

Abstract
There have been nearly 400genome-wide association studies published since 2005. The GWAS approach has been exceptionally successful in identifying common genetic variants that predispose to a variety of complex human diseases and biochemical and anthropometric traits. Although this approach is relatively new, there are many excellent reviews of different aspects of the GWAS method. Here, we provide a primer, an annotated overview of the GWAS method with particular reference to psychiatric genetics. We dissect the GWAS methodology into its components and provide a brief description with citations and links to reviews that cover the topic in detail.
Keywords: genome-wide association study, review, psychiatric genetics
Overview
The first genome-wide association study (GWAS, “jē’ wŏs”) of age-related macular degeneration appeared in 2005 (Klein et al., 2005). Since then, nearly 400GWAS studies have been published in the NHGRI GWAS catalog (http://www.genome.gov/26525384, accessed 20 September 2009). The GWAS approach has been exceptionally successful in identifying common genetic variants that predispose to a variety of complex human diseases and biochemical and anthropometric traits and was named the “breakthrough” of 2007 by the journal Science. Indeed, the GWAS method has performed beyond expectations.
Although the GWAS approach is relatively new, there have already been a large number of excellent reviews of various components of the GWAS method. Indeed, the GWAS review literature is of such singular quality that another review would be redundant. Instead of another review, our goal is to provide a primer, an annotated overview of the entire approach with particular reference to psychiatric genetics. Our goal is dissemination of information about this methodology in order for a motivate reader to become more expert in this method. We dissect the GWAS methodology into its components and, for each, provide a brief description of the component and provide citations and links to reviews that cover the topic in detail (Table 1).
Table 1.
Further primer information by topic.
An Introduction to GWAS Methodology
Basic Principles in Genetics
It is beyond the scope of this review to cover fundamental topics in genetics. A number of useful starting points are shown in Table 1.
Definition
A GWAS for a disease is usually a variant of a cross-sectional case-control study, the study design that is the familiar workhorse in biomedicine and epidemiology (Schlesselman, 1982). Another term for GWAS is whole-genome association study (WGAS, “dŭb’ əl-yōō găs”). Cases are defined as individuals who meet lifetime criteria for a disease (e.g., Crohn’s disease, type 2 diabetes mellitus, or schizophrenia). Controls should have never met criteria for the disease and, ideally, be through the period of risk. Moreover, for case-control comparisons to be as unbiased as possible, controls should be draws from the same population as cases particularly with respect to exposure to any potentially relevant risk factors (Rothman, 1986). Each individual in the sample is assayed (i.e., genotyped) for a comprehensive set of genetic markers scattered across the genome. The genetic markers are single nucleotide polymorphisms (SNPs, “snips”) which are relatively straight-forward to assay. The two major current GWAS technological platforms contain 906,000 (Affymetrix 6.0) and 1,199,187 SNPs (Illumina 1M) spaced across the 22 autosomes (chr1–chr22), the sex chromosomes (chrX and chrY) and the mitochondrial genome (chrM).
The key analysis in a GWAS for a disease is logistic regression with the dependent variable case-control status (1=case, 0=control) and a SNP genotype as an independent variable (coded as the number of copies of the minor or less frequent allele, 1 degree of freedom). The output of a logistic regression is identity of the reference allele and an odds ratio with its standard error (or confidence intervals) along with a statistic and p-value that test whether the odds ratio differs from unity.
Standard of Evidence
In a GWAS, logistic regressions are done for every SNP (i.e., a total of ~1 million regression models). Given the number of statistical tests, p-values that are very small by traditional standards are to be expected merely by the play of chance (e.g., 10 p-values < 0.00001 and 100 p-values < 0.0001). Thus, the standard of evidence that has emerged for a compelling GWAS finding is rigorous: (a) a strong association in an initial sample, (b) precise replication in one or more independent samples (i.e., the same SNP, allele, and direction of association), and (c) a cumulative p-value < 5×10−8 (Chanock et al., 2007). The 5×10−8 threshold is akin to a Bonferroni correction of the traditional 0.05 Type 1 error level for 1,000,000 statistical tests (although the full argument is more complex as some of these tests are not independent due to linkage disequilibrium) (Pe’er et al., 2008). P-values that are smaller than expected by chance and which replicate well in other samples highlight a genomic region associated with a disorder (and potentially causal).
Statistical Power
Because of the requirement to adjust for the large number of statistical tests in order to control Type 1 error, adequate statistical power (to minimize Type 2 error) is crucial particularly given the small genetic effect sizes typical for human GWAS findings (see below). Figure 1 shows power curves for four different sample sizes. Given the large number of statistical tests and as the genetic effects are likely to be quite subtle, power is inadequate unless very large numbers of cases and controls are studied.
Figure 1.
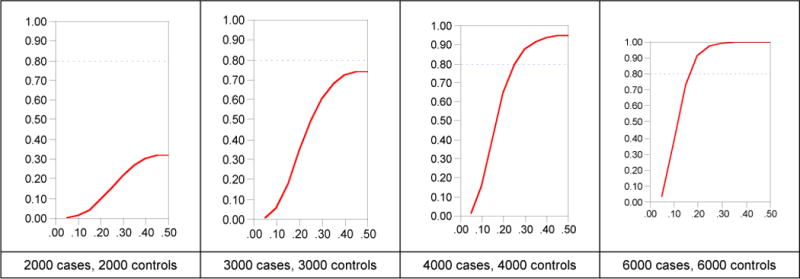
Statistical power in a GWAS.
Figure 1 illustrates statistical power for GWAS of four different sample sizes assuming a discrete trait with lifetime prevalence of 0.01 (similar to schizophrenia, bipolar disorder, or anorexia nervosa), a log additive genetic model, a genotypic relative risk of 1.25 (typical for GWAS for human complex diseases, and two-tailed α=5×10−8). The x-axis shows minor allele frequency and the y-axis statistical power
GWAS Statistics
We illustrate here some properties of published GWAS in biomedicine from the NHGRI GWAS catalog (accessed 20 September 2009). Of 396 published GWAS, there were 238 studies reporting 693 SNP associations with p < 5×10−8. These associations were for 59 human diseases and 61 other quantitative traits. The diseases with the greatest number of associations were Crohn’s disease, type 1 diabetes mellitus, type 2 diabetes mellitus, prostate cancer, and rheumatoid arthritis. The top quantitative traits were height, lipid levels (triglycerides and HDL and LDL cholesterol), QT interval, and body mass index.
Figure 2a shows the temporal trends in the publication dates for these studies. Figure 2b illustrates a number of properties of the findings from the literature. Note that only ~15% of the SNP-disease associations are detectible with 90% power with a sample of 1,000 cases and 1,000 controls whereas only about 4% would not be detected with 25,000 cases and 25,000 controls (the estimated number of GWAS samples available for schizophrenia and bipolar disorder by 2014). This point is crucial – based on power calculations and empirical findings for other disorders, “failure” to detect an association is meaningful only if the sample size is very large.
Figure 2.
