

Abstract
The transcription factor Sox4 plays an indispensable role in the development of early progenitor B cells from hematopoietic stem cells. However, its role in B-cell acute lymphoblastic leukemia, a malignant counterpart of normal progenitor B cells, is not fully understood. Here we show that SOX4 is highly expressed in human acute lymphoblastic leukemia cells. To systematically study the function of Sox4 in acute lymphoblastic leukemia, we established a genetically defined mouse leukemia model by transforming progenitor B cells carrying a floxed Sox4 allele and inducing deletion of the allele by the self-excising Cre recombinase. This model allowed us to work with two groups of leukemic cells that had either one copy or both copies of Sox4 deleted. We found that depletion of Sox4 in transformed cells in vitro reduced cell growth in vitro and the progression of leukemia in vivo. Moreover, depletion of Sox4 in leukemic cells in vivo prolonged the survival of the mice, suggesting that it could be a potential target in acute lymphoblastic leukemia therapy. Our microarray and bioChIP studies revealed that Tcf7l1 was the key gene directly regulated by Sox4. Knockdown of Tcf7l1 reduced cell proliferation, just as did knockout of Sox4, and ectopic expression of Tcf7l1 could reverse the effect of Sox4 knockout on cell proliferation. These data suggest that Sox4 and Tcf7l1 form a functional axis that promotes the progression of BCR-ABL-positive acute lymphoblastic leukemia.
Introduction
Sox4, a member of the C subgroup of the Sox transcription factor family, plays a critical role in diverse developmental processes such as formation of the cardiac outflow tract and endocrine islet, differentiation of thymocytes, and development of osteoblasts and neural and glial cells.1 Sox4 is also indispensable to early B-cell development.2 In our laboratory it was demonstrated that conditional inactivation of Sox4 in hematopoietic stem cells completely abrogates the development of progenitor B (pro-B) cells without having significant deleterious effects on other hematopoietic lineages. Sox4 is important in maintaining the survival of pro-B cells since deficiency in B-cell development caused by Sox4 inactivation could be partially rescued with transgenic expression of the anti-apoptotic protein Bcl2.3 On the basis of these findings, we hypothesized that Sox4 is involved in the malignant transformation of pro-B cells while functioning as a pro-proliferative and/or anti-apoptotic factor.
Increasing evidence shows that SOX4 is up-regulated in various human malignancies. However, the role of SOX4 in different tumor types remains controversial.4 For example, SOX4 has been shown to function as an oncogene in prostate, colorectal, and breast cancers, by inducing and maintaining cancer-initiating cells, supporting cancer cell survival, and promoting cancer cell invasion and metastasis. In contrast, increased SOX4 expression was also shown to correlate with prolonged survival and slower disease progression in patients with bladder carcinoma, gallbladder carcinoma, and medulloblastoma, suggesting that SOX4 can have a tumor-suppressor role.
Increased expression of Sox4 induced by retroviral insertional mutagenesis has been shown to be associated with leukemia and lymphoma.5,6 The role of Sox4 as an oncogene in leukemia transformation was also shown in mice that had received bone marrow cells infected with a Sox4-expressing virus and subsequently developed myeloid leukemia.7 At the molecular level in myeloid leukemogenesis, Sox4 was reported to cooperate with various factors, including Evi1, PU.1, AML1-ETO, NUP98-DDX10, p15INK4b loss, HOXA9, CREB, PML-RARa and miR129-2.8–16 However, little is known about the role of SOX4 in lymphoid leukemias. In adult T-cell leukemia/lymphoma, SOX4 was found to be downstream of FRA-2 and induced HDAC8 expression.17 Recently Ramezani-Rad et al.18 reported the role of SOX4 in PI3K/AKT and MAPK signaling in B-cell acute lymphoblastic leukemia (ALL). In the present study we used a genetically defined mouse BCR-ABL-positive ALL model and investigated the role of Sox4 in leukemia progression, the molecular mechanisms involved, and the potential role of Sox4 as a target in ALL therapy.
Methods
Mice
All animal experiments were performed according to the protocols approved by the Institutional Animal Care and Use Committee of The University of Texas M. D. Anderson Cancer Center. The generation of loxp-flanked Sox4 allele mice,19 Cre-ER mice20 and Rosa26eYFP mice21 (Jackson Laboratory, Bar Harbor, ME, USA) has been described elsewhere.
OP9 and pro-B cell co-culture
OP9 cells were maintained in alpha minimum essential medium (AMEM) (Cellgro, Manassas, VA, USA) supplemented with 20% fetal bovine serum (ATCC, Manassas, VA, USA), 2 mM glutamine (Gibco, Grand Island, NY, USA), 100 units/mL penicillin, and 100 μg/mL streptomycin (Gibco). For OP9 and pro-B cell co-culture, AMEM with 20% fetal bovine serum (Omega Scientific, Techview, Singapore), 2 mM glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin, 50 μM β-mercaptoethanol (Sigma, St. Louis, MO, USA), and 10 ng/mL interleukin-7 (Miltenyi Biotec, Bergisch Gladbach, Germany) was used.
