

The ETV6 gene (previously known as TEL) belongs to the ETS (E26 transformation specific) family of transcription factors characterized by 2 important domains: the C-terminal Ets domain responsible for specific DNA-binding activities and the N-terminal helix–loop–helix (HLH) oligomerization domain, also known as pointed (PNT) or sterile alpha motif (SAM), that mediates protein–protein interaction with Ets factors.1–3 The ETV6 protein plays a crucial role in the embryonic development and hematopoietic regulation.4 ETV6 is also a versatile element at the center of a network of genes involved in hematologic malignancies through diverse molecular mechanisms, such as fused with other genes and deletions.3,5–8 ETV6 was originally identified as a fusion partner of the gene that is fused to PDGFRB (platelet derived growth factor receptor beta) gene in chronic myelomonocytic leukemia (CMML) patients with t(5;12)(q33;p13).8 Subsequently, a growing number of genes have been identified as fusion partners of ETV6. At present, 30 partner genes of the ETV6 gene have been described in a broad spectrum of hematopoietic malignancies.9 Deregulation of the ETV6 gene through deletion is also recurrent in leukemia, especially in acute lymphoblastic leukemia patients with t(12;21)(p13;q22).10
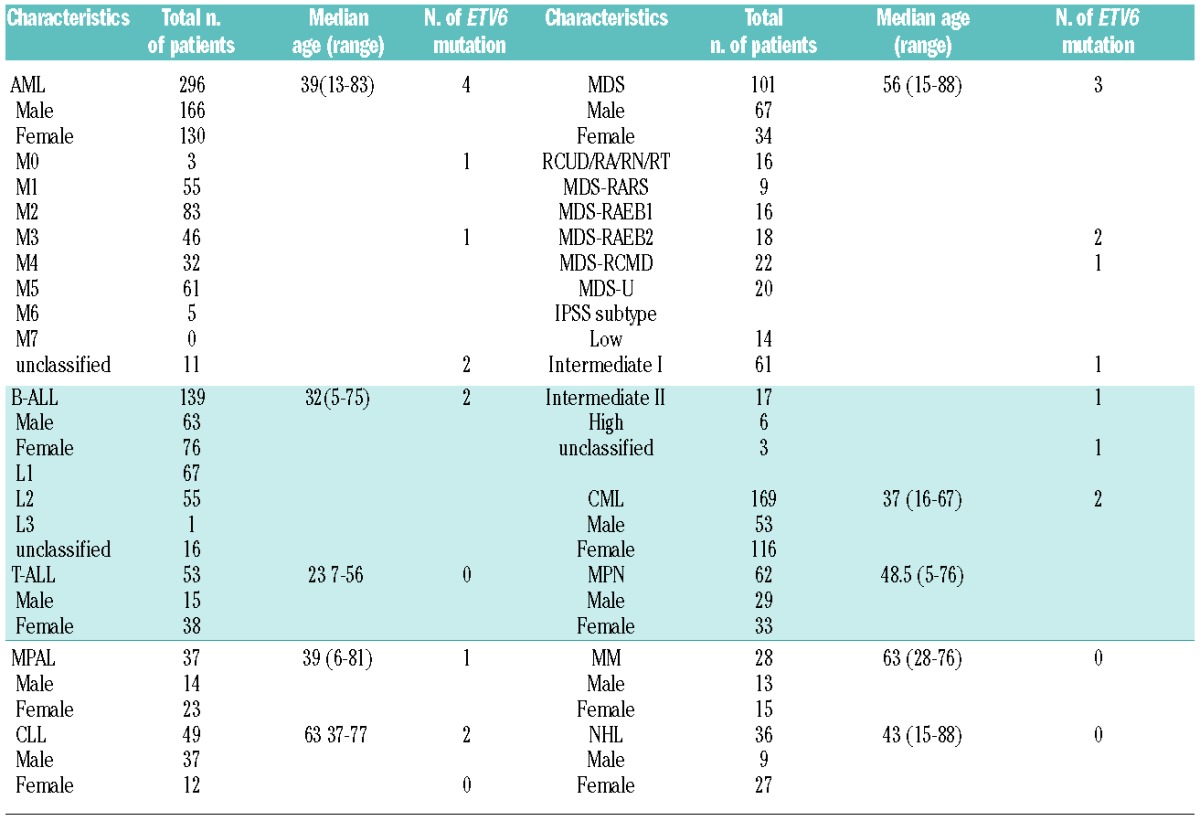
Recently, point mutations in the ETV6 gene have been reported in 2.7% cases of myelodysplastic syndromes (MDS),11 24–33% of early T-cell precursor ALL (ETP-ALL),12,13 and a few cases of acute myelogenous leukemia (AML).14 However, only few data are available on other entities of hematologic malignancies. In order to analyze the frequency of ETV6 mutations and their clinical impact, we investigated a total of 970 cases. In detail, we analyzed 296 de novo AML, 139 B-cell acute lymphoblastic leukemia (B-ALL), 53 T-cell acute lymphoblastic leukemia (T-ALL), 37 mixed-phenotype acute leukemia (MPAL), 169 chronic myeloid leukemia (CML), 101 MDS, 49 chronic lymphocytic leukemia (CLL), 62 myeloproliferative neoplasms (MPN), 28 multiple myeloma (MM), and 36 non-Hodgkin lymphoma (NHL) cases. There were 462 male and 501 female patients in this series; median age was 44 years (range 5–88 years). Main patients’ characteristics are summarized in Table 1.