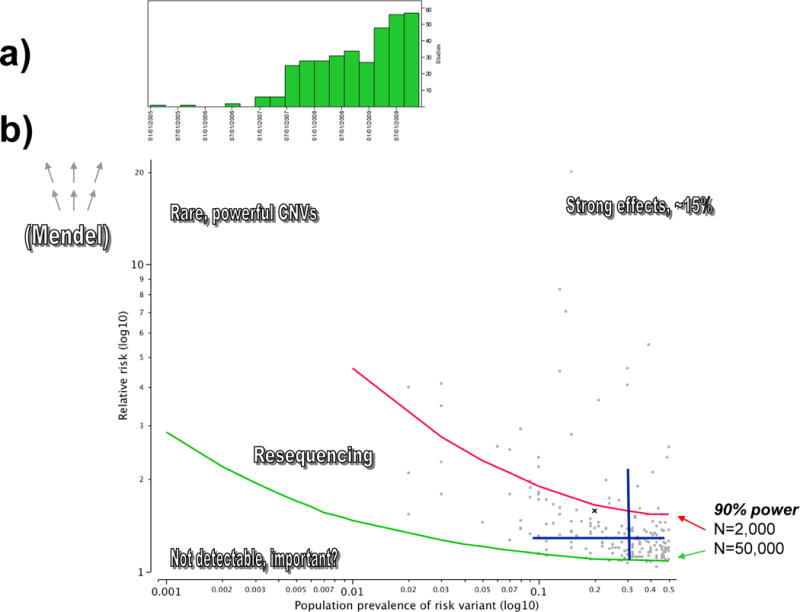
Properties of GWAS findings from the literature.
Figure 2a shows quarterly temporal trends in the publication of GWAS. Figure 2b shows the accumulated GWAS literature on human diseases. The x-axis if the population prevalence of a risk variant and the y-axis the relative risk conferred (both using a log10 scale in order to provide separation). The grey point show the prevalence-risk combination for all SNP associations for human complex diseases with p < 5×10−8. Power curves are shown for 1,000 cases/1,000 controls (red line) and 25,000 cases/25,000 controls (green line). The blue lines depict the 10th–90th percentiles from the GWAS literature for allele frequency (horizontal line) and relative risk (vertical line). The intersection of the blue lines is the median population prevalence (0.3) and relative risk (1.25).
Several intriguing trends were evident in these data on human diseases. First, with few exceptions (e.g., Alzheimer’s disease and APOE), the regions implicated by GWAS were not previously known. Candidate genes based on prior knowledge of pathophysiology or intuition have usually not been identified. Second, the majority of these findings (90%) were not in the coding region of a gene, and only 8% were non-synonymous variants (i.e., DNA variants that change the amino acid sequence of the corresponding protein). Indeed, 43% were not in a known gene and 23% were not within 20,000 bases of a known gene. Common variation underlying complex human diseases is dissimilar to that underlying Mendelian diseases where major changes to proteins are typical.
Meta-Analysis
Given the requirement for historically large sample sizes, it has become typical for primary studies to band together to form meta-analytic consortia. This has proven to be a crucially important step in achieving sufficient statistical power. For example, two primary type 2 diabetes mellitus GWAS were completely unremarkable individually and yet, after meta-analysis, multiple highly significant and replicated findings emerged (Saxena et al., 2007, Scott et al., 2007).
GWAS for Psychiatric Disorders
Multiple GWAS for psychiatric disorders are published, in progress, or planned. The disorders include anorexia nervosa, attention-deficit hyperactivity disorder, autism, bipolar disorder, drug use disorders (smoking behavior and alcohol dependence), major depressive disorder, obsessive-compulsive disorder, and schizophrenia. There are > 50 primary samples, mostly in subjects of European ancestry but with increasing numbers of subjects of African and East Asian ancestry. Prominent examples of GWAS findings for psychiatric disorders are described in Table 2. This area is moving quickly, and additional findings are known to be in the publication pipeline.
Table 2.
Notable psychiatric GWAS findings (as of September 2009)
| Disease | Citation | Locus | Subjects | MAF, OR† | Best SNP & P-value |
|---|---|---|---|---|---|
| Autism | (Wang et al., 2009) | CDH10-CDH9 intergenic | 12,834 | 0.38, 1.19 | rs4307059, 2×10−10 |
| Bipolar disorder | (Ferreira et al., 2008) |
ANK3 CACNA1C |
10,596 10,596 |
0.05, 1.45 0.32, 1.18 |
rs10994336, 9×10−9 rs1006737, 7×10−8 |
| Schizophrenia | (O’Donovan et al., 2008a, International Schizophrenia Consortium, 2009, Shi et al., 2009, Stefansson et al., 2009) |
MHC-NOTCH4 region MHC-histone cluster NRGN TCF4 ZNF804A |
47,536 47,536 47,536 47,536 20,142 |
0.85, 1.15 0.87, 1.19 0.83, 1.15 0.06, 1.23 0.59, 1.12 |
rs3131296, 2×10−10 rs6913660, 1×10−9 rs12807809, 2×10−9 rs9960767, 4×10−9 rs1344706, 2×10−7 |
Illustrative minor allele frequency (MAF) in controls and odds ratio (OR).
The Psychiatric GWAS Consortium (PGC) was formed in 2007 to conduct “mega-analysis” of individual genotype and phenotype data, and is described in detail elsewhere (Cross Disorder Phenotype Group of the Psychiatric GWAS Consortium, 2009, Psychiatric GWAS Consortium, 2009a, Psychiatric GWAS Consortium, 2009b). GWAS data for ADHD, autism, bipolar disorder, major depressive disorder, and schizophrenia from European subjects are being analyzed as of this writing in 9/2009. To our knowledge, this study of >59,000 independent cases and controls and >7,700 family trios will be the largest biological experiment conducted in psychiatry.
The PGC has two major goals. Aim 1 is to conduct five separate GWAS mega-analyses for ADHD, autism, bipolar disorder, major depressive disorder, and schizophrenia. The Aim 2 cross-disorder mega-analyses have two components. The “nosological” sub-aim takes cases as defined by DSM-IV criteria and looks for SNPs that are compellingly associated with two or more disorders and effectively searches for genomic regions with pleomorphic effects. The “heterogeneity” sub-aim reclassifies subjects according to pre-specified phenotypic characteristics (e.g., subjects with bipolar disorder with two manic and dozens of depressive episodes should be more major depression-like than bipolar-like). This is a convenient segue to the next crucial issue – are psychiatric phenotypes qualitatively different from other biomedical diseases?
Genetic Models
One of major unknowns for psychiatric disorders is the nature of the genetic models by which variation at the DNA level increases risk for the clinical phenotype. For Mendelian disorders, a genetic model can be hypothesized by examination of pedigrees (e.g., dominant, recessive, or sex-linked) and knowledge of prevalence. For psychiatric diseases, we assume complex inheritance (allowing for mixtures of genetic and environmental effects along with diagnostic imprecision). Two genetic models have received particular attention, that complex traits are caused by common versus rare genetic variation. In the former, psychiatric disease results from the cumulative effect of many genetic variants each of which is common in the population and which confer subtle genetic risk (common disease/common variant model, CDCV). In the latter, psychiatric disease results from many different mutations each of which is rare but of powerful effects (multiple rare variant model, MRV).