Viral vector constructs
Details regarding the SE-Cre vector are available elsewhere.22 To silence Tcf7l1 expression, human U6 promoter-directed shRNA expression vectors were generated as follows: the RNAi-Ready pSIREN-RetroQ-DsRed-Express vector (pSIN) was self-inactivated as described by Xu et al.23 and PGK promoter (from pLVX-Tight-Puro) (Clontech, Mountain View, CA, USA) and mCherry-coding sequences were cloned into BamHI- and EcoRV-digested pSIN vector. Regulatory sequences that flanked shRNA in the pGIPZ vector were synthesized as oligonucleotides with 5′-end phosphorylation modification and cloned downstream of the human U6 promoter in the pSIN-PGK-mCherry vector. Finally shRNA from the pGIPZ vector was excised and sub-cloned into the above vector.
bioChIP
The bioChIP system24 (BirA/biotin/biotin acceptor protein/streptavidin) was used to pull down Sox4-bound chromatin.25 In this system, biotin-conjugating enzyme, BirA ligase, and biotin acceptor peptide (BAP)-tagged Sox4 (BAP-Sox4) (BAP served as a control) were introduced into the p190 BCR-ABL-transformed pro-B cells that had Sox4fl/fl deletion (Sox4fl/flSE-Cre). In the presence of biotin, BirA catalyzes conjugation of biotin to BAP-Sox4 which can then be specifically pulled down, together with bound DNA fragments (Sox4 specific ChIP DNA), by magnetic beads conjugated with streptavidin (Dynabeads® MyOne™ Streptavidin T1; Invitrogen, Grand Island, NY, USA). The Tcf7l1 promoter sequences were detected by polymerase chain reaction (PCR) with the following primers: forward: 5′ ggcgatggggaaggagggag 3′; reverse: 5′ gaaggtgcaagcgagcagga 3′.
In vivo transplantation
NOD-SCID (NOD.CB17-Prkdcscid/J, Jackson laboratory) mice were sublethally irradiated (250 rads) for 6 to 18 hours before they were transplanted with transformed pro-B cells (1×106 cells per mouse) through the tail vein. At weekly intervals, each mouse was injected intraperitoneally with 150 mg/kg D-luciferin (Biosynth, Staad, Switzerland) and imaged in a Xenogen IVIS 100 imaging system (PerkinElmer, Waltham, MA, USA). For in vivo deletion of the floxed Sox4 gene, transplanted NOD/SCID mice were given peritoneal injections of tamoxifen for 5 consecutive days.
Immunoblot analysis
Mouse polyclonal anti-Sox4 (Abnova, Taipei City, Taiwan) at 1:3000 dilution and rabbit polyclonal anti-α-tubulin (Cell Signaling Technology, Danvers, MA, USA) at 1:3000 dilution were used for immunoblot analysis. Anti-rabbit or anti-mouse secondary antibodies conjugated to horseradish peroxidase (Sigma) were used at 1:3000 dilution and bands were detected using a chemiluminescence detection system (Pierce Biotechnology, Rockford, IL, USA).
Results
As an initial step, we determined the levels of SOX4 mRNA by real-time reverse transcriptase (RT)-PCR in various types of human cells. Results showed that SOX4 was expressed at relatively high levels in T-cell ALL cell lines (ranging from 30 to 66 times the level in pooled peripheral blood mononuclear cells, which was arbitrarily set as 1 for comparison) and B-cell ALL cell lines (ranging from 7.6 to 30 times), but at low levels in AML cell lines (ranging from 0.99 to 1.3 times), normal peripheral blood B cells (0.17 times) and T cells (0.16 times), and normal bone marrow CD34+ cells (2.1 times) (Figure 1A). We also determined the levels of SOX4 mRNA in patients’ leukemic cells by using real-time RT-PCR. Consistent with the results from the cell lines, SOX4 mRNA expression was significantly higher in patients’ B-cell ALL and T-cell ALL cells than in AML cells (P<0.05) (Figure 1B). High SOX4 expression in B-cell ALL and T-cell ALL was confirmed by immunohistochemical analysis in cases in which bone marrow sections were available (data not shown). These findings indicated that SOX4 may play an important role in human ALL. Since B-cell development, but not T-cell development, was severely impeded in Sox4-deficient mice,3 we focused our studies on B-cell ALL.
Figure 1.
