

Abstract
The use of corticosteroid prodrugs in pharmacokinetic studies poses the risk of overestimation of corticosteroid concentrations due to in vitro hydrolysis of prodrugs after sample collection. This study tests the effectiveness of the anticoagulant EDTA as a stabilizer for dexamethasone sodium phosphate (DSP) in rat plasma and provides simultaneous HPLC analysis of DSP and dexamethasone. An already developed ion-paired reversed-phase HPLC assay for simultaneous measurement of corticosteroid phosphate ester prodrugs and their active steroids was applied in this study. This assay was used for analyzing samples from an in vitro DSP hydrolysis study in rat plasma. In agreement with allometric principles, the prodrug hydrolysis occurred at a much faster rate (in vitro half-life of 1.75 h) in rat plasma as compared with previously reported prodrug hydrolysis half-life of 10–12 h in sheep and human plasma. The in vitro degradation of the prodrug in rat plasma was greatly minimized in plasma containing EDTA at the concentration commonly used an anticoagulant. This study demonstrates that artifacts in pharmacokinetic profiles of corticosteroids due to in vitro prodrug hydrolysis can be greatly minimized by collecting blood samples with EDTA as the anticoagulant.
Keywords: Corticosteroid, EDTA, HPLC, Pharmacokinetics, Prodrug, Stability in plasma
Dexamethasone is a potent corticosteroid with gluconeogenic, immunosuppressive and anti-inflammatory properties. Dexamethasone is used for treating various inflammatory conditions, lymphomas and for inducing precocious fetal lung maturation in threatened preterm labor. The poor solubility of dexamethasone is circumvented by the formulation of a phosphate ester prodrug, which allows rapid input after parenteral administration. However, an analytical issue associated with the use of DSP is its ex vivo hydrolysis to dexamethasone by plasma enzymes after sample collection. This causes overestimation of dexamethasone concentrations after parenteral administration at early time points. This artifact produces pharmacokinetic profiles with elevated biexponential character, leading to flawed pharmacokinetic parameter estimates (Samtani et al., 2004a). This problem is usually prevented by preserving samples with high concentrations (100 mM) of the phosphatase inhibitor sodium arsenate (Hare et al., 1975; Samtani et al., 2004b). This method is helpful if the objective is to measure total drug concentrations. However, it is often of interest to measure unbound corticosteroid concentrations during a variety of disease states (e.g. inflammation, pregnancy complications) that may be associated with changes in plasma protein binding (Mulay and Varma, 1984; Varma and Yue, 1984; Garg et al., 1994). The measurement of free steroid levels can be hampered by the presence of the high salt and alkaline milieu produced by sodium arsenate, which may disrupt protein binding. We therefore aimed to find a non-disruptive, simple and non-toxic method for preserving plasma samples. A survey of the literature indicated that other phosphate ester prodrugs, such as fosphenytoin and etoposide phosphate are stabilized in plasma samples by drawing blood in collection devices containing EDTA as an anticoagulant (Cwik et al., 1997; Reif et al., 2001). EDTA activity as a chelating agent interferes with the functioning of plasma hydrolysing enzymes, which require metal ions as cofactors. The concentration of EDTA tested as a sample stabilizer was 4 mM (1.5 mg/mL), which is the commonly used concentration as an anticoagulant. The most commonly employed animal model for dexamethasone pharmacokinetics is the rat and hence the stability of its prodrug was studied in blank rat plasma.
Fresh rat blood containing heparin was obtained, quickly centrifuged to obtain plasma and incubated with DSP. The concentration of the prodrug was 6000 ng/mL, which is the expected concentration maximum for corticosteroid prodrugs in pharmacokinetic studies (Samtani et al., 2004b). Incubations in the presence and absence of EDTA were performed in plasma from three animals and the incubation volume used was 600 μL. Aliquots of 50 μL were sampled from the incubation mix at 0, 30, 60, 120, 240 and 360 min.
The ability of EDTA to stabilize DSP during a freeze thaw cycle was also tested. EDTA treated plasma was spiked with DSP and six 50 μL aliquots were prepared. Three aliquots were analyzed immediately and served as a measure of the initial starting concentration. The remaining aliquots were stored at −20 °C for 1 week, thawed unassisted at room temperature (RT), allowed to stand at RT for 4 h and then assayed. The 1 week storage and 4 h RT step were incorporated to mimic sample processing delays that occur during pharmacokinetic studies.