Some commentators hold extremist views – e.g., that psychiatric diseases arise only from a MRV model (see the Controversies section below). However, empirical results to date are consistent with a place for both MRV and CDCV models. For schizophrenia, bipolar disorder, and autism, the data are consistent with the presence of multiple common variants of subtle effect in some patients and rare variants in others. More examples are likely to emerge with improved technologies and larger sample sizes.
One fascinating empirical development has been the emergence of the “profile score” concept, an extreme form of the CDCV model. In a recent Nature paper (International Schizophrenia Consortium, 2009), the authors developed a list of ~30,000 SNPs and their risk alleles in one large schizophrenia case-control sample. This list can be used to compute a risk profile for each person in independent samples (i.e., the number of schizophrenia risk alleles). The score from the initial sample significantly predicted schizophrenia risk in three independent samples (p-values 2×10−28, 5×10−11, and 0.008), bipolar risk in two independent samples (p-values 1×10−12 and 9×10−9), and, crucially, was not associated with risk of six non-psychiatric biomedical disorders (Crohn’s disease, T1DM, T2DM, coronary artery disease, hypertension, and rheumatoid arthritis). These data strongly support the CDCV model and also suggest genetic overlap between schizophrenia and bipolar disorder.
From these basic models, multiple elaborations are possible. For example, different genetic variants in the same gene could be associated with a disease in different populations.
As an example of MRV, copy number variation (CNV) has emerged as rare but powerful risk factor for neuropsychiatric disorders. CNVs are segments of the genome > 1000 bases where the number of copies of this segment is different from the expected number. Down’s syndrome is a very large example where three copies (instead of two) of chr21 is present. The chr22q11 hemideletion (one copy of chr22 from 17.3–20.3 million bases) is another example, and has been associated with multiple neuropsychiatric disorders. GWAS chips also contain many CNV probes, leading to increasing interest in this topic.
The Phenotype
The most crucial issue in a case-control study is how to define cases and controls, and this is particularly so in psychiatric genetics. This is more difficult to define and measure than for most non-psychiatric disorders. Further, we have less knowledge of the causes and mechanisms of pathogenesis. Our current official classification systems, DSM and ICD, are descriptive systems that were developed to have clinical utility, acceptable reliability, but with no expectation that the categories represented valid entities with respect to etiology. Although these phenotype definitions are moderately to highly heritable and hence sensible starting points for genetic research, it is generally agreed that the most useful biological categories and/or dimensional definitions and measures are still unknown. The strikingly high level of co-occurrence of different diagnoses within the same individual (“co-morbidity”) almost certainly reflects a substantial overlap in the underlying biology of currently defined syndromes. This is further evidenced by family studies demonstrating shared familial liability across diagnostic boundaries (e.g., schizophrenia and bipolar disorder) (Lichtenstein et al., 2009). It is interesting to note that some of the strongest association signals to emerge from GWAS of schizophrenia and bipolar disorder show an overlap across traditional disorder categories (International Schizophrenia Consortium, 2009).
In view of these observations, it can be expected that a range of approaches to the clinical phenotype may be required in order to maximize the potential from molecular genetic studies. This includes analyses across the traditional illness categories (“lumping”) and analyses of clinically-meaningful subsets within a category or set of categories (“splitting”). It is also possible to use approaches that are not based on any specific prior model of the clinical phenotype and to seek clinical entities (whether they are categories or dimensions) that would “make more sense” from a genetic perspective. For example, for a highly significant and consistently replicated genetic association, cases with and without the genetic variant can be investigated to attempt to identify the phenotypic consequences of the variant – do cases with the variant have earlier onset, more severe symptoms, worse response to treatment, or alter brain structure or function? This is also know as “reverse phenotyping” or “phenotype refinement”. Another analytic possibility, which will be particularly valuable if there is a high degree of polygenicity (i.e., hundreds or thousands of susceptibility alleles of small effect), will be to consider a large set of polymorphisms and use aggregate measures of their overall contribution to phenotypic susceptibility to seek to define “signatures” of genetic variants, the patterns of which could be compared across phenotypes.
Molecular genetics will certainly not provide a simple, gene-based classification of psychiatric illness. However, it can be expected that establishing the relationship between genotypes and psychiatric phenotypes will inform understanding of psychiatric nosology and move psychiatry towards a diagnostic classification that is much closer to the underlying pathophysiology than are the current descriptive classifications. This may well be a relatively early and, clinically important, “pay off” from the major research investment in molecular genetic research in psychiatry.
GWAS Genotyping
Source of DNA
DNA samples are readily obtainable from multiple sites although most studies use peripheral blood lymphocytes from venous samples. Some studies use samples from the oral cavity (buccal scrapings or epithelial cells in saliva) but these samples can be plagued by lesser DNA quantity, inferior DNA quality, and interference of DNA from oral microbial flora. Some samples are derived from lymphocytes transformed by Epstein-Barr virus into immortalized cell lines but such samples can have artifacts that complicate some analyses (e.g., trisomy 12 in copy number analyses). Although some investigators advocate using DNA pooling due to lower cost (i.e., genotyping aggregated cases and aggregated controls), this approach can have serious issues with accuracy and reliability and has not entered wide usage.
Genotyping
The cost of genotyping has decreased by a factor of 2000 in the past decade due to the development of reliable, robust, and highly multiplexed genotyping systems (meaning that many genetic markers are genotyped simultaneously) and because of competition between multiple companies. As of this writing in mid-2009, Affymetrix and Illumina are the main suppliers of GWAS genotyping platforms. Each uses different technologies and each has advantages and disadvantages in regard to genotyping accuracy, genomic coverage, ease of use, and total cost. Both platforms genotype a predefined set of SNPs, a crucial reason why cost has decreased. SNPs are genotyped as they are relatively common in the human genome and relatively straight-forward to assay.
For each platform, genotyping takes three or four days per sample, and most labs run dozens or even hundreds of samples simultaneously. Such high throughput means that even large-scale projects can be completed in under a year. Practically, there are always numerous issues to resolve such as subjects whose stated sex does not match patterns of chrX and chrY SNPs or samples that are unexpectedly identical.
Genotype Calling
For each SNP, GWAS platforms assess each of the two possible alleles with independent assays which can be viewed as a scatter plot. Figure 3a depicts scatter plots for two SNPs in a GWAS. The scatter plots show the intensity values for one SNP allele plotted by the intensity values for the other SNP allele with each point corresponding to one subject. In the scatter plot on the left, the points fall into three well-defined clusters, and individuals in each cluster are “called” as having the same genotype for that SNP (i.e., GG, AG, or AA). A genotype calling algorithm is used to assign these clusters into genotypes for each subject. The scatter plot on the right shows an example of poor cluster separation, and this SNP should be excluded from analysis.