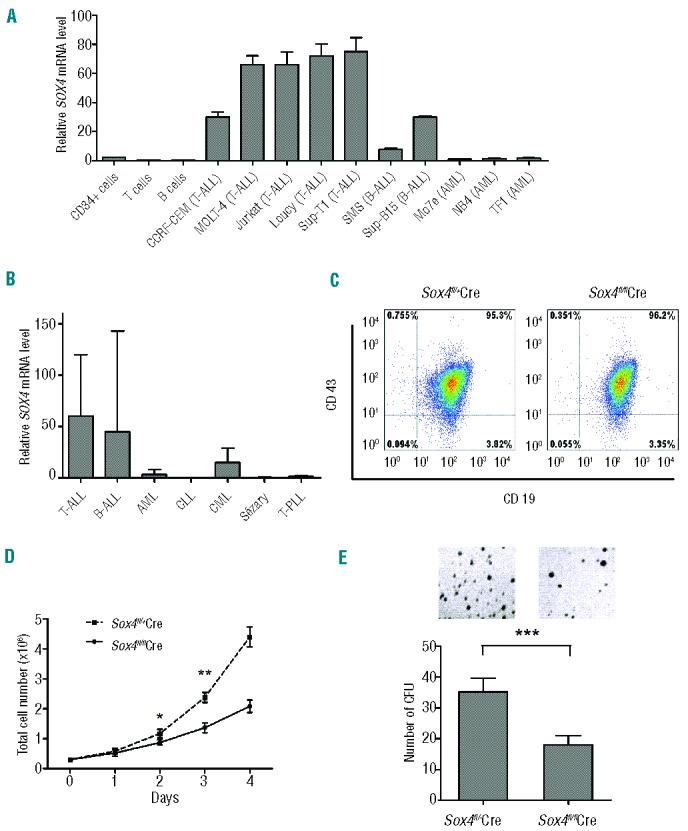
Role of Sox4 in ALL. (A) SOX4 mRNA expression in normal bone marrow CD34+ cells, normal peripheral blood T and B cells, and human ALL and AML cell lines as determined by real-time RT-PCR. The expression level in pooled peripheral blood mononuclear cells was set as 1. Expression of 18S rRNA was used for normalization. (B) SOX4 mRNA expression in leukemic cells from patients with T-cell ALL (n=21), B-cell ALL (n=34), AML (n=22), CLL (n=6), CML (n=7), Sézary disease (n=5), and T-cell prolymphocytic leukemia (T-PLL, n=5). The expression level in pooled peripheral blood mononuclear cells was set as 1. Expression of 18S rRNA was used for normalization. (C) Flow cytometry analysis of p190 BCR-ABL (mCherry+) transformed Sox4fl/flSE-Cre or Sox4fl/+SE-Cre cells for CD43 and CD19 expression. Purified B220+ B cells from bone marrow of Sox4fl/flRosa26-eYFP (experimental) and Sox4fl/+Rosa26-eYFP (control) mice were cultured with OP-9 primary bone marrow stromal cells in the presence of interleukin-7, which yielded pro-B cells of >95% purity.29 Subsequently, the pro-B cells were transformed with p190 BCR-ABL (mCherry+) and transduced with self-excising Cre (SE-Cre) retrovirus.22 Cell surface expression of CD43 and CD19 was examined by flow cytometry. (D) Effect of Sox4fl deletion on the proliferation of ALL cells. p190 BCR-ABL (mCherry+) transformed Sox4fl/flSE-Cre or Sox4fl/+SE-Cre cells sorted out by flow cytometry were cultured, and the cells were enumerated daily for up to 5 consecutive days. Data are representative of three independent experiments. (E) Effect of Sox4 depletion on the colony-forming ability of BCR-ABL-transformed pro-B cells. Ten thousand experimental and control cells, as described in (C), were seeded in semi-solid medium in 3.5-cm dishes and colonies were counted after 14 days. The mean colony numbers were 18 ± 2.6 and 35 ± 3.9 for experimental and control cells, respectively. Data are representative of three independent experiments. Values are means ± SD (n≥3). *P<0.05, **P<0.01, ***P<0.001
We established mouse ALL cell lines by transforming bone marrow and fetal liver pro-B cells from mice that had either one copy (Sox4fl/+) or two copies (Sox4fl/fl) of the floxed Sox4 allele19 with p190 BCR-ABL and then by deleting Sox4fl in vitro with self-excising Cre (SE-Cre). The BCR-ABL vector also carried mCherry and Cre activity was indicated by eYFP so that a pure population of cells could be sorted for analysis (Online Supplementary Methods). Since the SE-Cre was floxed, it would be deleted by the same Cre recombinase encoded by itself, which terminated further Cre expression and avoided the toxicity caused by accumulated Cre seen with other Cre vectors. As the Cre was expressed in both experimental and control cells, any other potential side effects of Cre recombinase were further minimized. The transformed cells retained a pro-B cell phenotype (CD43+CD19+, Figure 1C) and the cells with both Sox4fl alleles deleted (Sox4fl/flSE-Cre) exhibited a lower proliferation rate (Figure 1D and Online Supplementary Figure S1) and lower colony formation ability (Figure 1E) than did transformed cells with only one Sox4fl deleted (Sox4fl/+SE-Cre), suggesting that Sox4 promotes leukemic cell growth in vitro. On the other hand, Sox4 deletion did not result in significant difference in apoptosis in the transformed cells (Online Supplementary Figure S2). Since deletion of Sox4 is pro-apoptotic in pro-B cells during normal B-cell development, as shown by previous studies in our laboratory,3 our current findings indicate that the pro-apoptotic effect of Sox4 deletion could be overshadowed by a potent anti-apoptotic effect of BCR-ABL signaling in the transformed cells.
