

Abstract
Histone deacetylases (HDACs) have been implicated in the pathogenesis of kidney diseases including diabetic nephropathy (DN); however, the underlying mechanism is poorly understood. In this issue, Wang et. al. have unraveled the changes in expression of various HDACs in DN and demonstrated that HDAC4 specifically contributes to podocyte injury in this disease. Mechanistically, HDAC4 deacetylates STAT1 to suppress autophagy, an essential cellular process for the function and viability of podocytes. The development of HDAC isoform-specific inhibitors may provide efficacious therapeutics for DN and related renal diseases.
Diabetic nephropathy (DN) is the leading cause of end stage renal disease. Pathologically, DN is characterized by abnormalities in all main kidney compartments, including mesangial expansion, basement membrane thickening, podocyte injury, capillary defects, tubular hypertrophy, and interstitial fibrosis1. Recent studies have suggested an emerging role of histone deacetylase (HDAC)-mediated epigenetic modifications in DN 2–5. However, it is known that the function of HDACs is not limited to epigenetic regulation by deacetylating histones as they may also control the acetylation status of other proteins of patho-physiological significance. In this issue, Wang and colleagues provide evidence that HDAC4, by deacetylating of STAT1, suppresses autophagy in podocytes, contributing to podocyte injury and DN 6.
The HDAC family in mammalian cells consists of at least 18 deacetylating enzymes, which are grouped into four classes based on the similarity to their yeast orthologs. Among them, 11 HDACs are Zn2+-dependent and can be further divided into class I, II, and IV according to their structure, sequence homology, and domain organization. In comparison, the class III HDACs (also known as the sirtuins, SIRT1–7) are NAD+-dependent and related to the Sir2 gene. While HDACs were originally identified as the enzymes removing the acetyl groups from ε amino groups of lysine residues in histones, it is now clear that they also deacetylate a growing number of non-histone proteins. By controlling their acetylation status, HDACs may regulate the conformation, localization, molecular interaction, and function of their target proteins. In experimental models of DN, Sirt1, a class III HDAC, was reported to be renal protective through the suppression of Claudin-1 in podocytes 4. In contrast, class I, II and IV HDACs may be pathogenic in DN as suggested by the beneficial effects of HDAC inhibitors 2, 3, 5. Trichostatin A, a nonselective HDAC inhibitor, was shown to ameliorate extracellular matrix deposition and early proteinuria following streptozotocin (STZ)-induced DN in rats. Vorinostat (also known as suberanilohydroxamic acid, SAHA) reduced tubular and glomerular hypertrophy, resulting in the arrest of increase in kidney size in STZ-model of DN 3. In addition, vorinostat decreased albuminuria, mesangiolysis, and collagen deposition in related models. Of note, these inhibitors block the activity of multiple HDACs of class I, II and/or IV typically by binding to their Zn2+-containing catalytic domains. Therefore, it is unclear the inhibition of which HDAC(s) accounts for their renal protective effect. Moreover, whether HDACs exert the effect by epigenetic (via deacetylating histones) and/or non-epigenetic (via deacetylating non-histone proteins) mechanisms remains elusive.
Wang and colleagues analyzed the expression profile of HDACs in DN in a systemic manner 6. Among all HDACs examined, the expression levels of HDAC2, 4, and 5 were shown to negatively correlate with estimated glomerular filtration rate (eGFR) in individual patients. Consistently, these three HDACs were up-regulated in kidney tissues of STZ-induced rats as well as diabetic db/db mice. Notably, in response to hyperglycemia different cell types of renal parenchyma showed different HDAC expression patterns, suggesting that these HDACs may serve distinct roles in these cells in DN. Specifically, HDAC2 was elevated in proximal tubular cells, whereas HDAC4 was dramatically increased in podocytes and HDAC5 in mesangial cells, respectively. Of note, increased HDAC4 expression was not limited to DN, but was also observed in the kidney biopsies from patients with focal segmental glomerulosclerosis (FSGS) 6, supporting the possibility of HDAC4 up-regulation as a common feature of podocytopathy that may be targeted for therapy. Mechanistically, HDAC4 may contribute to glomerular lesion and proteinuria by inducing a pro-inflammatory response and apoptosis and notably, by suppressing autophagy in podocytes.