Concentrations were determined by a recently described HPLC assay (Samtani et al., 2004b) with a lower limit of quantification of 300 ng/mL for dexamethasone and 500 ng/mL for DSP. Assay accuracy (% error) and precision (CV%) were found to be under 10% for both analytes. The assay is linear in the concentration range of 300–4800 ng/mL for dexamethasone and 500–8000 ng/mL for DSP.
To allow simultaneous fitting of the four data sets from the incubation study concentrations were converted to nanomolar units and the equations used were:
| (1a) |
| (1b) |
| (2a) |
| (2b) |
| (3a) |
| (3b) |
| (4a) |
| (4b) |
where CDSP and CDEX represent concentrations of DSP and dexamethasone as a function of time (t) in blank plasma, CDSP(0) the initial spiked concentration of the prodrug in blank plasma and kh represents the first-order rate constant for the conversion of DSP to dexamethasone. Terms with the subscript EDTA represent concentrations of the steroid in plasma treated with EDTA and t50 represents the time point where 50% of the plasma enzyme activity is inhibited by EDTA. The kinetic modeling was performed using the maximum likelihood estimator within the ADAPT II software (D’Argenio and Schumitzky, 1997). The variance model was:
| (5) |
where intercept and slope are variance parameters, and Y(t) represents the model output function.
In blank plasma 92% of the prodrug was converted to dexamethasone by 6 h (Fig. 1A). In contrast the breakdown of the prodrug was reduced to less than 15% in the presence of EDTA (Fig. 1B). The peculiar feature of EDTA plasma enzyme inhibition is that it does not cause immediate DSP stabilization. Prodrug breakdown occurs for approximately half an hour and then the reaction appears to be completely inhibited. This initial degradation causes a drop in DSP concentration and early generation of dexamethasone, which stays unchanged for up to 6 h. The slight time delay in inhibiting enzyme activity was captured by the non-linear inhibition function 1 – t/(t + t50) in Eqs. (3) and (4). As time exceeds t50 the function along with kh approaches zero and hence the change is steroid concentrations approaches a steady plateau. Estimate (CV%) for t50 was computed to be 3.7 min (6.4%), which indicates that inhibition of plasma enzymes by EDTA is fairly rapid. Samples mimicking sample processing delays also exhibited a 13% decrease in DSP concentrations, which probably occurs due to the exposure to room temperature for 4 h after a freeze thaw cycle (Fig. 2). This drop in concentration is similar to the decrease in DSP concentration observed in freshly incubated plasma samples depicted in Fig. 1B. These results may indicate that there is minimal prodrug breakdown when the samples are frozen which is not surprising because DSP plasma hydrolysis is a temperature dependant enzymatic process (Derendorf et al., 1986; Samtani et al., 2004b). Furthermore, plasma hydrolysing enzymes seem to retain their activity during a freeze thaw cycle and remain susceptible to EDTA inhibition.
Fig. 1.
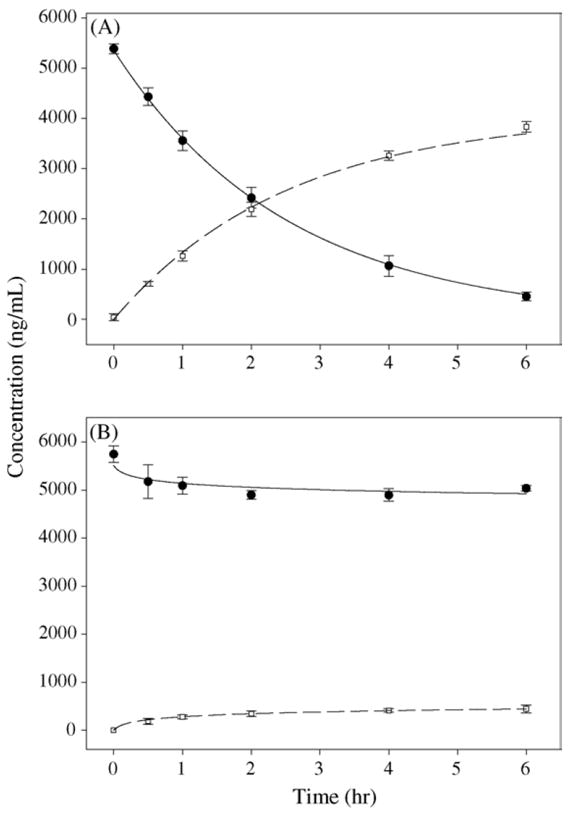
Time course for in vitro prodrug hydrolysis at 25 °C in (A) fresh rat plasma and (B) EDTA treated plasma. Closed circles indicate DSP concentrations, while open circles indicate dexamethasone concentrations. Data points with standard deviation bars represent mean concentrations in plasma from three animals. Solid and dashed curves indicate DSP and dexamethasone profiles fitted simultaneously using Eqs. (1)–(4).