Figure 3.
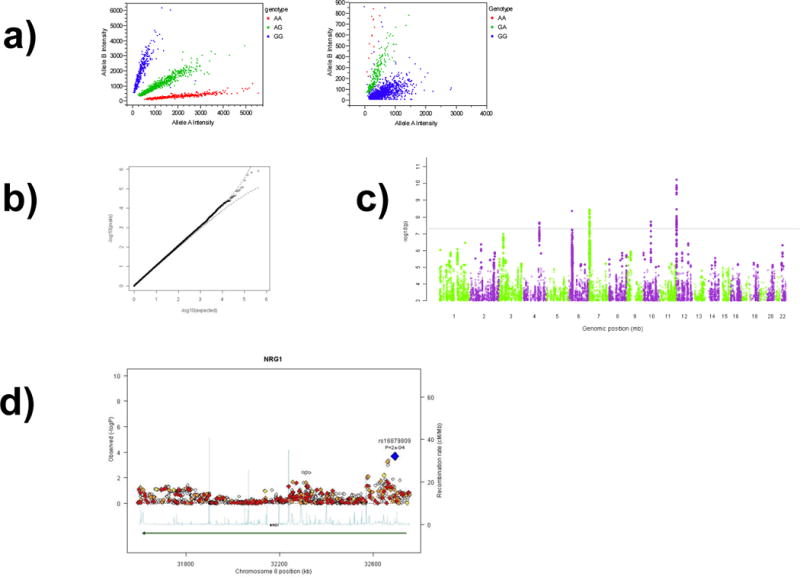
Images important for assessing a GWAS.
See text for description. These figures are from different studies. Figure 3a shows the allele intensity plots for two SNPs from which SNP genotype calls are generated. Figure 3b depicts a quantile-quantile plot in which the observed p-values are plotted against the p-value distribution expected by chance (on –log10 scale). Figure 3c shows a Manhattan plot. Figure 3d shows an expanded set of findings in the region of neuregulin 1 (NRG1).
GWAS Analysis
Quality Control (QC)
One of the most crucial and time-consuming steps in conducting a GWAS is QC, the removal of SNPs and subjects with unreliable data plus assessment of biases that might lead to spurious results. Excellent reviews of GWAS QC are available (McCarthy et al., 2008, Neale and Purcell, 2008, Attia et al., 2009b).
Individual SNPs are removed for any of the following reasons:
Imprecise mapping to the genome (some SNPs map to multiple places)
Excessive disagreement among duplicated samples
Excessive missing genotypes on subjects (e.g., > 5%)
Low minor allele frequency (e.g., < 1%)
Observed genotype frequencies deviate markedly from expectations (e.g., Hardy-Weinberg equilibrium p < 1 × 10−6)
After SNP removal, subjects are dropped due to any of the following:
Disagreement between chrX/chrY genotypes and phenotypic sex (usually indicating an unreliable link between genotype and phenotype data)
Excessive missing data (e.g., > 5%)
Inadvertent sample duplication or close relation (MZ twin or first- or second-degree) to some other subject
Ancestry outlier
Bias
Given hundreds of thousands of SNPs per subject, relatives are readily identified and excluded as their presence can lead to inflation of Type 1 error. Similarly, genome-wide data allow identification and control for the most infamous bias of a case-control study, inflation of Type 1 error due to population stratification. This occurs when cases and controls are mismatched by ancestry and disease prevalence differs by ancestry, and has been responsible for numerous false positive findings in the literature. With genome-wide SNP data, it is possible to identify individuals with divergent ancestry (even within a continental population); these individuals can be excluded or a statistical method used to control for this bias.
In addition, important bias can be introduced if samples from cases and controls were handled differently – e.g., if samples from cases are older, if DNA had been extracted with a different method from controls, or if cases were genotyped at a different place and time from controls. Careful assessment of these and other sources of bias is crucial to understanding the impact of a range of method artifacts.
Statistical Testing
Using the SNPs and subjects that passed QC, investigators generally use logistic regression with case-control status as the dependent variable and a single SNP as the predictor. The SNP is coded as 0, 1, or 2 (i.e., the number of copies of one of the two alleles) for an additive test with one degree of freedom. This analysis is repeated for each SNP for a million or more statistical tests. Some investigators include covariates in the logistic regression model like age, sex, or indicators of ancestry. In some instances, alternative genetic models are used (e.g., recessive or dominant) but most studies use a 1 degree of freedom additive test as the primary statistical test.
Multiple Testing
A typical GWAS for one disease includes one logistic regression per SNP, or 500,000 or more statistical tests. These tests are not all independent as SNPs that are located close to one another can be correlated due to linkage disequilibrium. Even so, with 105–106 statistical tests, very small p-values by conventional standards are expected by chance. As noted above, p-values < 5×10−8 (akin to a Bonferroni correction of the traditional 0.05 Type 1 error level for 1,000,000 statistical tests) (Pe’er et al., 2008) are generally required for significance. Experience suggests that findings more significant than this threshold tend to replicate well across studies. However, unless power is exceptional, it is generally incorrect to exclude a SNP from consideration if does not exceed this threshold. Indeed, some SNPs that are unimpressive in an initial study (e.g., p=0.001) can eventually replicate well and exceed the critical threshold. As emphasized above, replication is essential.
Visualization
The scale of a GWAS can be overwhelming, and many find it useful to use graphics to depict certain results. Figure 3b shows a quantile-quantile plot, a scatter plot of the p-values observed in a GWAS versus that expected by chance. To spread the graph out, the points are transformed using –log10(p-value) (e.g., 0.0001 or 10−4 becomes +4.0). In this instance, the plot shows that the observed p-values conform closely to the expected suggesting that no finding is individually impressive after accounting for multiple comparisons. Figure 3c shows a “Manhattan plot” (to some eyes, this resembles the night skyline of the Manhattan borough of New York City viewed from across the Hudson River), a depiction of all small p-values by genomic position. These results (from a different study than in Figure 3b), suggest that genomic regions on chromosomes 4, 6, 7, 10, and 12 exceed genome-wide significance. Figure 3d (again from a different study) shows an expanded view of a genomic region of interest (NRG1, neuregulin 1). The region of maximum signal on the right-hand side of the graph is quite far from the region suggested as a risk factor for schizophrenia (on the far left-hand side of the figure).
Imputation
Samples from the HapMap project have been genotyped for a very large number of SNPs. Under the assumption that these samples (e.g., the northern European subset) are comparable to members of a case-control collection, the combination of these datasets can be used to estimate (impute) genotypes in the case-control collection by treating it as a missing data problem. Thus, it is possible to increase the number of available genotypes from, for example, 500,000 directly assessed SNPs to 2 million directly genotyped and imputed SNPs. A major use of imputation is to allow direct comparison of case-control studies that were genotyped using different GWAS platforms. For many of the Affymetrix and Illumina platforms, the number of SNPs directly genotyped on both platforms is < 20%. Imputation is often an essential precursor for meta-analysis.