Table 1.
The clinical and cytogenetic characteristics of the 970 patients.
We examined ETV6 mutation by PCR amplification of the entire coding region followed by direct DNA sequencing. Genomic DNA was extracted from frozen bone marrow mononuclear cells (BMMCs) after Ficoll gradient centrifugation using standard procedures. Point mutations were confirmed in an independent second experiment. Primer sequences and PCR conditions are shown in Online Supplementary Table S1. Known single nucleotide polymorphisms are excluded based on the NCBI (Accession number NM_001987, Version NM_001987.4) or the 1000 Genomes databases.
In total, 14 ETV6 mutations were identified in our study, resulting in an overall frequency of 1.5% (14 of 970). ETV6 mutations were most frequently detected in CLL (2 of 49, 4.0%), followed by MDS (3 of 101, 2.97%), MPAL (one of 37, 2.7%), B-ALL (3 of 139, 2.2%), AML (4 of 296, 1.35%), and CML (2 of 169, 1.2%). No mutations were found in NHL, MM, T-ALL, or MPN. Among these, frameshift, missense, and nonsense mutations respectively accounted for 57.1% (8 of 14), 28.6% (4 of 14) and 14.3% (2 of 14) of all ETV6 mutations. These mutations are localized in exons 2, 7 and 8 (one case each), and exons 6, 5, 4, and 3 (4, 3, 2, and 2 cases, respectively). None of the 14 mutations have been reported previously. Characteristics of these patients are listed in Online Supplementary Table S2. In all available cases (4 of 14), analysis of matched newly diagnosed and remission genomic DNA confirmed the somatic origin of ETV6 mutations (R105GR, S139fsX152, D351fsX384, and N382fsX383). The ETV6 mutations detected in the present study are listed in Table 1 and graphically depicted in Figure 1A. We found that, except for the 4 missense mutations (I140V, E197K, F357Y, and K421E), all the mutations were predicted to cause the loss of either the ETS domain or the SAM-PNT domain. This mutation pattern was consistent with previous studies.12–14
Figure 1.
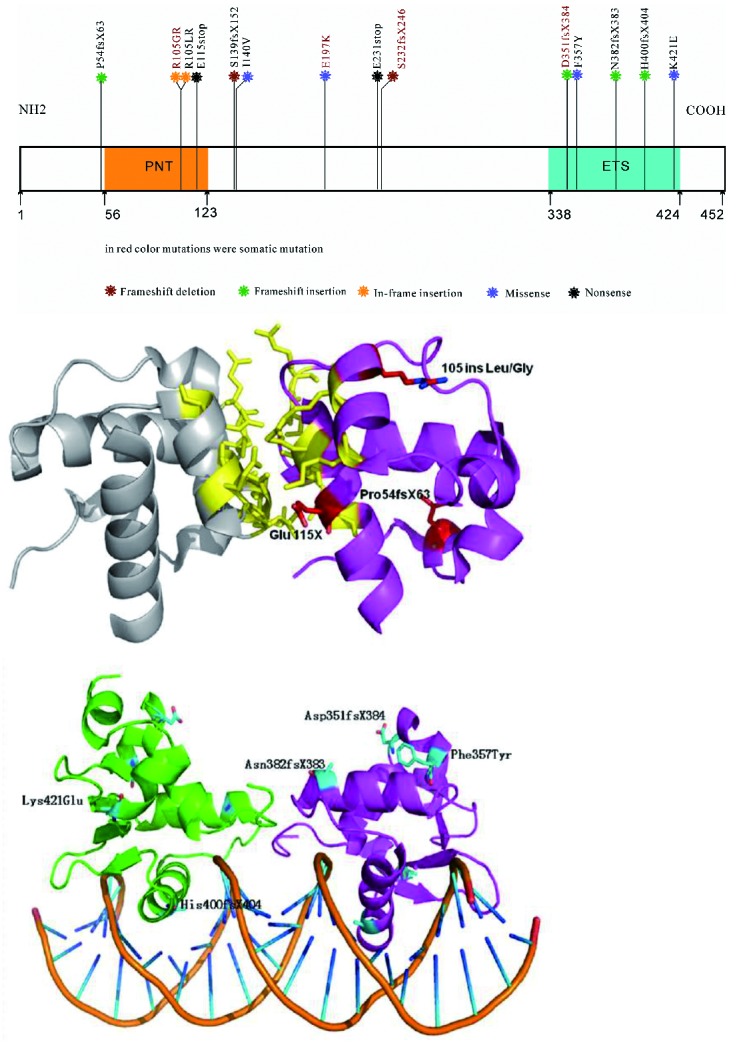
(A). Distribution of somatic sequence mutations in ETV6. Two conserved domains in ETV6 are shown: the PNT domain at its amino terminus which mediates homotypic oligomerization of ETV6 molecules and also heterotypic interaction with other protein and the DNA-binding ETS domain at amino terminus which mediates protein-protein interactions. In red color mutations were somatic mutation and all the mutations have not been reported previously. (B) ETV6 mutations in SAM domain. The structure of a heterodimer with an interface is illustrated by native interface of the SAM domain polymer of TEL. The main residuals from salt-bridges are shown in yellow. The mutation loci discovered in the SAM domain and its relation with the interface of heterodimer are labeled in red. (C). ETV6 mutations in ETS domain. ETS domain (334–437) 3D model of Tel dimmer DNA complex and the mutation residuals also were labeled.