To investigate the effect of Sox4 deletion on B-cell ALL development in vivo, we transduced the BCR-ABL–transformed cells with a luciferase-expressing viral vector and intravenously injected equal numbers of the cells into sublethally irradiated (250 cGy) NOD/SCID mice. Bioluminescence imaging indicated that the ALL cells homed to bone marrow immediately after injection and that their numbers expanded rapidly afterwards. Importantly, the onset of disease in mice injected with transformed Sox4fl/flSE-Cre cells was delayed compared to that in mice injected with transformed Sox4fl/+SE-Cre cells (Figure 2A). Kaplan-Meier analysis showed that recipient mice with Sox4fl/flSE-Cre cells had a significantly longer survival (median 33 days, n=10) than that of mice with Sox4fl/+SE-Cre cells (median 21 days, n=9) (Figure 2B; P<0.0001). In the terminal stage, ALL cells accounted for nearly 90% of bone marrow cells by flow cytometry analysis (data not shown). As expected, Sox4 mRNA was readily detected in ALL cells from the bone marrow of the mice engrafted with transformed Sox4fl/+SE-Cre cells but barely detectable in ALL cells from recipients with transformed Sox4fl/flSE-Cre cells (Figure 2C). The faster onset of the disease and shorter survival in mice transplanted with Sox4fl/+SE-Cre ALL cells suggested that Sox4 promotes leukemic cell growth in vivo.
Figure 2.

Role of Sox4 in ALL in vivo. (A) and (B) Effect of Sox4 depletion in vitro on the development of leukemia in a NOD/SCID transplantation model. Each mouse was injected via the tail vein with luciferase-labeled and p190 BCR-ABL-transformed Sox4fl/flSE-Cre or Sox4fl/+SE-Cre pro-B cells (106 cells/mouse) and development of leukemia was monitored weekly by bio-imaging. Overall survival was analyzed by the Kaplan-Meier method. (C) Analysis of Sox4 mRNA expression by real-time RT-PCR in ALL cells from bone marrow of mice transplanted with BCR-ABL+ Sox4fl/+Cre or Sox4fl/flCre cells. (D) and (E) Effect of Sox4 depletion in vivo on the progression of leukemia in a NOD/SCID transplantation model. Sox4fl/fl;Cre-ER;eYFP pro-B cells that were transformed with BCR-ABL and labeled with luciferase were transplanted into NOD/SCID mice (3×106 cells/mouse) and the deletion of floxed Sox4 was induced with tamoxifen (TAM) after the onset of leukemia. Leukemia progression was monitored weekly by bio-imaging. (F) Analysis of Sox4 mRNA expression by real-time RT-PCR in ALL cells from bone marrow of mice transplanted with Sox4fl/fl;Cre-ER;eYFP pro-B cells and treated with TAM or vehicle. The relative mRNA levels were normalized to the level of Gapdh mRNA. Values are means ± SD (n=3). *P<0.05, **P<0.01, ***P<0.001
To investigate whether Sox4 could be used as a target in B-cell ALL treatment, we sought to induce Sox4 deletion in vivo after the establishment of leukemia. We first crossed floxed Sox4 mice with Cre-ER mice and eYFP reporter mice and obtained Sox4fl/fl;Cre-ER;eYFP mice. We then transformed pro-B cells from these mice with p190 BCR-ABL and transduced them with luciferase-expressing retroviral vector in vitro. Flow cytometry-sorted BCR-ABL-transformed and luciferase-expressing cells were transplanted into sublethally irradiated NOD/SCID mice, and leukemia development was monitored by bioluminescence imaging. Upon detection of the expansion of transplanted ALL cells, we induced Sox4 deletion by peritoneal injection of tamoxifen on day 5 post-transplantation for 5 consecutive days. Our results showed that mice that received tamoxifen survived significantly longer (median survival, 126 days; n=7) than did the control mice that received the vehicle only (median survival, 46 days; n=6) (Figure 2D,E; P=0.0002). Consistent with floxed Sox4 gene deletion, Sox4 mRNA was barely detectable by real-time PCR in ALL cells from the mice that had received tamoxifen (Figure 2F). These results suggest that targeting Sox4 is potentially effective in the treatment of ALL.
To identify genes regulated by Sox4 in B-cell ALL, we performed gene expression microarray analysis with BCR-ABL-transformed Sox4fl/flSE-Cre and Sox4fl/+SE-Cre cells of fetal liver or bone marrow origin. We identified Sox4 and 17 other differentially expressed genes that all showed decreased expression in Sox4fl/flSE-Cre cells (Figure 3A). These genes are known to be involved in the regulation of cell growth, the cell cycle, B-cell differentiation, and other cellular processes. We confirmed the microarray results by real-time RT-PCR for the expression of the differentially expressed genes in Sox4fl/+SE-Cre and Sox4fl/flSE-Cre cells (data not shown). To test whether expression of the down-regulated genes in Sox4fl/flSE-Cre cells could be reversed by ectopic Sox4 expression, we introduced Sox4-expressing retrovirus into the Sox4fl/flSE-Cre cells. Sox4 expression was significantly higher in Sox4fl/flSE-Cre;Sox4 cells than in Sox4fl/+SE-Cre cells (Figure 3B). We found that the ectopic Sox4 expression in Sox4fl/flSE-Cre cells restored mRNA levels of most of these genes (Figure 3C). Notably, among the genes tested, the greatest difference in expression level was observed for Tcf7l1, whose mRNA levels in Sox4fl/flSE-Cre;Sox4 cells were nearly 30 times higher than those in Sox4fl/flSE-Cre cells.