Bioinformatics
Two web resources for investigating psychiatric genetics findings are shown in Table 1 (Allen et al., 2008, Konneker et al., 2008). GWAS analyses described above take an agnostic approach to GWAS data. Experience gleaned from other diseases indicates that SNPs identified and confirmed by replication are not necessarily those with the smallest p-values in an initial study. Bioinformatics approaches can be useful in annotating and organizing GWAS SNP data to identify SNPs for replication. SNPs may be prioritized based on many additional types of information: previous genetic association data; by location in exons, putative functional regions of the genome, or in brain-expressed genes; or on the basis that the identified SNP allele has an effect on gene expression in brain (Xu and Taylor, 2009).
Pathway Analysis
Pathway analysis represents an alternative analytical approach to interrogating GWAS data. A number of formal pathway-based analytical methods have been described (Hong et al., 2009). Essentially, these methods attempt to establish if SNPs mapping to genes in a pathway show more evidence of association with a disorder than other SNPs in the GWAS study, or SNPs mapping to other pathways. Pathway refers to groups of genes that are similar in some way – e.g., highly expressed in a tissue like prefrontal cortex, crucial to a biological process like neuronal differentiation, etc. The approach can be applied to test for involvement of specific pathways, to perform a hypothesis-free test of many different pathways, or to investigate whether pre-identified risk genes may be involved in the same molecular pathway or process. Investigating at the level of molecular pathways rather than individual risk variants may offer several potential advantages by being robust to the effects of genetic heterogeneity or in reducing the total multiple testing burden in analysis. However, this approach is dependent on the quality of annotation of the pathways being investigated (which can be uncertain) and assumes that risk variation falls within genes. As mentioned in our description of GWAS studies for 37 human diseases, a large subset of identified genetic risk variation (43%) fell outside gene boundaries. Arguably the principal advantage of this approach is to establish additional information relating to function over and above the statistical SNP GWAS data. Implicating a molecular pathway in a disease process is likely to be more biologically informative than interpreting evidence of involvement of an anonymous genetic marker.
Meta-Analysis
Conducting meta-analysis – the combined analysis of summary results from multiple primary studies – is now known to be crucial in the identification of robust genetic signals. This general principle has been identified on multiple occasions as evidenced by studies of Crohn’s disease, type 2 diabetes mellitus, and type 1 diabetes mellitus (Barrett et al., 2008, Zeggini et al., 2008, Barrett et al., 2009). As discussed above, the PGC is conducting such analyses for psychiatric disorders.
However, a high-quality meta-analysis must confront and surmount numerous conceptual and technical issues. These issues include: the comparability of samples and phenotype definitions; quality control; imputation to a common genotype set with attention to strand and allele issues; statistical methods to combine data; visualization; bioinformatics; and follow-up strategies. De Bakker and colleagues provide a practical treatment of these issues (de Bakker et al., 2008).
Follow-Up Strategies
Assume that a GWAS identifies a highly reproducible and consistently replicated association with a genomic region: what next? The implications are discussed in the next section (ELSI) and additional follow-up experiments are described here (Ioannidis et al., 2009). The fundamental idea is to design experiments to develop a detailed understanding of how changes at the genetic level act and interact with the environment to alter risk of a psychiatric disorder. These experiments should be at multiple levels – DNA, RNA, protein, biological process, cell, local cell systems, organ, organ system, organism, and community levels are all potentially relevant.
These associations could be direct (i.e., the identified variant is the causal variant) but are more likely to be associated indirectly in that the identified variant is correlated with some other genomic variant. For indirect association, the causal variant could be some other SNP, a set of interacting SNPs, a haplotype, an insertion/deletion polymorphism, a CNV, or a more complex type of genetic variant. It is also wise to leave open the possibility of a causal genetic mechanism that is currently unknown. The genetic effects are highly likely to be subtle and probabilistic (and even conditionally dependent on external influences) rather than deterministic as with classical Mendelian disorders.
DNA
Broadly, genetic follow-up aims to validate and refine notable SNP associations to identify underlying causal variants and map their relation to clinical phenotypes. One approach, beyond simple replication, is to investigate an implicated genomic region at higher marker density to refine the association signal (fine-mapping). For other complex diseases this approach has met with mixed results, suggesting that in many cases the impact of risk variants on common disease phenotypes is complex and not necessarily related to obvious effects on gene function, such as alteration of protein structure. This may relate to the limited coverage achieved by these studies, but it has been estimated that by direct genotyping and imputation a large percentage (>85%) of common SNP variation is already being assayed by GWAS studies although this can vary markedly by genomic region.
Many investigators would conduct regional “deep” re-sequencing of large numbers of cases and controls to discover previously unknown genetic variants. The emerging technology of genome resequencing has shown that there are usually an array of undiscovered genetic variants. A more detailed understanding of genetic variation in human populations will soon be available through the 1000 Genomes Project (http://www.1000genomes.org) which is performing genomic resequencing of > 1000 people from around the world. This is likely to prove very informative in guiding fine-mapping studies and potentially untangling more complex effects on phenotype.
Alternative genetic mechanisms may also contribute to disease and disruption of the same genes or molecular pathways by different mechanisms are likely to be relevant to the consequent phenotype. Follow-up strategies are increasingly using GWAS results to test other genetic risk mechanisms such as involvement of copy-number variation (CNV), the cumulative impact of CNV burden (e.g., the number of CNVs), and the cumulative impact of hundreds or thousands of SNP genotypes. In addition, investigators are actively working to assess the cumulative impact of individually rare risk alleles and epigenetic phenomena such as methylation.
RNA
A potentially useful gene annotation is whether a genetic variant leads to changes in RNA abundance or structure. This so-called expression QTL approach is in its early stages, but refinement and larger studies could give investigators a useful set of initial hypotheses should an associated region be shown to alter messenger RNA for a nearby gene. These data can also be used to answer a crucial question – to what gene does an associated SNP “belong”? Investigators usually assume that a SNP exerts its immediate effect on a gene it is in or near. In general, this assumption may be reasonable, but there are examples where this assumption is incorrect (e.g., lactase persistence is due to MCM6 intronic variation, ~14kb from the lactase gene). Moreover, 23% of GWAS hits are >20 kb from known genes. Fascinating examples include the 8q24 “gene desert” (30–500 kb from MYC) that is robustly associated with multiple different cancers and a 5p14 region with replicated associations with autism but ~1 Mb from the nearest gene.
Molecular and cellular biology
There are many powerful technologies that could be brought to bear. These approaches are too numerous to describe succinctly and their choice depends critically on the details of a genomic variant. In many instances, use of transgenic manipulation (knock-out or humanizing knock-in approaches) of non-human model organisms (mouse or worm in particular) might be used to gain greater understanding of the impact of a genomic variant.
