

Abstract
General anesthesia is a neurophysiological state that consists of unconsciousness, amnesia, analgesia, and immobility along with maintenance of physiological stability. Despite use of general anesthesia in the United States for more than 166 years, how these drugs act in the brain and central nervous system to create this state remains incompletely understood. Propofol is one of the most widely used and the most widely studied anesthetics. When administered for general anesthesia or sedation, the electroencephalogram (EEG) under propofol shows highly structured, rhythmic activity that is strongly associated with changes in the patient’s level of arousal. These highly structured oscillations lend themselves readily to mathematical descriptions using dynamical systems models. We review recent model descriptions of the commonly observed EEG patterns associated with propofol: paradoxical excitation, strong frontal alpha oscillations, anteriorization and burst suppression. Our analysis suggests that propofol’s actions at GABAergic networks in the cortex, thalamus and brainstem induce profound brain dynamics that are one of the likely mechanisms through which this anesthetic induces altered arousal states from sedation to unconsciousness. Because these dynamical effects are readily observed in the EEG, the mathematical descriptions of how propofol’s EEG signatures relate to its mechanisms of action in neural circuits provide anesthesiologists with a neurophysiologically-based approach to monitoring the brain states of patients receiving anesthesia care.
Introduction
General anesthesia is a fascinating man-made, neurophysiological phenomenon that has been developed empirically to enable safe and humane performance of surgical and non-surgical procedures. The state consists of unconsciousness, amnesia, analgesia, and immobility along with maintenance of physiological stability. In the United States, more than 60,000 patients receive general anesthesia daily.[1,2] Despite use of general anesthesia in this country for more than 166 years, how these drugs act in the brain and central nervous system to create this state remains incompletely understood. Mathematical modeling has been used in anesthesiology to study the dynamics of anesthetic binding at specific receptor sites, to provide pharmacokinetic and pharmacodynamic descriptions of anesthetic behavior within the body, [3–5] and to describe specific brain states such as burst suppression. [▪6,7] There is now growing interest in using modeling studies to describe anesthetic actions in neural circuits. [8–18]
Propofol, 2,6 di-isopropyl-phenol, is one of the most widely used anesthetics. This drug is administered for induction of general anesthesia, maintenance of sedation, and in combination with a narcotic and a muscle relaxant for maintenance of general anesthesia. Propofol acts enhance inhibition at GABAA receptors, which are widely present throughout the brain and central nervous system.[19] Binding of propofol to the post-synpatic GABAA receptors on a pyramidal neuron helps maintain chloride channels in the open state, thus enhancing the inward chloride current, which hyperpolarizes the post-synaptic cell and leads to inhibition [19]. The behavioral effects of propofol depend critically on how much and how rapidly the anesthetic is administer, in addition to the physiological state of the patient, i.e. age, weight and co-morbidities [20], and the presence of other arousal potentiating medications.
Propofol’s behavioral effects are strongly associated with highly structured oscillatory patterns in the electroencephalogram (EEG), local field potentials and in neural spiking activity [▪21–23]. The highly reproducible nature of these patterns suggest that they relate to the mechanisms through which propofol’s binding at GABAA receptors leads to strong coordinated network activity throughout major portions of the brain. Because much is known about brain circuit architecture, the highly rhythmic features in these patterns suggests that mathematical modeling research can make important contributions to our understanding of propofol’s actions in neural circuits, and as a consequence, how this anesthetic produces its altered states of arousal.
In this review, we summarize recent work using mathematical models to investigate the dynamical effects of propofol on brain circuits.
1. Paradoxical Excitation
Propofol is well-known to induce paradoxical behavioral and electrophysiological manifestations of excitation, rather than sedation, at low doses [24–27]. A common EEG marker for this paradoxical excitation is elevated power in the Beta (12.5–25Hz) frequency band [25]. Despite these characterizations, the neuronal mechanisms that underlie the low-dose effects of propofol were not well understood.