Figure 3.
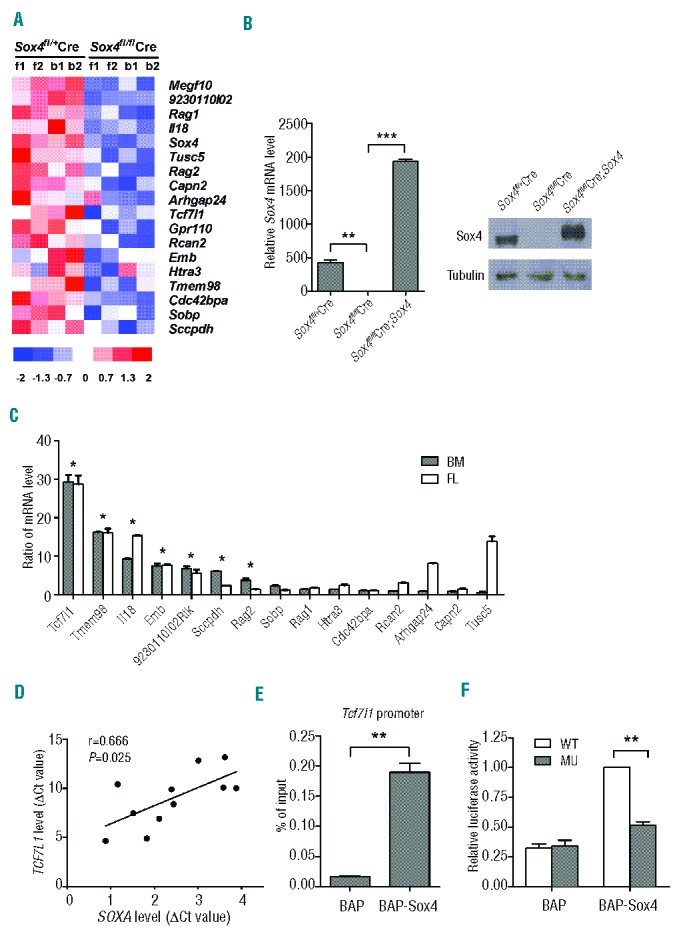
Identification of Sox4-regulated genes in ALL. (A) Genes differentially expressed in transformed Sox4fl/flSE-Cre and Sox4fl/+SE-Cre cells. Fetal liver (FL) or bone marrow (BM) pro-B cells from Sox4fl/fl or Sox4fl/+ mice were transformed with p190 BCR-ABL and transduced with SE-Cre. Gene expression microarray experiments were performed and results were compared between the two types of cells. Genes with a Sox4fl/flSE-Cre/Sox4fl/+SE-Cre signal intensity ratio of <1/3 and P<0.0001 were listed. (B) Analysis of Sox4 mRNA expression by real-time RT-PCR (left) and protein by western blotting (right) in transformed Sox4fl/+SE-Cre, Sox4fl/flSE-Cre, and Sox4fl/flSE-Cre;Sox4 pro-B cells. (C) Ratio (Sox4fl/flSE-Cre;Sox4 to Sox4fl/flSE-Cre) of mRNA expression levels of differentially expressed genes in both bone marrow (BM) and fetal liver (FL) derived transformed cells by real-time RT-PCR. The relative mRNA levels of specific genes were normalized to the level of Gapdh mRNA. Note that the ratios for most genes tested were substantially above 1, suggesting that expression of these genes was reversed upon ectopic Sox4 expression. (D) Scatter diagram demonstrating the correlation between Tcf7l1 and Sox4 expression. Tcf7l1 mRNA level is correlated with Sox4 mRNA level in leukemic cells from patients with ALL (n=11; r=0.666; P=0.0253). Expression of Gapdh was used for normalization of the RT-PCR results. (E) Enrichment of Tcf7l1 promoter sequence in Sox4-specific ChIP DNA by quantitative PCR in a bioChIP analysis. Biotin-conjugating enzyme, BirA ligase, and biotin acceptor peptide (BAP)-tagged Sox4 (BAP-Sox4) (BAP served as control) were introduced into the p190 BCR-ABL-transformed pro-B cells that had Sox4fl/fl deletion (Sox4fl/flSE-Cre). Chromatin was pulled down by magnetic beads conjugated with streptavidin (Dynabeads® MyOne™ Streptavidin T1; Invitrogen, Grand Island, NY, USA). The Sox4-specific and the control ChIP DNA was purified and subjected to real-time PCR for expression of the Tcf7l1 promoter sequence. (F) Mutational analysis of a potential Sox4 binding sequence in the Tcf7l1 promoter. The 460 bp fragment upstream of the transcription start site contains the potential Sox4 binding sequence in which mutations were introduced (-28 to -23bp: ‘ctttgt’ to ‘tgctag’) by PCR (Online Supplementary Methods). This fragment was used to construct mutant reporter pSIN-luc plasmid (MU). BCR-ABL-transformed BAP and BAP-Sox4 pro-B cells were transduced with retrovirus carrying wild-type sequence (WT), MU vector, or empty vector and luciferase activities were determined 2 days after transduction using empty vector as the background control. Data are representative of three independent experiments. Values are means ± SD (n=3). *P<0.05, **P<0.01, ***P<0.001.