Clinical
Risk variants identified by GWAS are individually likely to be of modest effect which poses challenges for clinical follow up studies. These are not insurmountable, but are at the present time dependent on the availability of detailed phenotypic information from subjects involved in GWAS studies or the ability to re-contact subjects for additional studies. Recent efforts in schizophrenia, demonstrate the application of a phenotype refinement approach, in this case identifying a disturbed neural connectivity phenotype in carriers of the risk allele at ZNF804A using a neuroimaging approach (Esslinger et al., 2009). If disorders are highly polygenic it could be possible to group participants into classes based on total burden of risk variation or contribution from different functional pathways. Such groupings could than be used within a disorder, or across current diagnostic boundaries, to investigate clinical profiles, cognitive functioning, drug response or clinical outcome. By extension, such approaches could also be applied to investigate gene-environment interaction in risk. The optimum approach would be integration of genetic and epidemiological research to investigate, prospectively, the effects of risk genes and gene-environment interaction in prospective studies or within high-risk groups.
Controversies
GWAS efforts have been subject to multiple criticisms, both for the method generally and with respect to psychiatric disorders. Criticism has been welcomed, particularly those that are based on empirical data and not opinion. One initial criticism – that GWAS will not work in the sense of identifying any replicable associations – has been robustly disproved as GWAS clearly “works” for a broad range of biomedical disorders. The crucial question for our field is whether GWAS will “work” for psychiatric disorders (as discussed above, there is positive evidence that it has). Common criticisms of GWAS are listed below. All have been articulated at length and strong counter-arguments to each have been articulate (see Table 1 for citations).
Phenotype criticisms – the clinically-derived DSM and ICD systems are merely descriptive. The disorders are too heterogeneous and imprecisely defined – i.e., investigating “schizophrenia” is like studying “cancer” or “fever”.
Genetic model criticisms. The vast majority of GWAS employ perhaps the simplest conceivable model, a test for the additive effect of a single, relatively common SNP variant on the phenotype. Some have argued that this model is completely wrong, that risk for psychiatric disease is entirely something else – i.e., risk is entirely due to rare variants, epigenetic modifications, etc.
The “so what” criticism. Some have argued that robust GWAS findings cannot contribute to individualized medicine and thus do not matter.
An empirically-based criticism of GWAS is that the current genotyping technologies miss potentially important genetic variation (e.g., a subset of common variants, a large proportion of rare variants, non-SNP genetic variants like insertion-deletion polymorphisms, and are not optimal for CNV detection).
Ethical, Legal, & Social Implications (ELSI)
Major scientific advances in the molecular genetic understanding of psychiatric illness are associated with important ethical issues that must be considered carefully. Although many issues in psychiatric genetics are no different from those for other complex disorders, this combination of genetics and mental illness justifiably receives close scrutiny of ethical and psychosocial issues. It is well known that behavior genetics research has been misused in the past, most notoriously to support Nazi claims of racial superiority which had an important role in the Holocaust. It is, therefore, extremely important that relevant issues are considered and debated as early as possible and, where appropriate, ethical guidance and legal frameworks put in place to protect individuals and society against potential misuse of the new technologies and data. In recognition of its major importance, ELSI was an integral component of the Human Genome Project from its inception.
Key ethical issues under current debate include the need for new approaches to informed consent for large-scale genetic studies and consideration of the legal issues relating to confidentiality and use of genetic data. For example, under what circumstances (if any) might it be useful or appropriate to use genetic data in a court case to support an argument about responsibility for a behavior? Should insurance companies or employers have access to genetic data that inform risk of mental illness? How can we prevent genetic results being used to reify racist, sexist, or other stigmatizing biases? Quite apart from these potential non-medical uses of genetic data, there is the important question of whether and when genetic tests may be useful clinically – to help in confirming diagnosis; to direct management in a patient with signs of illness; or to predict risk in a person without signs of illness. At present risk variants have not been robustly established that would provide clinically useful individual predictive power and it may well be many years in the future before this is possible. Nonetheless we need to think through the issues in advance of the scientific and technical reality. It is highly desirable that the clinical usefulness of any genetic test is demonstrated before it is made widely available. “Direct to consumer” genetic tests of spurious clinical usefulness are already available commercially so there is an urgent need to develop frameworks and guidelines for best practice.
As well as the continuing public debate, consultation and education on these issues, there is a need for scrupulous integrity by scientists in the way they present research findings. Reports should be appropriately cautious, balanced and free from “hype”, “spin” or commercial bias.
The exciting challenge for psychiatry in the coming years is to ensure that a revolution in understanding of the biology of mental illness is translated into a revolution in clinical care. The important challenge for society is to ensure that new knowledge and powerful technologies are not misused.
Acknowledgments
Funding was from the US National Institute of Mental Health (MH085520, MH080403, MH077139, MH081802, and MH074027).
Footnotes
Conflicts of Interest
In the interests of full disclosure, Dr. Sullivan reports receiving unrestricted research funding from Eli Lilly for genetic research in schizophrenia. The other authors report no conflicts.
References
- Allen N, Bagade S, McQueen M, Ioannidis J, Kavvoura F, Khoury M, Tanzi R, Bertram L. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: The szgene database. Nature Genetics. 2008;40:827–34. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- Arranz MJ, Kapur S. Pharmacogenetics in psychiatry: Are we ready for widespread clinical use? Schizophrenia Bulletin. 2008;34:1130–44. doi: 10.1093/schbul/sbn114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attia J, Ioannidis JP, Thakkinstian A, McEvoy M, Scott RJ, Minelli C, Thompson J, Infante-Rivard C, Guyatt G. How to use an article about genetic association: A: Background concepts. Journal of the American Medical Association. 2009a;301:74–81. doi: 10.1001/jama.2008.901. [DOI] [PubMed] [Google Scholar]
