O, 0.026 mM EDTA, and 5.5 mM glucose. Endothelium was removed and arterial segments were incubated in Eagle's minimum essential medium containing 1% L-glutamine, 10% fetal bovine serum (FBS), 0.5% penicillin and streptomycin, and incubated with vehicle (10 µL/mL methanol) or 100 µM PugNAc for 24 h. Following incubation, thoracic aortas (4 mm in length) were carefully mounted as ring preparations in standard organ chambers for recording of isometric tension by a PowerLab 8/SP data acquisition system (ADInstruments Pty Ltd., Australia). Tissues were immersed in solutions that were continuously bubbled with a mix of 95% O2 and 5% CO2 and were maintained at 37°C, under a resting tension of 30 mN. After a 60-min equilibration period, aorta integrity was assessed first by stimulation of vessels with a solution high in potassium chloride (120 mM KCl); and, after washing and a new stabilization period, the absence of the endothelium was verified by contracting the segments with 1 µM PE followed by stimulation with 10 µM acetylcholine.
Concentration-responses to 1 nM to 100 µM PE were measured in the presence and absence, for 40 min, of: 10 µL/mL methanol (vehicle for PugNAc), 0.5 µL/mL dimethylsulfoxide (DMSO, vehicle for AICAR and ML-9), an MLCP inhibitor (10 nM calyculin A), a selective Ca2+-calmodulin-dependent MLCK inhibitor (1 µM ML-9), or 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (20 mM AICAR), a pharmacological activator of AMPK. Response curves were calculated in each case.
VSMCs isolation and culture
VSMCs were isolated from rat thoracic aortas by explantation, as previously described (19). Cultures were maintained in Dulbecco's modified Eagle's medium (Gibco-BRL, USA) supplemented with 10% FBS (Invitrogen, USA). Only fourth-passage cells were used. Immunoreactivity assays were used to characterize the VSMCs and to confirm the absence of other cell types in the cultures. The cells expressed α-smooth muscle actin and calponin, which are contractile proteins indicative of VSMCs. No positive immunoreactivity to von Willebrand factor VIII or CD31 (PECAM-1), which are markers of endothelial cells, was detected (data not shown).
After reaching maximum confluence, and 24 h after removal of serum, cells were incubated with vehicle (methanol) or 100 μM PugNAc (for 15 or 30 min, or 1, 6, or 24 h).
Western blot analysis
Proteins (60 µg) extracted from aortas or VSMCs were separated by electrophoresis on 10% polyacrylamide gels and transferred to nitrocellulose membranes. Nonspecific binding sites were blocked with 5% skim milk in Tris-buffered saline solution with 10% Tween for 1 h at 24°C. Membranes were then incubated with antibodies overnight at 4°C. Anti-O-GlcNAc (CTD 110.6, 1:2000; Pierce Biotechnology, USA), anti-AMPK (#80039, 1:1000; Abcam, USA), anti-protein kinase CPI-17 (#32213, 1:1000; Abcam, USA), anti-MYPT-1 (#2634), anti-rho-kinase (ROCK)-α (#8236), anti-ROCK-β (#4035), anti-MLC (#8505) and anti-RhoA (#2117) (all 1:1000; Cell Signaling, USA, or BD Biosciences Transduction Laboratories, USA) were used. Immunoblots for nonphosphoproteins were carried out on the same membranes used to evaluate the phosphorylated (phospho-) forms: phospho-MYPT-1 (Thr853), phospho-CPI-17 (Thr38), phospho-MLC (Thr18/Ser19), and phospho-AMPK (Thr172), (1:500; Cell Signaling, USA). After incubation with secondary antibodies, signals were developed for chemiluminescence, visualized by autoradiography, and quantified densitometrically. Results were normalized to beta-actin protein (#A5316, 1:10000; Sigma-Aldrich, Inc., USA), or to the total form of each phosphorylated protein, and reported as arbitrary units.