The observation of HDAC4-mediated suppression of autophagy is particularly interesting, because autophagy has been recently shown to be essential to the maintenance of cellular homeostasis in podocytes 7. Autophagy is an intracellular process of the degradation of cytoplasmic components including protein aggregates, lipid drops as well as dysfunctional organelles. Although originally discovered as a mechanism of recycling nutrients during starvation, autophagy is now recognized to be a general cellular response to a variety of stress that helps the cells to cope with ensuing injury. A complex molecular machinery centered on a multitude of autophagy-related genes (Atg) is responsible for autophagic induction, autophagosome formation, lysosomal fusion, and finally lysosomal degradation. Podocytes have a remarkably high level of basal autophagy and deletion of Atg5 in these cells leads to pathological protein aggregation and ER stress, resulting in podocyte loss, proteinuria and characteristic glomerulopathy in aging mice. Together, these finding suggest that autophagy is a critical mechanism for the maintenance of the homeostasis, function and viability of podocytes under both physiological and pathological conditions 7. Interestingly, there is recent evidence for a decline of autophagy in kidney cells and tissues in DN 8. In STZ-induced diabetic mice, kidney tissues showed a time-dependent decrease in several Atgs, including beclin-1, LC3 and Atg5-Atg12 complex. Similar observations were shown during high glucose incubation of cultured podocytes. Rapamycin, which promotes autophagy by inhibiting mTOR, was shown to be partially effective in protecting against podocytes injury during high glucose incubation 8. Wang and colleagues have now further verified the defective autophagy in podocytes in experimental models of DN. Together, these and other related studies support a therapeutic potential of enhancing autophagy in DN. Despite these findings, definitive evidence for the role of autophagy in DN remains to be demonstrated and the mechanism whereby autophagy becomes defective in podocytes in DN is largely unknown. By demonstrating a role of HDAC4 in autophagy suppression in podocytes in DN, Wang and colleagues have solved one piece of the puzzle 6.
HDACs have been implicated in the regulation of autophagy in cancer models. For example, HDAC6 was shown to promote autophagy by deacetylating LC3B-II, resulting in tumor suppression 9. In this regard, Wang and colleagues have demonstrated the first evidence for regulation of autophagy by HDACs in renal diseases. Specifically, they showed that inhibition of HDAC4 can prevent autophagic defects in cultured podocytes and kidneys under diabetic conditions. Functionally, the maintenance of autophagy by HDAC4 inhibition was accompanied by amelioration of podocyte injury/death and attenuation of glomerulopathy, including albuminuria, podocytes loss and mesangial expansion 6.
How does HDAC4 suppress autophagy? HDACs, as implied by the name, have a predominant function of histone deacetylation, allowing histones to wrap DNA tightly to prevent gene transcription. While this function is important for epigenetic regulation, HDACs are also known to play non-epigenetic roles by deacetylating non-histone proteins. A growing number of non-histone proteins, like p53 and LC3II-B as mentioned above, have been identified as substrates of HDACs and deacetylation of these proteins may regulate their stability, localization, and activity or function. In their study 6, Wang and colleagues demonstrated a molecular interaction between HDAC4 and STAT1, a signaling module that was recently suggested to inhibit autophagy in the heart 10. STAT1 is a member of the STAT family of transcription factors, which are activated by cytokine binding to their cell surface receptors, leading to STAT phosphorylation, dimerization and translocation to the nucleus to initiate gene transcription. Recent research has further discovered a pivotal role of deacetylation in the regulation of STAT1. Essentially, STAT1 has to be deacetylated to permit its phosphorylation and activation. In kidneys, STAT1 signaling contributes to renal injury in DN. Putting these observations together, it is suggested that HDAC4-mediated deacetylation enhances the phosphorylation of STAT1, which then dimerizes and translocates into the nucleus to initiate the transcription of a variety of genes that may induce apoptosis and inflammation, and suppress autophagy, culminating in podocyte injury (Fig. 1).
Figure 1. Schematic diagram of HDAC4/STAT1 pathway in podocytes injury.
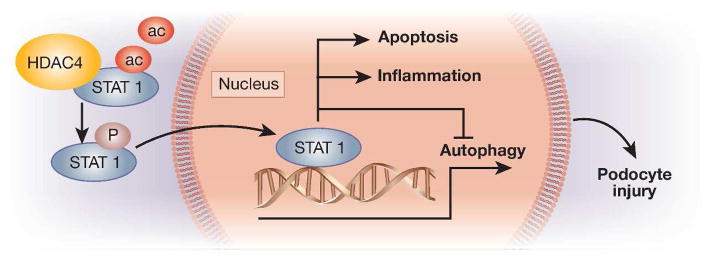
HDAC4 interacts with and de-acetylates STAT1 to promote the phosphorylation and activation of STAT1, which then transolcates into the nucleus to induce gene expression, leading to the induction of inflammation and apoptosis and the suppression of autophagy.