To verify the potential relationship of Tcf7l1 and Sox4 expression, we examined their mRNA levels in leukemic cells from patients with ALL and found that the expression of Tcf7l1 mRNA was positively correlated with that of Sox4 mRNA with a Pearson correlation coefficient of 0.666 (Figure 3D; n=11, P=0.0253). To test whether Tcf7l1 was directly regulated by Sox4, we performed a bioChIP assay. Biotin-conjugating enzyme, BirA ligase, and BAP-Sox4 (BAP served as control) were introduced into the p190 BCR-ABL-transformed pro-B cells that had Sox4fl/fl deletion (Sox4fl/flSE-Cre). In the presence of biotin, BirA would catalyze conjugation of biotin to BAP-Sox4 which could then be specifically pulled down, together with bound DNA fragments, by magnetic beads conjugated with streptavidin. The bioChIP DNA samples were examined for the presence of the Tcf7l1 promoter sequence by real-time PCR and the Sox4-specific ChIP DNA showed significant enrichment of the Tcf7l1 promoter sequence (Figure 3E). Mutation of the potential Sox4 binding sequence25 (-28 to -23bp: ‘ctttgt’ to ‘tgctag’) reduced luciferase expression by approximately 50% (Figure 3F). These findings indicated that the Tcf7l1 gene is directly regulated by Sox4.
We further studied the role of Tcf7l1 in ALL by shRNA knockdown in transformed pro-B cells. Knockdown of Tcf7l1 resulted in a significant reduction in cell proliferation and colony formation (Figure 4A–C and Online Supplementary Figure S3), but no significant difference in cell apoptosis (Online Supplementary Figure S4), as seen with Sox4 knockout. To determine whether the effect of knockdown was Tcf7l1- specific and not due to off-target effects of knockdown, we introduced retrovirus-mediated ectopic Tcf7l1 into transformed Sox4fl/flSE-Cre cells. In contrast to the knockdown effect, ectopic expression of Tcf7l1 restored cell proliferation and colony formation (Figure 4D–F). Together, these data suggest that Tcf7l1 mediates the oncogenic role of Sox4 in ALL.
Figure 4.
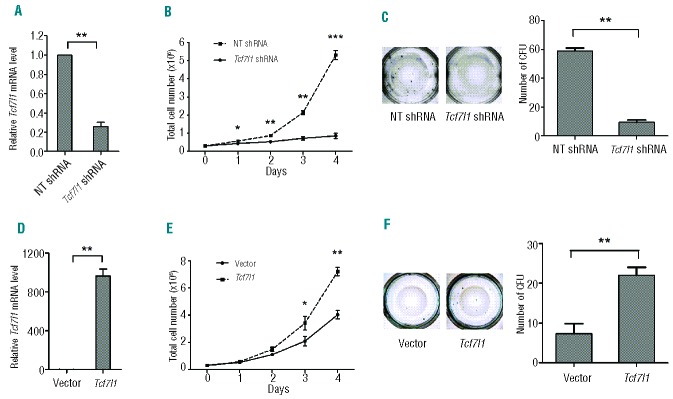
Effect of Tcf7l1 in ALL. (A) Decreased Tcf7l1 mRNA expression in Tcf7l1 shRNA cells compared with NT shRNA cells. The relative mRNA levels of Tcf7l1 gene were normalized to the level of Gapdh mRNA. (B) and (C) Effect of Tcf7l1 mRNA knockdown on the proliferation and colony formation of BCR-ABL–transformed wild-type pro-B cells. The mean colony numbers were 10±2.12 and 59±2.83 for Tcf7l1 knockdown and control cells, respectively. Tcf7l1 shRNA: Tcf7l1 specific shRNA; NT shRNA: non-targeting shRNA. (D) Analysis of Tcf7l1 mRNA expression in transformed Sox4fl/flSE-Cre cells transfected with Tcf7l1 overexpression vector or control vector. (E) and (F) Effect of Tcf7l1 overexpression on cell proliferation and colony formation of transformed Sox4fl/flSE-Cre pro-B cells. The mean colony numbers were 7±2.51 and 22±2.0 for control cells and cells with Tcf7l1 overexpression, respectively. Data are representative of three independent experiments. Values are means ± SD (n=3). *P< 0.05, **P<0.01, ***P<0.001
Discussion
Sox4 is required for normal development of B lineage cells.2 We previously showed that deletion of the Sox4 gene in hematopoietic stem cells causes severe deficiency in pro-B cells and later stage B cells.3 In this study, we further showed that Sox4 is also required for the growth of BCR-ABL-positive B-cell ALL cells, the malignant counterpart of normal pro-B cells. We showed that SOX4 is highly expressed in human ALL cell lines and in leukemic cells from patients with ALL. Using BCR-ABL-induced B-cell ALL as a model, we showed that mice that received ALL cells in which Sox4 had been deleted developed leukemia slower and survived longer than did the mice that had received ALL cells without Sox4 deletion. By using Sox4fl/fl;Cre-ER;eYFP cells, we were able to demonstrate that depletion of Sox4 in vivo after establishment of leukemia in recipient mice attenuated progression of the leukemia and prolonged the animals’ survival, suggesting that targeting Sox4 or a key Sox4-regulated gene might be effective in the treatment of BCR-ABL-positive ALL. Furthermore, our Sox4fl/flSE-Cre and Sox4fl/+SE-Cre experimental system allowed us to identify a list of potential Sox4 downstream genes, including Tcf7l1 which is directly regulated by Sox4 and is one of the important downstream effectors of Sox4 in ALL.