Data analysis
The results are reported as means±SE for the number of animals (n) used in the experiments. Contractions were recorded as changes in the displacement (mN) from baseline. Concentration-response curves were fitted using a nonlinear interactive fitting program (Graph Pad Prism 5.0; GraphPad Software Inc., USA) for two pharmacological parameters, the maximal effect (Emax) generated by the agonist, and -log EC50 (pD2). Statistical analyses of Emax or pD2 values were performed using one-way ANOVA or the Student t-test. Post hoc comparisons were performed using the Newman-Keuls test. Western blot data were analyzed by one-sample t-tests and P values were computed from the t-ratio and the numbers of degrees of freedom. Values of P<0.05 were considered to be statistically significant.
Results
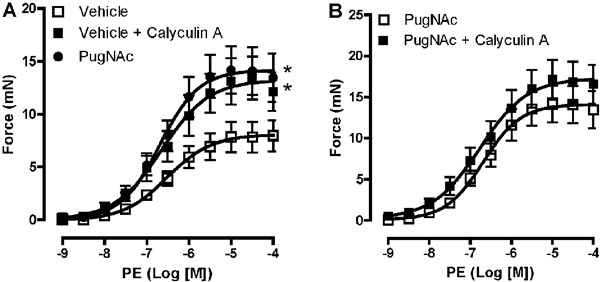
Vascular contraction in response to the alpha 1-adrenergic receptor agonist, PE, was augmented in rat aortas incubated with PugNAc. In addition, treatment of aortic segments with MLCP inhibitor (10 nM calyculin A) augmented the contractile response to PE, with contractile levels comparable to those induced by PugNAc (Figure 1A). No differences were observed in vascular reactivity to PE between aortas treated with PugNAc or calyculin A, and coincubation with both drugs did not further increase the contractile responses (Figure 1B).
Figure 1. Calyculin A and PugNAc augment vascular reactivity to phenylephrine (PE). PugNAc and calyculin A produced similar increases in vascular contraction induced by PE in rat thoracic aorta (A) and co-incubation did not modify this response (B). Data are reported as means±SE (n=7). *P<0.05 vs vehicle (10 µL/mL methanol, vehicle to PugNAc, or 0.5 µL/mL DMSO, vehicle to calyculin A) (one-way ANOVA).
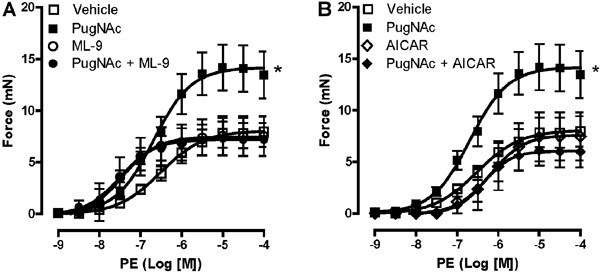
Since MLCK, which phosphorylates MLC leading to VSMC contraction, is a substrate for AMPK, we decided to evaluate the vascular effect of PugNAc in the presence of MLCK inhibitor (ML-9) or with the activator of AMPK (AICAR). The effect of PugNAc was not observed when vessels were previously incubated with ML-9 (Figure 2A). Furthermore, our data showed that differences in the PE-induced contractile response between the groups were abolished with AICAR treatment (Figure 2B).
Figure 2. ML-9 and AICAR incubation abolished the differences between the groups. The effect of PugNAc was not observed when vessels were previously incubated with myosin light chain kinase inhibitor (ML-9) (A) or with an AMPK activator (AICAR) (B). Treatment with ML-9 or AICAR, itself, did not modify PE-induced contractile response. Data are reported as means±SE (n=5-7). *P<0.05 vs vehicle (10 µL/mL methanol, vehicle to PugNAc, or 0.5 µL/mL DMSO, vehicle to ML-9 and AICAR) (one-way ANOVA).
Considering that O-GlcNAcylation is a highly dynamic post-translational modification that plays a key role in signal transduction pathways, we determined whether PugNAc changes the expression and activity (indicated by phosphorylation levels) of proteins that are involved in the regulation of MLCK and MLCP activity.