Recently, a detailed computational model was developed in order to elucidate these mechanisms [9]. The model attributes the generation of Beta-band activity at low doses of propofol to cortical dynamics involving the interaction of pyramidal neurons with two type of inhibitory interneurons. The model specifically focusses on the role of the M-current, a slow potassium current, in low-threshold spiking (LTS) interneurons [28]. Propofol is modeled as a potentiation of the conductance and time constant of the GABAA synaptic current [29]. A low dose of propofol is modeled as inducing up to a twofold increase in each of these parameters. At such levels, the interaction between the GABAA synaptic current and the M-current creates a dynamical transition from synchrony to antisynchrony in networks of cortical interneurons (see Figure 1A, B). At the population level, this antisynchrony manifests as an increase in the spiking frequency of pyramidal neurons into the Beta range. By modeling the collective activity of these pyramidal neurons as a surrogate for the EEG [30], the model thus predicts paradoxical excitation as a collective state of cortical interneuron antisynchrony mediated principally by LTS cells. A detailed mathematical analysis of this model was subsequently performed in [31]. There, in a highly reduced version of [9], the authors used geometric singular perturbation theory to show how the M-current and GABAA interplay is highly specific to the low dose regime. The essential dynamical mechanism in this regime was revealed to be the creation of post-inhibitory rebound spiking in LTS interneurons.
Figure 1.

Thalamic and cortical dynamics for low- and surgical- dose levels. (A) In a baseline regime, the thalamocortical system functions through interactions between four principle cell types: Cortical pyramidal neurons (PY), Interneurons (LTS/FS), Thalamic Reticular Neurons (RE) and Thalamocortical neurons (TC). (B) At low doses of propofol, a moderate increase in GABAA interacts with the hyperpolarization-activated current Ih in interneurons to create a primarily cortical Beta oscillation [9]. (C) At higher (surgical) dose levels, the effects on GABAA increase in both cortical and thalamic networks, creating alpha (10–13Hz) time-scales paced by the decay-rate of the inhibition ■[10]. The reverberant connections between thalamic and cortex reinforces this oscillation, leading to broad and coherent alpha expression in the EEG. Figure adapted from ■[10].
Other modeling efforts on propofol have used a mean-field models that describe neuronal dynamics at the scale of cortical macrocolumns [32,33]. These approaches focus on electrophysiological phenomena at deeper levels of general anesthesia. They do not include mechanisms of spike-frequency adaptation, such as those created by the M-current, that may be essential in low-dose paradoxical excitation [34]. A recent exception is [35], in which a linear mean-field model was developed that exhibits increases in frequency at certain levels of GABAA inhibition due to an oscillatory instability in the model dynamics.
2. Thalamocortical Networks and Alpha Oscillations
At deeper levels of propofol, the paradoxical effects described above give way to the more well-known behavioral endpoints of sedation and, eventually, unconsciousness [1]. Classically, these states have been associated with the manifestation of low-frequency delta (1–4 Hz) activity in the EEG [25,26]. Recent evidence, however, suggests that the point at which propofol induces unconsciousness is well-characterized by the appearance of a highly coherent alpha (9–13 Hz) rhythm in frontal cortices [▪21–23].
Building on the work of [9], a model was developed to explain the genesis of this alpha rhythm [10]. The model explicitly takes into account the thalamus, a structure that is well known to mediate oscillatory activity in the alpha range during sleep [36]. Several models have been developed to describe these thalamic dynamics, centered on the so-called spindle oscillation [37–39]. The model in [▪10] includes both thalamic and cortical components. A high dose of propofol is modeled as a 300–400% increase in GABAA conductance and time-constant. It is shown that at such levels, the cortical dynamics exit the paradoxical Beta regime of [9] and enter an alpha frequency regime mediated by the time-constant of inhibition. Meanwhile, the increased levels of GABAA in the thalamus lead to enhanced post-inhibitory rebound spiking mediated by the hyperpolarization activated current (h-current). This enhancement strengthens the already present alpha mechanisms of [37–39]. The reciprocal coupling between thalamus and cortex serves to reinforce this oscillation, leading to prolonged periods of alpha activity (see Figure 1A, C). The model further shows that this activity synchronizes between cortical regions via the thalamus, leading to high spatial coherence. Figure 2A, C demonstrates the Beta and Alpha phenomenology described above in an EEG recording of a human subject receiving an increasing dose of propofol (from [18]). This phenomenology is well-matched by the model output in Figure 2B, D.
Figure 2.