In normal hematopoiesis, TCF7L1 is predominantly expressed in human hematopoietic stem cells and multi-potent progenitors, but is undetectable in pro-B cells.26 Our findings that Tcf7l1 is expressed in transformed pro-B cells and that the expression is controlled by Sox4 suggest that gene expression profiles in transformed pro-B cells recapitulate those in early precursors and that Sox4 may play an important role in the reprograming. In our study, both Sox4 and Tcf7l1 had a pro-proliferation function in p190 BCR-ABL-transformed pro-B cells, suggesting that Tcf7l1 executes the role of Sox4 in leukemic cell proliferation. Consistent with this, Tcf7l1 is required for the proliferation of spinal progenitors and for the maintenance of pluripotency in embryonic stem cells.27,28 However, the precise molecular cues of the role of Tcf7l1 in ALL are still largely unknown and await further in-depth study.
While the animal experiments of this study were being finalized, Ramezani-Rad et al. published their work suggesting that Sox4 enabled oncogenic survival signals in BCR-ABL-positive ALL.18 Notably, nearly half of the potential Sox4 downstream genes identified by the gene expression microarray in their study were those known to be involved in apoptosis, which were not identified by the gene expression microarray in our study. This discrepancy might have resulted from the difference in the cells used. In our system, the transformed experimental cells (Sox4fl/flSE-Cre) and control cells (Sox4fl/+SE-Cre) both had Cre recombinase. Moreover, we used SE-Cre to avoid the potential toxicity of Cre recombinase that might cause cell apoptosis. These designs were expected to lead to results that specifically show the effect of Sox4 on leukemic cells.
The BCR-ABL gene is a strong oncogene and the transformed pro-B cells could be cultured indefinitely in vitro, produce leukemia in vivo and readily cause the death of recipient mice. Depletion of Sox4, whether in vitro prior to transplantation or in vivo after the establishment of leukemia, could significantly prolong survival, which is indicative of Sox4 having an important role in leukemia progression. Nevertheless, the fact that the transformed cells still grew in vitro in the absence of Sox4 and recipient mice transplanted with Sox4-depleted cells eventually died of leukemia suggest that Sox4 is not absolutely essential for the initiation and progression of BCR-ABL-positive ALL. The role of Sox4 in other types of mouse leukemia and in human leukemia warrants further investigation.
Acknowledgments
We thank Dr. Véronique Lefebvre at Cleveland Clinic for the Sox4 floxed mice.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by a Research Scholar Grant to XS from the American Cancer Society, a Physician Scientist Program Award to XS from The University of Texas MD Anderson Cancer Center, a research grant to XS from the Ladies Leukemia League, an Institutional Research Grant to XS from MD Anderson Cancer Center, and the MD Anderson Support Grant CA016672.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Dy P, Penzo-Mendez A, Wang H, Pedraza CE, Macklin WB, Lefebvre V. The three SoxC proteins–Sox4, Sox11 and Sox12–exhibit overlapping expression patterns and molecular properties. Nucleic Acids Res. 2008;36(9):3101–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schilham MW, Oosterwegel MA, Moerer P, Ya J, de Boer PA, van de Wetering M, et al. Defects in cardiac outflow tract formation and pro-B-lymphocyte expansion in mice lacking Sox-4. Nature. 1996;380(6576):711–4 [DOI] [PubMed] [Google Scholar]
- 3.Sun B, Mallampati S, Gong Y, Wang D, Lefebvre V, Sun X. Sox4 is required for the survival of pro-B cells. J Immunol. 2013;190(5):2080–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vervoort SJ, van Boxtel R, Coffer PJ. The role of SRY-related HMG box transcription factor 4 (SOX4) in tumorigenesis and metastasis: friend or foe? Oncogene. 2013;32(29):3397–409 [DOI] [PubMed] [Google Scholar]
- 5.Li J, Shen H, Himmel KL, Dupuy AJ, Largaespada DA, Nakamura T, et al. Leukaemia disease genes: large-scale cloning and pathway predictions. Nat Genet. 1999;23(3):348–53 [DOI] [PubMed] [Google Scholar]
- 6.Suzuki T, Shen H, Akagi K, Morse HC, Malley JD, Naiman DQ, et al. New genes involved in cancer identified by retroviral tagging. Nat Genet. 2002;32(1):166–74 [DOI] [PubMed] [Google Scholar]