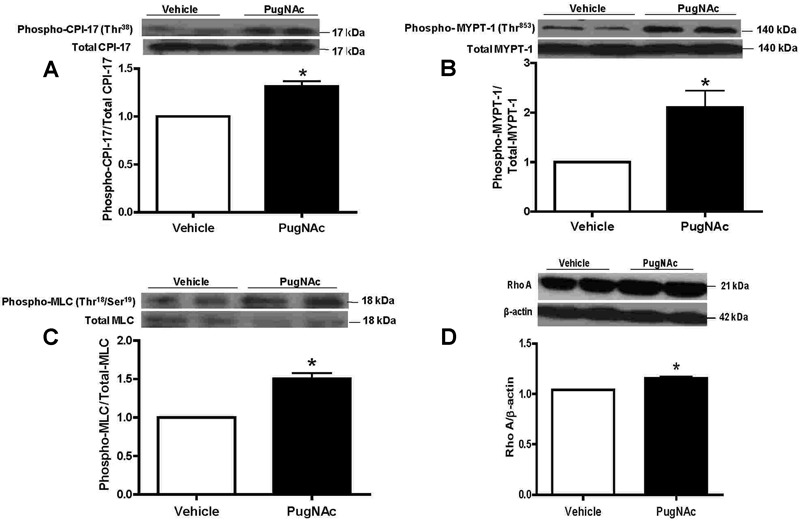
Our data showed that incubation of aortic segments with PugNAc did not change expression of ROCK-α, ROCK-β (data not shown), total CPI-17 (Figure 3A), or total MYPT-1 (Figure 3B). However, PugNAc increased expression of the phosphorylated forms of MYPT-1 (Thr853) and CPI-17 (Thr38), which are directly involved in RhoA/ROCK-mediated inhibition of myosin phosphatase. Furthermore, PugNAc incubation also augmented phosphorylation levels of MLC (Thr18/Ser19) (Figure 3C), which correlate with smooth muscle contraction. In addition, our data showed that RhoA expression in rat aortas was increased by PugNAc (Figure 3D).
Figure 3. PugNAc increased expressions of phospho-CPI-17 (Thr38), phospho-MYPT-1 (Thr853), phospho-MLC (Thr18/Ser19) and RhoA in rat aorta. PugNAc incubation did not change expression of total CPI-17 (A), MYPT-1 (B), or MLC (C), but increased expression of the phosphorylated forms of CPI-17 (Thr38) (A), MYPT-1 (Thr853) (B), MLC (Thr18/Ser19) (C), and RhoA expression (D). Data are reported as means±SE of the relative expression of phosphorylated forms of CPI-17 (Thr38), MYPT-1 (Thr853), and MLC (Thr18/Ser19) after normalization to the corresponding total protein expression (A, B, C) or the relative expression of RhoA after normalization to β-actin expression (D) (n=6). *P<0.05 vs vehicle (10 µL/mL methanol, vehicle to PugNAc) (Student t-test).
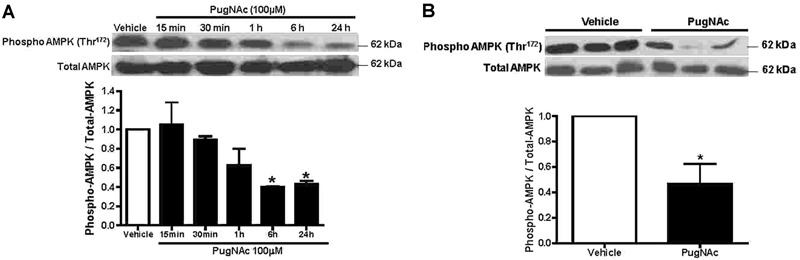
In order to better determine whether PugNAc elicits changes in phosphorylation levels of MLC (Thr18/Ser19), rat aortic VSMCs were exposed to PugNAc at various times. PugNAc produced a time-dependent increase in phosphorylated MLC (Thr18/Ser19) in VSMCs (Figure 4). The greatest effect on phosphorylation levels of MLC (Thr18/Ser19) by PugNAc was observed at 24 h. Interestingly, PugNAc incubation produced a time-dependent decrease in levels of phosphorylated AMPK (Thr172) in VSMCs (Figure 5A). In addition, as shown in Figure 5B, incubation of aortas with PugNAc for 24 h decreased phosphorylation levels of AMPK compared with aortas treated with vehicle. This result suggested that augmented O-GlcNAcylation increased vascular reactivity to constrictor stimuli, due possibly in part to its effects on AMPK, and consequently this increased MLCK activity.