To determine whether these mutations might disrupt the ETS domain or the SAM domain-mediated transcriptional repression activity, structural homology modeling of the etv6 mutant ETS domain DNA binding complex and the SAM domain self-associated oligomerization interface were performed (Figure 1B and C). The structural homology modeling illustrated that the four mutations in the SAM domain (P54fsX63, R105LR, R105GR, and E115X) were near the interface and may disorder Etv6 functions significantly by interfering with the forming of oligomer. While 5 mutations in the ETS domain (D351fsX384, F357Y, N382fsX383, H400fsX404 and K421E) might impede DNA-binding activities of ETV6. Furthermore, 3 out of 5 mutations located in the link region are nonsense (E231X) or out-of-frame mutations (S232fsX246, S139fsX152) that lead to loss of ETS domain.
In cases with translocations involving ETV6, deletion of the non-rearranged ETV6 allele has been identified as an important secondary event that occurs in up to 60% of childhood ALL with ETV6-RUNX1 fusion gene.15 In a search for additional genetic abnormalities involving ETV6, we applied genome-wide array-based comparative genomic hybridization (array-CGH) or fluorescence in situ hybridization (FISH) technique to patients with and patients without ETV6 mutations. However, no ETV6 deletion was found in those patients with ETV6 mutation, while in 73 cases of hemotologic malignancies FISH showed 7 with ETV6 translocation (9.6%) and 12 with haploid deletion (16.4%). Furthermore, 11 patients with deletions were identified in 59 patients by array-CGH. Among these deletions, 2 were in AML (n=7, 28.6%), 6 in ALL (n=25, 24.0 %), 2 in MPAL (n=15,13.3%) and one in CML (n=2).
To evaluate the consequences of mutations on ETV6 expression, quantitative RT-PCR (QRT-PCR) was performed on samples from 12 patients with and 20 patients without ETV6 mutations. There was no significant difference in the expression of ETV6 between patients with and patients without ETV6 mutations (P>0.05).
In order to identify genetic defects that might co-operate with ETV6 mutations in the pathogenesis of leukemia, we sequenced ASXL1, CBL, DNMT3A, EZH2, FLT3, IDH1, IDH2, IKZF1, K-RAS, NPM1, NRAS, P53 RUNX1, TET2, and WT1 in ETV6-mutated leukemia samples. We identified the IKZF1 deletion in one B-ALL patient and the RUNX1 mutation in one patient with CML in blast crisis. No association of ETV6 mutations with other molecular abnormalities was found in this study.