- Attia J, Ioannidis JP, Thakkinstian A, McEvoy M, Scott RJ, Minelli C, Thompson J, Infante-Rivard C, Guyatt G. How to use an article about genetic association: B: Are the results of the study valid? Journal of the American Medical Associaton. 2009b;301:191–7. doi: 10.1001/jama.2008.946. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, Plagnol V, Pociot F, Schuilenburg H, Smyth DJ, Stevens H, Todd JA, Walker NM, Rich SS. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nature Genetics. 2009 doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ. Genome-wide association defines more than 30 distinct susceptibility loci for crohn’s disease. Nature Genetics. 2008 doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis G, Altshuler D, Bailey-Wilson JE, Brooks LD, Cardon LR, Daly M, Donnelly P, Fraumeni JF, Jr, Freimer NB, Gerhard DS, Gunter C, Guttmacher AE, Guyer MS, Harris EL, Hoh J, Hoover R, Kong CA, Merikangas KR, Morton CC, Palmer LJ, Phimister EG, Rice JP, Roberts J, Rotimi C, Tucker MA, Vogan KJ, Wacholder S, Wijsman EM, Winn DM, Collins FS. Replicating genotype-phenotype associations. Nature. 2007;447:655–60. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- Collier DA. Schizophrenia: The polygene princess and the pea. Psychological Medicine. 2008;38:1687–91. doi: 10.1017/S0033291708003668. discussion 1818–20. [DOI] [PubMed] [Google Scholar]
- Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–23. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- Craddock N, O’Donovan MC, Owen MJ. Phenotypic and genetic complexity of psychosis. British Journal of Psychiatry. 2007;190:200–3. doi: 10.1192/bjp.bp.106.033761. [DOI] [PubMed] [Google Scholar]
- Cross Disorder Phenotype Group of the Psychiatric GWAS Consortium. Dissecting the phenotype in genome-wide association studies of psychiatric illness. British Journal of Psychiatry. 2009;195:97–9. doi: 10.1192/bjp.bp.108.063156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow TJ. Schizophrenia: The polygene emperors have no clothes. Psychological Medicine. 2008 doi: 10.1017/S0033291708003395. [DOI] [PubMed] [Google Scholar]
- de Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Human Molecular Genet. 2008;17:R122–8. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esslinger C, Walter H, Kirsch P, Erk S, Schnell K, Arnold C, Haddad L, Mier D, Opitz von Boberfeld C, Raab K, Witt SH, Rietschel M, Cichon S, Meyer-Lindenberg A. Neural mechanisms of a genome-wide supported psychosis variant. Science. 2009;324:605. doi: 10.1126/science.1167768. [DOI] [PubMed] [Google Scholar]
- Ferreira M, O’Donovan M, Meng Y, Jones I, Ruderfer D, Jones L, Fan J, Kirov G, Perlis R, Green E, Smoller J, Grozeva D, Stone J, Nikolov I, Chambert K, Hamshere M, Nimgaonkar V, Moskvina V, Thase M, Caesar S, Sachs G, Franklin J, Gordon-Smith K, Ardlie K, Gabriel S, Fraser C, Blumenstiel B, Defelice M, Breen G, Gill M, Morris D, Elkin A, Muir W, McGhee K, Williamson R, MacIntyre D, McLean A, St Clair D, VanBeck M, Pereira A, Kandaswamy R, McQuillin A, Collier D, Bass N, Young A, Lawrence J, Ferrier I, Anjorin A, Farmer A, Curtis D, Scolnick E, McGuffin P, Daly M, Corvin A, Holmans P, Blackwood D, Gurling H, Owen M, Purcell S, Sklar P, Craddock N. Collaborative genome-wide association analysis of 10,596 individuals supports a role for ankyrin-g (ank3) and the alpha-1c subunit of the l-type voltage-gated calcium channel (cacna1c) in bipolar disorder. Nature Genetics. 2008 doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB. Common genetic variation and human traits. New England Journal of Medicine. 2009;360:1696–8. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- Hardy J, Singleton A. Genomewide association studies and human disease. New England Journal of Medicine. 2009;360:1759–68. doi: 10.1056/NEJMra0808700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn JN. Genomewide association studies–illuminating biologic pathways. New England Journal of Medicine. 2009;360:1699–701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- Holmans P, Green EK, Pahwa JS, Ferreira MA, Purcell SM, Sklar P, Owen MJ, O’Donovan MC, Craddock N. Gene ontology analysis of gwa study data sets provides insights into the biology of bipolar disorder. American Journal of Human Genetics. 2009;85:13–24. doi: 10.1016/j.ajhg.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong MG, Pawitan Y, Magnusson PK, Prince JA. Strategies and issues in the detection of pathway enrichment in genome-wide association studies. Human Genetics. 2009 doi: 10.1007/s00439-009-0676-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009 doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis JP, Thomas G, Daly MJ. Validating, augmenting and refining genome-wide association signals. Nature Reviews Genetics. 2009;10:318–29. doi: 10.1038/nrg2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye J. The regulation of direct-to-consumer genetic tests. Human Molecular Genetics. 2008;17:R180–3. doi: 10.1093/hmg/ddn253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS. Reflections on the relationship between psychiatric genetics and psychiatric nosology. American Journal of Psychiatry. 2006;163:1138–46. doi: 10.1176/ajp.2006.163.7.1138. [DOI] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, Sangiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor h polymorphism in age-related macular degeneration. Science. 2005;308:385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konneker T, Barnes T, Furberg H, Losh M, Bulik CM, Sullivan PF. A searchable database of genetic evidence for psychiatric disorders. American Journal of Medical Genetics (Neuropsychiatric Genetics) 2008;147:671–5. doi: 10.1002/ajmg.b.30802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft P, Hunter DJ. Genetic risk prediction–are we there yet? New England Journal of Medicine. 2009;360:1701–3. doi: 10.1056/NEJMp0810107. [DOI] [PubMed] [Google Scholar]
- Lichtenstein P, Yip B, Bjork C, Pawitan Y, Cannon TD, Sullivan PF, Hultman CM. Common genetic influences for schizophrenia and bipolar disorder: A population-based study of 2 million nuclear families. Lancet. 2009;373:234–9. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunshof JE, Chadwick R, Vorhaus DB, Church GM. From genetic privacy to open consent. Nature Reviews Genetics. 2008;9:406–11. doi: 10.1038/nrg2360. [DOI] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nature Reviews Genetics. 2008;9:356–69. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- Mitchell K, Porteus D. Gwas for psychiatric disease: Is the framework built on a solid foundation? Molecular Psychiatry. 2009;14:740–1. doi: 10.1038/mp.2009.17. [DOI] [PubMed] [Google Scholar]
- Neale BM, Purcell S. The positives, protocols, and perils of genome-wide association. American Journal of Medical Genetics (Neuropsychiatric Genetics) 2008;147B:1288–94. doi: 10.1002/ajmg.b.30747. [DOI] [PubMed] [Google Scholar]
- Nussbaum R, McInnes R, Willard HF. Thompson & thompson genetics in medicine. Elsevier Science; New York: 2007. [Google Scholar]