Figure 4. PugNAc-incubation produces a time-dependent increase in expression of phosphorylated myosin light chain (MLC) in vascular smooth muscle cells (VSMCs). VSMCs cultured from rat thoracic aortas were incubated with PugNAc at different times (15, 30 min, 1, 6, or 24 h). Western blot analysis was performed to evaluate expression of phosphorylated MLC in comparison to VSMCs that were incubated with vehicle (methanol, for 24 h). Data are reported as means±SE of the relative expression of the phosphorylated form of MLC (Thr18/Ser19) after normalization to the corresponding total protein expression (n=6). *P<0.05 vs vehicle (10 µL/mL methanol, vehicle to PugNAc) (Student t-test).
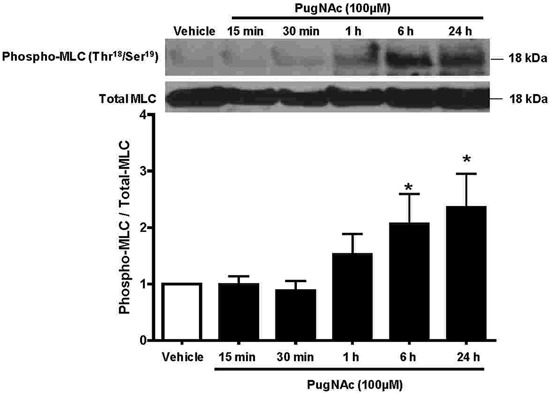
Figure 5. PugNAc incubation produced a time-dependent decrease in phosphorylated levels of AMP-activated protein kinase (AMPK). Vascular smooth muscle cells (VSMCs) cultured from rat thoracic aortas were incubated with PugNAc at different times (15, 30 min, 1, 6 or 24 h). Western blot analysis was performed to evaluate expression of phosphorylated AMPK in comparison to VSMCs that were incubated with vehicle (methanol, for 24 h) (A). Thoracic aortas incubated with PugNAc for 24 h also decreased phosphorylation levels of AMPK compared to aortas treated with vehicle (24 h) (B). Data are reported as means±SE of the relative expression of the phosphorylated form of AMPK (Thr172) after normalization to the corresponding total protein expression (n=6). *P<0.05 vs vehicle (10 µL/mL methanol, vehicle to PugNAc) (Student t-test).
Discussion
O-GlcNAcylation is an innovative way to think about signaling events both in physiological conditions and in disease states. O-GlcNAc cycling in proteins is controlled by two highly conserved enzymes, O-GlcNAc transferase (OGT, or uridine diphospho-N-acetylglucosamine: polypeptide β-N-acetylglucosaminyl transferase; UDP-NAc transferase), and β-N-acetylglucosaminidase (O-GlcNAcase). Whereas OGT catalyzes the addition of O-GlcNAc to the hydroxyl group of serine or threonine residues of a target protein, O-GlcNAcase catalyzes the hydrolytic cleavage of O-GlcNAc from post-translationally modified proteins (10,12,14,18). Accordingly, PugNAc, an inhibitor of O-GlcNAcase, significantly increased the content of O-GlcNAc proteins in arteries from Wistar rats (10-12,15,18,20,21).
This post-translational modification interferes with vascular processes, mainly vascular reactivity, under conditions where ET-1 levels are augmented (e.g., salt-sensitive hypertension, ischemia/reperfusion, and stroke) (12,20). Recent evidence from our laboratory suggests that ET-1 increases O-GlcNAcylation levels in VSMCs, which leads to augmented RhoA/Rho kinase activation, consequently increasing vascular reactivity to constrictor stimuli (11). Although it is known that augmented O-GlcNAcylation increases vascular reactivity to constrictor stimuli, there is a paucity of information on the vascular effects of O-GlcNAcylation, especially whether this post-translational modification directly alters MLC function.
We have previously demonstrated that PugNAc incubation, which increased vascular content of O-GlcNAc-proteins, augments responses to contractile stimuli (10,15,20). Our data show that calyculin A, an MLCP inhibitor, mimics PugNAc effects on vascular reactivity. In addition, calyculin A incubation had a minor and nonsignificant effect on arteries incubated with PugNAc. Based on these observations, one may speculate that elevated levels of O-GlcNAc augment vascular reactivity to constrictor stimuli via a decrease in MLCP activity, thereby contributing to phosphorylation of MLC and a contracted state of VSMCs.