Fig. 2.

Stability of DSP during a freeze thaw cycle in EDTA treated plasma. Bars with standard deviations represent concentrations in plasma from three aliquots.
The prodrug break down half-life in blank plasma was computed to be 1.75 h (CV%: 1.53). The in vitro half-life of corticosteroid phosphate prodrugs in plasma from sheep and humans has been reported to be 10–12 h (Derendorf et al., 1986; Samtani et al., 2004b). The allometric equation for time parameters is:
| (6) |
This equation predicts that the half-life (t1/2) for in vitro prodrug breakdown in rat plasma would be 2.4–2.9 h. Thus, the in vitro t1/2 observed in this study is in reasonable agreement with allometric principles and meets the expectation that processes occur at a much faster rate in rodents.
The observations indicate that ex vivo prodrug hydrolysis will be controlled by two competing forces. DSP has an in vivo half-life of 5–10 min in humans (Mollmann et al., 1995; Samtani et al., 2004b) and this process will be greatly hastened in rats. The faster clearance processes in rodents will cause rapid activation of the prodrug in vivo. Thus, only those samples collected at the earliest time points in rats will contain DSP. However, these rat samples will also be difficult to stabilize completely because of the faster and efficient enzymatic processes. Thus, DSP break down will continue to occur even after sample collection but will be greatly minimized in the presence of EDTA.
Based on our experience with rat studies the ex vivo prodrug hydrolysis can be minimized by administering the prodrug via the intramuscular route. After intramuscular administration a good fraction of the prodrug is activated at the injection site and the steroid can be absorbed in the active form directly (Petersen et al., 1984). This is the preferable administration route since it is used clinically and produces almost complete and rapid dexamethasone input (Samtani and Jusko, 2005). Blood samples should be collected in EDTA, kept on ice during processing delays, rapidly spun down at −4 °C and stored immediately at −20 to −70 °C. Prodrug hydrolysis is an enzymatic process and is highly temperature dependant (Derendorf et al., 1986; Samtani et al., 2004b). Cooling the samples, therefore, reduces ex vivo generation of dexamethasone.
It is interesting to consider the previously reported meta-analysis of dexamethasone disposition in rats and earlier work from our laboratory on dexamethasone pharmacokinetics (Ramakrishnan, 2001; Mager et al., 2003). In all early work, the commonly reported pharmacokinetic function for dexamethasone was the biexponential function. These profiles exhibited marked multi-compartment character, which was attributed to dexamethasone distribution and the problem of ex vivo prodrug hydrolysis was ignored. To demonstrate the utility of the above recommended procedures we call attention to our recently reported dexamethasone pharmacokinetic study (Samtani and Jusko, 2005). This most recent work took into consideration the above procedural details and the pharmacokinetic function that was sufficient to describe dexamethasone pharmacokinetics was a one-compartment mammillary equation. We, thus, believe that biexponential profiles of corticosteroids commonly reported in the literature after administration of corticosteroid prodrugs are probably often artifacts due to improper sample handling. Such problems can be prevented by careful sample processing and study design.
Acknowledgments
This work was supported by NIH Grant No. GM 24211.