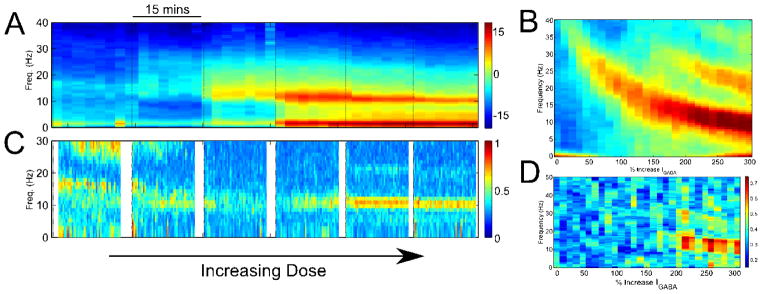
EEG phenomena in the transition from baseline to surgical levels of propofol. (A) Spectrogram of human EEG showing the emergence, sequentially, of diffuse Beta- and narrow Alpha- band oscillations. (B) The output of the thalamocortical model ■[10] matches this phenomenology. The same is true when comparing the EEG coherence between the actual data (C) and model (D). Figure adapted from ■[10].
Other thalamocortical models of neural activity during general anesthesia include [14], which focused primarily on delta and theta frequencies. Very detailed models of the thalamocortical system have also been developed to study the transition from awake to natural sleep [13]. Mean-field type models of propofol anesthesia, as discussed above, generally do not include the thalamus, focusing instead on cortical dynamics. The general focus of these models has been on explaining the origin of slower frequency delta oscillations [11,15–17,40,41], due to a range of anesthetic drug effects beyond GABA-ergic increase. In the absence of full thalamocortical interactions, however, the alpha-rhythm regime for propofol may be difficult to completely model [16,18].
3. Anteriorization
The alpha oscillations described above at deep levels of propofol have a distinct spatial structure. Namely, these oscillations manifest predominantly in frontal regions [▪21,23,42]. This lies in contrast to the classical occipital alpha rhythm associated with eye closure during wakefulness [36,43]. Indeed, while the frontal alpha emerges at propofol-induced unconcsciousness, the posterior occipital alpha is attenuated [▪21,23,44]. Collectively, the gain in anterior power, concurrently with loss in posterior power, has been described as a phenomenon of anteriorization [▪▪1,23,45].
A recent model has been developed that describes neuronal mechanisms that may underlie this anteriorization of alpha rhythms [▪46]. The model combines thalamocortical mechanisms in frontal [▪10] and posterior [47] projecting thalamic nuclei. The primary mechanism involves propofol-mediated reduction in the conductance of the hyperpolarization activated current (h-current) [48,49], in addition to its GABAergic effects. The h-current attenuation alters the dynamics of the high-threshold thalamocortical neurons thought to mediate the generation of the occipital alpha [47], leading to an overall attenuation in posterior alpha rhythms. Meanwhile, the reduction in h-current does not significantly affect the GABA-mediated alpha genesis in frontal thalamocortical circuits [▪10,46]. Consequently, anteriorization can be understood as a combined effect of propofol on GABA and the h-current, with specific effects on different thalamic nuclei based on cell-type (see Figure 3A).
Figure 3.
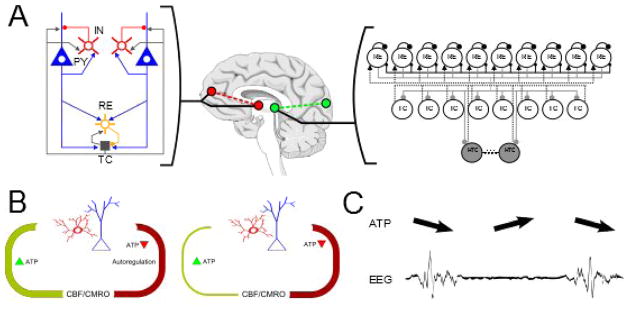
Secondary effects of propofol lead to anteriorization and burst suppression. (A) The differential structure and cell types (in particular, high-threshold thalamocortical neurons) of fronto-thalamic versus posterior-thalamic networks leads to the phenomenon of Anteriorization ■[23,46,47]. (B) At very high doses, propofol leads to secondary effects on cerebral metabolism which can be modeled as a disruption to the rate of ATP regeneration in local circuits. (C) This disruption causes periodic cessation of neuronal activity when ATP levels drop below threshold-levels. This cessation, in turn, manifests as the phenomenon of EEG burst suppression ■[6]. Figure adapted from ■[6,46].