Considering that calyculin A effects on PE reactivity were not observed in the presence of PugNAc, we determined whether increased levels of O-GlcNAcylation modify the proteins that regulate MLCP activity.
Rho-kinase is a serine/threonine protein kinase that contains an N-terminal catalytic kinase domain. As mentioned, the small G protein RhoA and its downstream target Rho kinase play an important role in the regulation of MLCP activity (4,22). We observed that increased vascular O-GlcNAc levels by PugNAc augmented phosphorylation of CPI-17 and RhoA expression, whereas expression of Rho-kinase isoforms was unchanged. A study by Wang et al. (23) reported that silencing of both ROCK isoforms leads to reduced MYPT-1 and MLC phosphorylation. On the other hand, functional studies have demonstrated that CPI-17 can be phosphorylated by PKC as well as by Rho-kinase. PKC phosphorylates CPI-17, which then inhibits MLCP, increasing MLC phosphorylation and enhancing vascular smooth muscle contraction (9,24,25). Interestingly, immunoprecipitation assays in vascular segments have shown that different PKC isoforms, including PKC-α, -βII, and -ε, are targets for O-GlcNAc modification (18). Together, these results suggest that increased vascular O-GlcNAcylation potentiates the activation of PKC, which may contribute to augmented phosphorylation levels of CPI-17.
Several recent studies suggest a role for additional regulators of MLCK and MLCP (26,27). Similar to CPI-17, Rho-kinase was first identified to phosphorylate MYPT-1, inhibiting its activity. Later, many kinases were found to phosphorylate MYPT-1, thus promoting the phosphorylated state of MLC (4,9). Our data showed that PugNAc increased the expression of phosphorylated MYPT-1. Accordingly, Cheung et al. (28) have shown that changes in protein activity, such as MYPT-1, as well as changes in cell physiology, are associated with transient and dramatic alterations in O-GlcNAcylation levels (28,29).
The calmodulin-dependent protein kinase promotes VSMC relaxation by decreasing the sensitivity of MLCK for Ca2+ (4). AMPK is activated by phosphorylation of Thr172 in a number of physiological and pathological states in which an increase in the AMP/ATP ratio occurs in the cell (30,31). It has been shown that AMPK, when phosphorylated, decreased the affinity of MLCK for Ca2+/calmodulin (1). Our data showed that the effect of PugNAc was not observed when vessels were pre-incubated with ML-9. In addition, treatment with an AMPK activator augmented vascular responses to PE and abolished the differences in PE-response between the groups. Furthermore, PugNAc incubation produced a time-dependent decrease in phosphorylated levels of AMPK (Thr172) in VSMCs. Interestingly, the greatest effect on phosphorylation levels of MLC by PugNAc was correlated with decreased AMPK phosphorylation by PugNAc. These results suggest that increased vascular O-GlcNAcylation attenuates phosphorylated forms of AMPK, allowing the MLC to remain phosphorylated, thereby promoting contraction.
One potential mechanism by which O-GlcNAcylation may change protein activity includes the complex interplay between O-GlcNAcylation and phosphorylation (32). O-GlcNAc is similar to protein phosphorylation (O-phosphate attachment); both modifications occur on serine and threonine residues, both are dynamically added and removed from proteins in response to cellular signals, and both alter the function and association of the modified protein (18,33).
As mentioned, VSMC contraction is principally regulated by Ca2+-calmodulin-activated MLC phosphorylation, which enables the molecular interaction of myosin with actin (4,34). We observed that increased O-GlcNAc levels augmented phosphorylation of MLC (Thr18/Ser19) and that PugNAc incubation changed expression of the phosphorylated forms of MYPT-1 (Thr853), and AMPK (Thr38), which are directly involved in the inhibition of myosin phosphatase and myosin kinase, respectively. Thus, we speculate that an imbalance between phosphorylation/O-GlcNAcylation may alter phosphorylation levels of proteins that play an important role in the regulation of MLCK and MLCP activity, prolonging phosphorylated MLC levels and maintaining muscle contraction.