References
- Cwik MJ, Liang M, Deyo K, Andrews C, Fischer J. Simultaneous rapid high-performance liquid chromatographic determination of phenytoin and its prodrug, fosphenytoin in human plasma and ultrafiltrate. J Chromatogr B: Biomed Sci Appl. 1997;693:407–414. doi: 10.1016/s0378-4347(97)00057-1. [DOI] [PubMed] [Google Scholar]
- D’Argenio DZ, Schumitzky A. Biomedical Simulations Resource. Los Angeles: 1997. ADAPT II User’s Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software. [Google Scholar]
- Derendorf H, Rohdewald P, Hochhaus G, Mollmann H. HPLC determination of glucocorticoid alcohols, their phosphates and hydrocortisone in aqueous solutions and biological fluids. J Pharm Biomed Anal. 1986;4:197–206. doi: 10.1016/0731-7085(86)80042-5. [DOI] [PubMed] [Google Scholar]
- Garg V, Hon YY, Jusko WJ. Effects of acute and chronic inflammation on the pharmacokinetics of prednisolone in rats. Pharm Res. 1994;11:541–544. doi: 10.1023/a:1018966516195. [DOI] [PubMed] [Google Scholar]
- Hare LE, Yeh KC, Ditzler CA, McMahon FG, Duggan DE. Bioavailability of dexamethasone. II. Dexamethasone phosphate. Clin Pharmacol Ther. 1975;18:330–337. doi: 10.1002/cpt1975183330. [DOI] [PubMed] [Google Scholar]
- Mager DE, Pyszczynski NA, Jusko WJ. Integrated QSPR-pharmacodynamic model of genomic effects of several corticosteroids. J Pharm Sci. 2003;92:881–889. doi: 10.1002/jps.10343. [DOI] [PubMed] [Google Scholar]
- Mollmann H, Balbach S, Hochhaus G, Barth J, Derendorf H. Pharmacokinetic–pharmacodynamic correlations of corticosteroids. In: Derendorf H, Hochhaus G, editors. Handbook of Pharmacokinetic/Pharmacodynamic Correlation. CRC Press Inc; Boca Raton: 1995. pp. 323–361. [Google Scholar]
- Mulay S, Varma DR. Influence of streptozotocin-diabetes on the pharmacokinetics, placental transfer and tissue localization of dexamethasone in rats. Br J Pharmacol. 1984;83:139–144. doi: 10.1111/j.1476-5381.1984.tb10128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, Ashley JJ, McBride WG, Nation RL. Disposition of betamethasone in parturient women after intramuscular administration. Br J Clin Pharmacol. 1984;18:383–392. doi: 10.1111/j.1365-2125.1984.tb02480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan R. PhD Thesis. State University of New York at Buffalo; USA: 2001. Pharmacodynamics of receptor-mediated complex pharmacological responses . [Google Scholar]
- Reif S, Kingreen D, Kloft C, Grimm J, Siegert W, Schunack W, Jaehde U. Bioequivalence investigation of high-dose etoposide and etoposide phosphate in lymphoma patients. Cancer Chemother Pharmacol. 2001;48:134–140. doi: 10.1007/s002800100280. [DOI] [PubMed] [Google Scholar]
- Samtani MN, Schwab M, Nathanielsz PW, Jusko WJ. Area/moment and compartmental modeling of pharmacokinetics during pregnancy: applications to maternal/fetal exposures to corticosteroids in sheep and rats. Pharm Res. 2004a;21:2279–2292. doi: 10.1007/s11095-004-7681-7. [DOI] [PubMed] [Google Scholar]
- Samtani MN, Schwab M, Nathanielsz PW, Jusko WJ. Stabilization and HPLC analysis of betamethasone sodium phosphate in plasma. J Pharm Sci. 2004b;93:726–732. doi: 10.1002/jps.10577. [DOI] [PubMed] [Google Scholar]
- Samtani MN, Jusko WJ. Comparison of dexamethasone pharmacokinetics in female rats after intravenous and intramuscular administration. Biopharm Drug Dispos. 2005;26:85–91. doi: 10.1002/bdd.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma DR, Yue TL. Influence of protein-calorie malnutrition on the pharmacokinetics, placental transfer and tissue localization of dexamethasone in rats. Br J Pharmacol. 1984;83:131–137. doi: 10.1111/j.1476-5381.1984.tb10127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]