- O’Donovan M, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, Nikolov I, Hamshere M, Carroll L, Georgieva L, Dwyer S, Holmans P, Marchini J, Spencer C, Howie B, Leung H-T, Hartmann A, Möller H-J, Morris D, Shi Y, Feng G, Hoffmann P, Propping P, Vasilescu C, Maier W, Rieschel M, Zammit S, Schumacher J, Quinn E, Schulze T, Williams N, Giegling I, Iwata N, Ikeda M, Darvasi A, Shifman S, He L, Duan J, Sanders A, Levinson D, Gejman P, Cichon S, Nöthen M, Gill M, Corvin A, Rujescu D, Kirov G, Owen M. Identification of novel schizophrenia loci by genome-wide association and follow-up. Nature Genetics. 2008a;40:1053–5. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- O’Donovan MC, Craddock N, Owen MJ. Schizophrenia: Complex genetics, not fairy tales. Psychological Medicine. 2008b;38:1697–9. doi: 10.1017/S0033291708003802. discussion 1818–20. [DOI] [PubMed] [Google Scholar]
- Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genetic Epidemiology. 2008;32:381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- Psychiatric GWAS Consortium. A framework for interpreting genomewide association studies of psychiatric disorders. Molecular Psychiatry. 2009a;14:10–7. doi: 10.1038/mp.2008.126. [DOI] [PubMed] [Google Scholar]
- Psychiatric GWAS Consortium. Genome-wide association studies: History, rationale, and prospects for psychiatric disorders. American Journal of Psychiatry. 2009b;166:540–6. doi: 10.1176/appi.ajp.2008.08091354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman KJ. Modern epidemiology. Little, Brown and Company; Boston: 1986. [Google Scholar]
- Rothstein MA. Science and society: Applications of behavioural genetics: Outpacing the science? Nature Reviews Genetics. 2005;6:793–8. doi: 10.1038/nrg1687. [DOI] [PubMed] [Google Scholar]
- Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson K, Isomaa B, Lettre G, Lindblad U, Lyon HN, Melander O, Newton-Cheh C, Nilsson P, Orho-Melander M, Rastam L, Speliotes EK, Taskinen MR, Tuomi T, Guiducci C, Berglund A, Carlson J, Gianniny L, Hackett R, Hall L, Holmkvist J, Laurila E, Sjogren M, Sterner M, Surti A, Svensson M, Tewhey R, Blumenstiel B, Parkin M, Defelice M, Barry R, Brodeur W, Camarata J, Chia N, Fava M, Gibbons J, Handsaker B, Healy C, Nguyen K, Gates C, Sougnez C, Gage D, Nizzari M, Gabriel SB, Chirn GW, Ma Q, Parikh H, Richardson D, Ricke D, Purcell S. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–6. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- Scherer SW, Lee C, Birney E, Altshuler DM, Eichler EE, Carter NP, Hurles ME, Feuk L. Challenges and standards in integrating surveys of structural variation. Nature Genetics. 2007;39:S7–15. doi: 10.1038/ng2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesselman JJ. Case-control studies: Design, conduct, analysis. Oxford University Press; New York: 1982. [Google Scholar]
- Schulze TG, McMahon FJ. Defining the phenotype in human genetic studies: Forward genetics and reverse phenotyping. Human Heredity. 2004;58:131–8. doi: 10.1159/000083539. [DOI] [PubMed] [Google Scholar]
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M. A genome-wide association study of type 2 diabetes in finns detects multiple susceptibility variants. Science. 2007;316:1341–5. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, Dudbridge F, Holmans PA, Whittemore AS, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Buccola NG, Byerley WF, Black DW, Crowe RR, Oksenberg JR, Mirel DB, Kendler KS, Freedman R, Gejman PV. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009 doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, Werge T, Pietilainen OP, Mors O, Mortensen PB, Sigurdsson E, Gustafsson O, Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Suvisaari J, Lonnqvist J, Paunio T, Borglum AD, Hartmann A, Fink-Jensen A, Nordentoft M, Hougaard D, Norgaard-Pedersen B, Bottcher Y, Olesen J, Breuer R, Moller HJ, Giegling I, Rasmussen HB, Timm S, Mattheisen M, Bitter I, Rethelyi JM, Magnusdottir BB, Sigmundsson T, Olason P, Masson G, Gulcher JR, Haraldsson M, Fossdal R, Thorgeirsson TE, Thorsteinsdottir U, Ruggeri M, Tosato S, Franke B, Strengman E, Kiemeney LA, Group Melle I, Djurovic S, Abramova L, Kaleda V, Sanjuan J, de Frutos R, Bramon E, Vassos E, Fraser G, Ettinger U, Picchioni M, Walker N, Toulopoulou T, Need AC, Ge D, Lim Yoon J, Shianna KV, Freimer NB, Cantor RM, Murray R, Kong A, Golimbet V, Carracedo A, Arango C, Costas J, Jonsson EG, Terenius L, Agartz I, Petursson H, Nothen MM, Rietschel M, Matthews PM, Muglia P, Peltonen L, St Clair D, Goldstein DB, Stefansson K, Collier DA, Kahn RS, Linszen DH, van Os J, Wiersma D, Bruggeman R, Cahn W, de Haan L, Krabbendam L, Myin-Germeys I. Common variants conferring risk of schizophrenia. Nature. 2009 doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachen T, Read AP. Human molecular genetics. John Wiley and Sons; NY: 2003. [Google Scholar]
- Sullivan PF. The dice are rolling for schizophrenia genetics. Psychological Medicine. 2008 doi: 10.1017/S003329170800367X. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Gejman PV. Response to mitchell & porteus, mol psych (2009) 14, 740-1. Molecular Psychiatry. doi: 10.1038/mp.2009.106. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, Kim CE, Hou C, Frackelton E, Chiavacci R, Takahashi N, Sakurai T, Rappaport E, Lajonchere CM, Munson J, Estes A, Korvatska O, Piven J, Sonnenblick LI, Alvarez Retuerto AI, Herman EI, Dong H, Hutman T, Sigman M, Ozonoff S, Klin A, Owley T, Sweeney JA, Brune CW, Cantor RM, Bernier R, Gilbert JR, Cuccaro ML, McMahon WM, Miller J, State MW, Wassink TH, Coon H, Levy SE, Schultz RT, Nurnberger JI, Haines JL, Sutcliffe JS, Cook EH, Minshew NJ, Buxbaum JD, Dawson G, Grant SF, Geschwind DH, Pericak-Vance MA, Schellenberg GD, Hakonarson H. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–33. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WTCCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Taylor JA. Snpinfo: Integrating gwas and candidate gene information into functional snp selection for genetic association studies. Nucleic Acids Research. 2009 doi: 10.1093/nar/gkp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PI, Abecasis GR, Almgren P, Andersen G, Ardlie K, Bostrom KB, Bergman RN, Bonnycastle LL, Borch-Johnsen K, Burtt NP, Chen H, Chines PS, Daly MJ, Deodhar P, Ding CJ, Doney AS, Duren WL, Elliott KS, Erdos MR, Frayling TM, Freathy RM, Gianniny L, Grallert H, Grarup N, Groves CJ, Guiducci C, Hansen T, Herder C, Hitman GA, Hughes TE, Isomaa B, Jackson AU, Jorgensen T, Kong A, Kubalanza K, Kuruvilla FG, Kuusisto J, Langenberg C, Lango H, Lauritzen T, Li Y, Lindgren CM, Lyssenko V, Marvelle AF, Meisinger C, Midthjell K, Mohlke KL, Morken MA, Morris AD, Narisu N, Nilsson P, Owen KR, Palmer CN, Payne F, Perry JR, Pettersen E, Platou C, Prokopenko I, Qi L, Qin L, Rayner NW, Rees M, Roix JJ, Sandbaek A, Shields B, Sjogren M, Steinthorsdottir V, Stringham HM, Swift AJ, Thorleifsson G, Thorsteinsdottir U, Timpson NJ, Tuomi T, Tuomilehto J, Walker M, Watanabe RM, Weedon MN, Willer CJ, Illig T, Hveem K, Hu FB, Laakso M, Stefansson K, Pedersen O, Wareham NJ, Barroso I, Hattersley AT, Collins FS, Groop L, McCarthy MI, Boehnke M, Altshuler D. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nature Genetics. 2008;40:638–45. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]