The cross-talk between O-GlcNAcylation and phosphorylation may occur by steric competition (35). For example, increased O-GlcNAc modification of endothelial nitric oxide synthase (eNOS) is associated with decreased phosphorylation of eNOS at Ser1177 (eNOS- Ser1177) and decreased eNOS activity (10,36). The steric competition between phosphorylation and O-GlcNAcylation occurring in eNOS is an example of competition that is occurring in the same site of occupancy. However, in other proteins, the competition occurs in proximal or distant sites (35), such as that observed in the tumor suppressor p53 (37).
Several specific kinases and phosphatases, encoded by distinct genes, regulate phosphorylation. In contrast, and as previously discussed, O-GlcNAcylation is controlled exclusively by two enzymes, OGT and O-GlcNAcase that are encoded in animals by a single highly conserved gene (35). The interplay between O-GlcNAc and O-phosphate is also underscored by interaction between enzymes that regulate these two post-translational modifications. Few studies have determined the effects of kinase and phosphatase inhibitors on the levels of O-GlcNAc of specific proteins (38). Furthermore, Wells et al. (39) have shown that OGT and protein phosphatase 1 (PP1c) β and γ exist together in a complex. As OGT, O-GlcNAcase has been shown to interact with specific proteins, including protein phosphatase-2B (40). Interestingly, the binding of MYPT-1 to PP1c inhibits the enzymatic activity of MLCP, allowing MLC to remain phosphorylated, thereby promoting contraction (4). In addition, the small protein CPI-17 also binds to the catalytic subunit of MLC phosphatase (PP1c) to inhibit the enzyme's activity (4,9). Together, these results suggest that interplay between O-GlcNAc and O-phosphate may alter actin-myosin interactions and myosin assembly, and may therefore favor vascular contraction.
The major finding of this study was that augmented O-GlcNAc modification seemed to compromise AMPK activation in vascular smooth muscle, favoring the activity of MLCK and augmenting vascular responses to contractile stimuli. In addition, increased O-GlcNAc in key proteins of the RhoA-Rho kinase pathway seems to augment the activation of this Ca2+ sensitization pathway, contributing to greater contractile responsiveness. We speculate that an imbalance between phosphorylation/O-GlcNAcylation may alter actin-myosin interactions and modify myosin assembly, favoring vascular contraction.
Acknowledgments
Research supported by FAPESP (#2008/58142-7), Fundação de Amparo è Pesquisa do Estado de Mato Grosso (FAPEMAT, #155007/2012-0), CAPES and CNPq (#471675/2013-0).
Footnotes
First published online August 15, 2014.
References
- 1.Horman S, Morel N, Vertommen D, Hussain N, Neumann D, Beauloye C, et al. AMP-activated protein kinase phosphorylates and desensitizes smooth muscle myosin light chain kinase. J Biol Chem. 2008;283:18505–18512. doi: 10.1074/jbc.M802053200. [DOI] [PubMed] [Google Scholar]
- 2.Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 3.Somlyo AP, Somlyo AV. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol. 2000;522((Part 2)):177–185. doi: 10.1111/j.1469-7793.2000.t01-2-00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hilgers RH, Webb RC. Molecular aspects of arterial smooth muscle contraction: focus on Rho. Exp Biol Med. 2005;230:829–835. doi: 10.1177/153537020523001107. [DOI] [PubMed] [Google Scholar]
- 5.Masuo M, Reardon S, Ikebe M, Kitazawa T. A novel mechanism for the Ca(2+)-sensitizing effect of protein kinase C on vascular smooth muscle: inhibition of myosin light chain phosphatase. J Gen Physiol. 1994;104:265–286. doi: 10.1085/jgp.104.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ichikawa K, Ito M, Hartshorne DJ. Phosphorylation of the large subunit of myosin phosphatase and inhibition of phosphatase activity. J Biol Chem. 1996;271:4733–4740. doi: 10.1074/jbc.271.9.4733. [DOI] [PubMed] [Google Scholar]