Despite these interesting findings and ideas, a number of questions remain to be answered. For example, the genes and signaling pathways that are responsible for HDAC4/STAT1-mediated autophagy suppression need to be delineated. Several autophagy genes, such as beclin-1 and LC3, were shown to be down-regulated in DN. Are they direct transcriptional targets of STAT1? Moreover, autophagy is carried out by a complex molecular machinery involving multiple protein complexes. Can HDAC4 suppress autophagy by directly deacetylating some of the autophagy proteins? In addition, while the non-epigenetic regulation of autophagy by HDAC4 is important, it would be interesting to know whether epigenetic mechanism via HDAC4-mediated histone deacetylation also contributes to podocytes injury and DN and, if yes, to what extents. Finally, how is HDAC4 up-regulated in podocytes in DN and related disease conditions?
In addition to HDAC4 in podocytes, Wang and colleagues also revealed the up-regulation of HDAC2 and HDAC5 by DN-related conditions in tubular and mesangial cells, respectively 6. Although podocytes injury is important to glomerulopathy, DN is characterized by pathological changes in most, if not all, renal compartments. It is therefore intriguing that HDAC2 may be specifically up-regulated in renal tubular cells and HDAC5 in mesangial cells. In DN, tubular hypertrophy is one of the earliest pathological features, which may be associated with the subsequent glomerular changes 1. Furthermore, HDAC2 up-regulation has been detected in renal tubular cells in a variety of kidney injury models including DN, unilateral ureteral obstruction, and septic acute kidney injury2. HDAC2 has been shown to regulate cell proliferation and the STAT3 signaling pathway in renal interstitial fibroblasts11, but whether and how HDAC2 regulates tubular pathology under disease conditions such as DN remain to be investigated. Similarly, it would be interesting and potentially important to determine the role of HDAC5 in the pathogenesis of DN, including mesangial expansion. Pan-HDAC inhibitors, such as vorinostat and trichostatin A, showed beneficial effects in experimental models of DN 2, 3, 5. However, considering the different expression patterns of HDAC isoforms in different kidney cells, it is imperative to develop inhibitors with high specificity towards individual HDACs for therapeutic purpose. Such inhibitors may show lower side effects and exhibit significantly higher therapeutic efficacy by specifically targeting the pathogenic HDAC(s).
Acknowledgments
We thank Dr. Fan Yi at Shandong University School of Medicine (Jinan, China) for suggestions. The work was supported in part by grants from National Basic Research Program of China 973 Program No. 2012CB517600 (No. 2012CB517601), National Natural Science Foundation of China [81370791], and the National Institutes of Health and Department of Veterans Administration of USA.
Footnotes
Disclosure: The authors declared no competing interests.
References
- 1.Kanwar YS, Sun L, Xie P, et al. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Ann Rev Pathol. 2011;6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noh H, Oh EY, Seo JY, et al. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am J Physiol Renal Physiol. 2009;297:F729–739. doi: 10.1152/ajprenal.00086.2009. [DOI] [PubMed] [Google Scholar]
- 3.Gilbert RE, Huang Q, Thai K, et al. Histone deacetylase inhibition attenuates diabetes-associated kidney growth: potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 2011;79:1312–1321. doi: 10.1038/ki.2011.39. [DOI] [PubMed] [Google Scholar]
- 4.Hasegawa K, Wakino S, Simic P, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nature Med. 2013;19:1496–1504. doi: 10.1038/nm.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Advani A, Huang Q, Thai K, et al. Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am J Pathol. 2011;178:2205–2214. doi: 10.1016/j.ajpath.2011.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Liu J, Zhen J, et al. HDAC4 selectively contributes to podocyte injury in diabetic nephropathy: Mechanism and therapeutic potential. Kidney Int. 2014 doi: 10.1038/ki.2014.111. [DOI] [PubMed] [Google Scholar]
- 7.Fang L, Zhou Y, Cao H, et al. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS One. 8(4):e60546. doi: 10.1371/journal.pone.0060546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartleben B, Godel M, Meyer-Schwesinger C, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu KP, Zhou D, Ouyang DY, et al. LC3B-II deacetylation by histone deacetylase 6 is involved in serum-starvation-induced autophagic degradation. Biochem Biophys Res Com. 2013;441:970–975. doi: 10.1016/j.bbrc.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 10.McCormick J, Suleman N, Scarabelli TM, et al. STAT1 deficiency in the heart protects against myocardial infarction by enhancing autophagy. J Cell Mol Med. 2012;16:386–393. doi: 10.1111/j.1582-4934.2011.01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pang M, Ma L, Liu N, et al. Histone deacetylase 1/2 mediates proliferation of renal interstitial fibroblasts and expression of cell cycle proteins. J Cell Biochem. 2011;112:2138–2148. doi: 10.1002/jcb.23135. [DOI] [PMC free article] [PubMed] [Google Scholar]