- 7.Du Y, Spence SE, Jenkins NA, Copeland NG. Cooperating cancer-gene identification through oncogenic-retrovirus-induced insertional mutagenesis. Blood. 2005;106(7):2498–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aue G, Du Y, Cleveland SM, Smith SB, Dave UP, Liu D, et al. Sox4 cooperates with PU.1 haploinsufficiency in murine myeloid leukemia. Blood. 2011;118(17):4674–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong KY, Yim RL, Kwong YL, Leung CY, Hui PK, Cheung F, et al. Epigenetic inactivation of the MIR129–2 in hematological malignancies. J Hematol Oncol. 2013;6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyd KE, Xiao YY, Fan K, Poholek A, Copeland NG, Jenkins NA, et al. Sox4 cooperates with Evi1 in AKXD-23 myeloid tumors via transactivation of proviral LTR. Blood. 2006;107(2):733–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tonks A, Pearn L, Musson M, Gilkes A, Mills KI, Burnett AK, et al. Transcriptional dysregulation mediated by RUNX1-RUNX1T1 in normal human progenitor cells and in acute myeloid leukaemia. Leukemia. 2007;21(12):2495–505 [DOI] [PubMed] [Google Scholar]
- 12.Yassin ER, Abdul-Nabi AM, Takeda A, Yaseen NR. Effects of the NUP98-DDX10 oncogene on primary human CD34+ cells: role of a conserved helicase motif. Leukemia. 2010;24(5):1001–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bies J, Sramko M, Fares J, Rosu-Myles M, Zhang S, Koller R, et al. Myeloid-specific inactivation of p15Ink4b results in monocytosis and predisposition to myeloid leukemia. Blood. 2010;116(6):979–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y, Sitwala K, Bronstein J, Sanders D, Dandekar M, Collins C, et al. Identification and characterization of Hoxa9 binding sites in hematopoietic cells. Blood. 2012;119(2):388–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandoval S, Kraus C, Cho EC, Cho M, Bies J, Manara E, et al. Sox4 cooperates with CREB in myeloid transformation. Blood. 2012;120(1):155–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Omidvar N, Maunakea ML, Jones L, Sevcikova S, Yin B, Himmel KL, et al. PML-RARalpha co-operates with Sox4 in acute myeloid leukemia development in mice. Haematologica. 2013;98(3):424–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higuchi T, Nakayama T, Arao T, Nishio K, Yoshie O. SOX4 is a direct target gene of FRA-2 and induces expression of HDAC8 in adult T-cell leukemia/lymphoma. Blood. 2013;121(18):3640–9 [DOI] [PubMed] [Google Scholar]
- 18.Ramezani-Rad P, Geng H, Hurtz C, Chan LN, Chen Z, Jumaa H, et al. SOX4 enables oncogenic survival signals in acute lymphoblastic leukemia. Blood. 2013;121(1):148–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Penzo-Mendez A, Dy P, Pallavi B, Lefebvre V. Generation of mice harboring a Sox4 conditional null allele. Genesis. 2007;45(12):776–80 [DOI] [PubMed] [Google Scholar]
- 20.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–5 [DOI] [PubMed] [Google Scholar]
- 21.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silver DP, Livingston DM. Self-excising retroviral vectors encoding the Cre recombinase overcome Cre-mediated cellular toxicity. Mol Cell. 2001;8(1):233–43 [DOI] [PubMed] [Google Scholar]
- 23.Xu W, Russ JL, Eiden MV. Evaluation of residual promoter activity in gamma-retroviral self-inactivating (SIN) vectors. Mol Ther. 2012;20(1):84–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J, Cantor AB, Orkin SH, Wang J. Use of in vivo biotinylation to study protein-protein and protein-DNA interactions in mouse embryonic stem cells. Nat Protoc. 2009;4(4):506–17 [DOI] [PubMed] [Google Scholar]
- 25.Mallampati S, Sun B, Lu Y, Ma H, Gong Y, Wang D, et al. Integrated genetic approaches identify the molecular mechanisms of Sox4 in early B cell development: intricate roles for RAG1/2 and CK1ε. Blood. 2014;123(26):4064–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laurenti E, Doulatov S, Zandi S, Plumb I, Chen J, April C, et al. The transcriptional architecture of early human hematopoiesis identifies multilevel control of lymphoid commitment. Nat Immunol. 2013;14(7):756–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim HS, Dorsky RI. Tcf7l1 is required for spinal cord progenitor maintenance. Dev Dyn. 2011;240(10):2256–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tam WL, Lim CY, Han J, Zhang J, Ang YS, Ng HH, et al. T-cell factor 3 regulates embryonic stem cell pluripotency and self-renewal by the transcriptional control of multiple lineage pathways. Stem Cells. 2008;26(8):2019–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakano T, Kodama H, Honjo T. Generation of lymphohematopoietic cells from embryonic stem cells in culture. Science. 1994;265(5175):1098–101 [DOI] [PubMed] [Google Scholar]