- 7.Sward K, Dreja K, Susnjar M, Hellstrand P, Hartshorne DJ, Walsh MP. Inhibition of Rho-associated kinase blocks agonist-induced Ca2+ sensitization of myosin phosphorylation and force in guinea-pig ileum. J Physiol. 2000;522((Part 1)):33–49. doi: 10.1111/j.1469-7793.2000.0033m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitazawa T, Takizawa N, Ikebe M, Eto M. Reconstitution of protein kinase C-induced contractile Ca2+ sensitization in triton X-100-demembranated rabbit arterial smooth muscle. J Physiol. 1999;520((Part 1)):139–152. doi: 10.1111/j.1469-7793.1999.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nunes KP, Rigsby CS, Webb RC. RhoA/Rho-kinase and vascular diseases: what is the link? Cell Mol Life Sci. 2010;67:3823–3836. doi: 10.1007/s00018-010-0460-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lima VV, Giachini FR, Carneiro FS, Carneiro ZN, Fortes ZB, Carvalho MH, et al. Increased vascular O-GlcNAcylation augments reactivity to constrictor stimuli - Vasoactive Peptide Symposium. J Am Soc Hypertens. 2008;2:410–417. doi: 10.1016/j.jash.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lima VV, Giachini FR, Carneiro FS, Carvalho MH, Fortes ZB, Webb RC, et al. O-GlcNAcylation contributes to the vascular effects of ET-1 via activation of the RhoA/Rho-kinase pathway. Cardiovasc Res. 2011;89:614–622. doi: 10.1093/cvr/cvq338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lima VV, Giachini FR, Hardy DM, Webb RC, Tostes RC. O-GlcNAcylation: a novel pathway contributing to the effects of endothelin in the vasculature. Am J Physiol Regul Integr Comp Physiol. 2011;300:R236–R250. doi: 10.1152/ajpregu.00230.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hedou J, Cieniewski-Bernard C, Leroy Y, Michalski JC, Mounier Y, Bastide B. O-linked N-acetylglucosaminylation is involved in the Ca2+ activation properties of rat skeletal muscle. J Biol Chem. 2007;282:10360–10369. doi: 10.1074/jbc.M606787200. [DOI] [PubMed] [Google Scholar]
- 14.Laczy B, Hill BG, Wang K, Paterson AJ, White CR, Xing D, et al. Protein O-GlcNAcylation: a new signaling paradigm for the cardiovascular system. Am J Physiol Heart Circ Physiol. 2009;296:H13–H28. doi: 10.1152/ajpheart.01056.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lima VV, Giachini FR, Carneiro FS, Carneiro ZN, Saleh MA, Pollock DM, et al. O-GlcNAcylation contributes to augmented vascular reactivity induced by endothelin 1. Hypertension. 2010;55:180–188. doi: 10.1161/HYPERTENSIONAHA.109.143818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitworth GE, Macauley MS, Stubbs KA, Dennis RJ, Taylor EJ, Davies GJ, et al. Analysis of PUGNAc and NAG-thiazoline as transition state analogues for human O-GlcNAcase: mechanistic and structural insights into inhibitor selectivity and transition state poise. J Am Chem Soc. 2007;129:635–644. doi: 10.1021/ja065697o. [DOI] [PubMed] [Google Scholar]
- 17.Kim EJ, Amorelli B, Abdo M, Thomas CJ, Love DC, Knapp S, et al. Distinctive inhibition of O-GlcNAcase isoforms by an alpha-GlcNAc thiolsulfonate. J Am Chem Soc. 2007;129:14854–14855. doi: 10.1021/ja076038u. [DOI] [PubMed] [Google Scholar]
- 18.Lima VV, Rigsby CS, Hardy DM, Webb RC, Tostes RC. O-GlcNAcylation: a novel post-translational mechanism to alter vascular cellular signaling in health and disease: focus on hypertension. J Am Soc Hypertens. 2009;3:374–387. doi: 10.1016/j.jash.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ross R. The smooth muscle cell. II. Growth of smooth muscle in culture and formation of elastic fibers. J Cell Biol. 1971;50:172–186. doi: 10.1083/jcb.50.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lima VV, Giachini FR, Choi H, Carneiro FS, Carneiro ZN, Fortes ZB, et al. Impaired vasodilator activity in deoxycorticosterone acetate-salt hypertension is associated with increased protein O-GlcNAcylation. Hypertension. 2009;53:166–174. doi: 10.1161/HYPERTENSIONAHA.108.116798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lima VV, Giachini FR, Webb RC, Tostes RC. ET-1-induced vascular contraction is mediated by RhoA/Rho-kinase pathway activation via O-GlcNAcylation. FASEB J. 2010;23:603–612. [Google Scholar]