In summary, the present study describes the frequency and spectrum of the somatic mutations of ETV6 in a variety of hematologic malignancies. Our results, together with previous reports in the literature, suggest that somatic mutations of ETV6 are infrequent but recurrent genetic abnormalities in a wider range of myeloid or lymphoid malignancies, including MDS, AML, ETP-ALL, CLL, MPAL, B-ALL, and CML. Structural homology modeling analysis showed that ETV6 mutations might disrupt the Etv6 structure and impede its function. In addition, it will be necessary to conduct further genetic studies with the novel genomics technologies, such as next generation sequencing, to determine the mutation landscape of ETV6 in other hematologic malignancies.
Acknowledgments
the authors would like to thank the National Key Scientific Projects of China (2011CB933501), Provincial Special Program of Medical Science (BL2012005), Jiangsu Province’s Key Medical Center (ZX201102), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Jiangsu Province’s Key Provincial Talents Program, the National Natural Science Foundation of China (81070416), and Jiangsu Province Natural Science Foundation for Distinguished Young Scholars (BK2012006).
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Golub TR, Barker GF, Stegmaier K, Gilliland DG. The TEL gene contributes to the pathogenesis of myeloid and lymphoid leukemias by diverse molecular genetic mechanisms. Curr Top Microbiol Immunol. 1997;220:67–79 [DOI] [PubMed] [Google Scholar]
- 2.Lopez RG, Carron C, Oury C, Gardellin P, Bernard O, Ghysdael J. TEL is a sequence-specific transcriptional repressor. J Biol Chem. 1999;274(42):30132–8 [DOI] [PubMed] [Google Scholar]
- 3.Bohlander SK. ETV6: a versatile player in leukemogenesis. Semin Cancer Biol. 2005;15(3):162–74 [DOI] [PubMed] [Google Scholar]
- 4.Hock H, Meade E, Medeiros S, Schindler JW, Valk PJ, Fujiwara Y, et al. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev. 2004;18(19):2336–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Attarbaschi A, Mann G, Strehl S, Konig M, Steiner M, Jeitler V, et al. Deletion of 11q23 is a highly specific nonrandom secondary genetic abnormality of ETV6/RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia. 2007;21(3):584–6 [DOI] [PubMed] [Google Scholar]
- 6.Wall M, Rayeroux KC, MacKinnon RN, Zordan A, Campbell LJ. ETV6 deletion is a common additional abnormality in patients with myelodysplastic syndromes or acute myeloid leukemia and monosomy 7. Haematologica. 2012;97(12):1933–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Odero MD, Carlson K, Calasanz MJ, Lahortiga I, Chinwalla V, Rowley JD. Identification of new translocations involving ETV6 in hematologic malignancies by fluorescence in situ hybridization and spectral karyotyping. Genes Chromosomes Cancer. 2001;31(2):134–42 [DOI] [PubMed] [Google Scholar]
- 8.Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell. 1994;77(2):307–16 [DOI] [PubMed] [Google Scholar]
- 9.De Braekeleer E, Douet-Guilbert N, Morel F, Le Bris MJ, Basinko A, De Braekeleer M. ETV6 fusion genes in hematological malignancies: a review. Leuk Res. 2012;36(8):945–61 [DOI] [PubMed] [Google Scholar]
- 10.Cave H, Cacheux V, Raynaud S, Brunie G, Bakkus M, Cochaux P, et al. ETV6 is the target of chromosome 12p deletions in t(12;21) childhood acute lymphocytic leukemia. Leukemia. 1997;11(9):1459–64 [DOI] [PubMed] [Google Scholar]
- 11.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, Haydu JE, Rigo I, Hadler M, et al. ETV6 mutations in early immature human T cell leukemias. J Exp Med. 2011;208(13):2571–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barjesteh van Waalwijk van Doorn-Khosrovani S, Spensberger D, de Knegt Y, Tang M, Lowenberg B, Delwel R. Somatic heterozygous mutations in ETV6 (TEL) and frequent absence of ETV6 protein in acute myeloid leukemia. Oncogene. 2005;24(25):4129–37 [DOI] [PubMed] [Google Scholar]
- 15.Attarbaschi A, Mann G, Konig M, Dworzak MN, Trebo MM, Muhlegger N, et al. Incidence and relevance of secondary chromosome abnormalities in childhood TEL/AML1+ acute lymphoblastic leukemia: an interphase FISH analysis. Leukemia. 2004;18(10):1611–6 [DOI] [PubMed] [Google Scholar]