- 22.Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, et al. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) J Biol Chem. 1996;271:20246–20249. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Zheng XR, Riddick N, Bryden M, Baur W, Zhang X, et al. ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ Res. 2009;104:531–540. doi: 10.1161/CIRCRESAHA.108.188524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Budzyn K, Paull M, Marley PD, Sobey CG. Segmental differences in the roles of rho-kinase and protein kinase C in mediating vasoconstriction. J Pharmacol Exp Ther. 2006;317:791–796. doi: 10.1124/jpet.105.100040. [DOI] [PubMed] [Google Scholar]
- 25.Woodsome TP, Eto M, Everett A, Brautigan DL, Kitazawa T. Expression of CPI-17 and myosin phosphatase correlates with Ca(2+) sensitivity of protein kinase C-induced contraction in rabbit smooth muscle. J Physiol. 2001;535:553–564. doi: 10.1111/j.1469-7793.2001.t01-1-00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solaro RJ. Myosin light chain phosphatase: a Cinderella of cellular signaling. Circ Res. 2000;87:173–175. doi: 10.1161/01.RES.87.3.173. [DOI] [PubMed] [Google Scholar]
- 27.Somlyo AP, Wu X, Walker LA, Somlyo AV. Pharmacomechanical coupling: the role of calcium, G-proteins, kinases and phosphatases. Rev Physiol Biochem Pharmacol. 1999;134:201–234. doi: 10.1007/3-540-64753-8_5. [DOI] [PubMed] [Google Scholar]
- 28.Cheung WD, Sakabe K, Housley MP, Dias WB, Hart GW. O-linked beta-N-acetylglucosaminyltransferase substrate specificity is regulated by myosin phosphatase targeting and other interacting proteins. J Biol Chem. 2008;283:33935–33941. doi: 10.1074/jbc.M806199200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zachara NE, Hart GW. Cell signaling, the essential role of O-GlcNAc! Biochim Biophys Acta. 2006;1761:599–617. doi: 10.1016/j.bbalip.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 30.Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. 2001;23:1112–1119. doi: 10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- 32.Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 33.Hu P, Shimoji S, Hart GW. Site-specific interplay between O-GlcNAcylation and phosphorylation in cellular regulation. FEBS Lett. 2010;584:2526–2538. doi: 10.1016/j.febslet.2010.04.044. [DOI] [PubMed] [Google Scholar]
- 34.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z, Gucek M, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc Natl Acad Sci U S A. 2008;105:13793–13798. doi: 10.1073/pnas.0806216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. 2001;108:1341–1348. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, et al. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol. 2006;8:1074–1083. doi: 10.1038/ncb1470. [DOI] [PubMed] [Google Scholar]
- 38.Griffith LS, Schmitz B. O-linked N-acetylglucosamine levels in cerebellar neurons respond reciprocally to perturbations of phosphorylation. Eur J Biochem. 1999;262:824–831. doi: 10.1046/j.1432-1327.1999.00439.x. [DOI] [PubMed] [Google Scholar]
- 39.Wells L, Kreppel LK, Comer FI, Wadzinski BE, Hart GW. O-GlcNAc transferase is in a functional complex with protein phosphatase 1 catalytic subunits. J Biol Chem. 2004;279:38466–38470. doi: 10.1074/jbc.M406481200. [DOI] [PubMed] [Google Scholar]
- 40.Wells L, Gao Y, Mahoney JA, Vosseller K, Chen C, Rosen A, et al. Dynamic O-glycosylation of nuclear and cytosolic proteins: further characterization of the nucleocytoplasmic beta-N-acetylglucosaminidase, O-GlcNAcase. J Biol Chem. 2002;277:1755–1761. doi: 10.1074/jbc.M109656200. [DOI] [PubMed] [Google Scholar]