4. Burst Suppression
At deeper still levels of propofol, the EEG exhibits the phenomenon of burst suppression, in which high amplitude activity alternates with periods of isoelectric quiescence [1]. It is a transitional state that occurs between the levels of propofol described above and a state of complete EEG flatline. The amount of suppression increases as a function of anesthetic depth this flatline is achieved. The electrophysiological properties of burst suppression include a broad spatial expression across the cortex and quasiperiodicity in the onset and offset of bursts [50,51].
Efforts to model burst suppression have been limited, due to an inability to account for the many etiologies in which the phenomenon arises. These include propofol anesthesia, but also pathological states such as hypothermia [52], hypoxic/ischemic coma [53], and certain infantile encephalopathies [54]. To account for this diversity of etiologies, a recent model has described burst suppression as arising from an interaction of neuronal and metabolic dynamics [▪6]. The model postulates that each of the conditions associated with burst suppression is linked by a significant reduction in cerebral metabolic rate [55]. To describe this, the model introduces an ATP sensitive potassium channel into all neurons. When ATP levels decrease below a critical threshold, these channels open, causing neurons to cease production of action potentials [56]. In the model, the reduction in cerebral metabolic rate is introduced via a parameter that governs the rate of ATP regeneration. The Sodium-ATP pump is the primary consumer of ATP via production of action potentials. In a normal metabolic regime, the regeneration of ATP is sufficient to sustain action potential production at rates dictates by the natural dynamics of the circuit in question. When ATP production is depressed, however, the ATP sensitive potassium channel is activated due to insufficient supply and spiking activity stops until such time as sufficient ATP levels are restored. In this sense, the model describes the periods of suppression as transient phases of metabolic recovery when the overall energetic state of the brain is depressed (see Figure 3B, C). Many of the predictions garnered from this model have recently been verified in human intracranial recordings [▪57], which additionally provides a more detailed spatial characterization of burst and suppression dynamics.
The phenomenon of burst suppression has also recently been modeled at a mean-field theory representations.[7] This model extends the mean field descriptions of brain dynamics under anesthesia [▪12,58] by introducing more abstract slow variables associated with synaptic resource depletion during sustained periods of population activity. The rate of depletion is related to the level of anesthesia. While a biophysical mechanism for this rate modulation is not explicitly given, it is suggested that, as in [▪6], this could be related to reductions in cerebral metabolism. The dimensionality of the field modeling approach in [7] makes tractable dynamical analyses over larger scale that are harder for the detailed biophysical approach, at the loss of specific mechanistic explanatory capability. A combination of modeling approaches is likely to prove most effective for describing completely the dynamics of propofol and other anesthetics.
5. Conclusion and Future Directions
The altered states of arousal induced by propofol are strongly coupled to highly structured EEG oscillations. Mathematical modeling can be used to gain insight into the mechanisms of these oscillations, and as a consequence, the mechanisms through which propofol induces its altered arousal states. Administering propofol for general anesthesia or sedation is equivalent to applying simultaneously strong inhibitory inputs to the brainstem, thalamus and cortex. However, because the brain is a high-dimensional dynamical system, with myriads of interconnections and feedback loops, this inhibition does not turn the brain off, but rather, induces highly structured oscillatory dynamics. Most of these oscillations are several times larger than the oscillations observed in the resting EEG of an eyes open subject. The profound nature of the oscillations burst suppression supports the idea that propofol entrains large numbers of brain circuits to act in a coordinated manner. These coordinated dynamics are likely responsible for propofol’s altered states of arousal.
Further modeling work with propofol tightly coupled with experimental studies and analyses will lead to deeper insights into the characteristics of its effects on brain dynamics. For example, anteriorization and strong frontal alpha oscillations show that there are different dynamics between the anterior and posterior parts of the brain during propofol-induced unconsciousness. The timing of this difference in dynamics is consistent with the timing of loss of fronto-parietal functional connectivity that has been identified marker of propofol-induced unconsciousness. Hence, the mechanisms underlying anteriorization and the strong frontal alpha oscillations likely also contribute to loss of fronto-parietal connectivity [59–61].
Combined modeling and experimental research applied to other anesthetics, such as ketamine and dexmedetomidine would also be beneficial. Ketamine and dexmedetomidine act in the same circuits as propofol yet, they target primarily NMDA and alpha 2 adrenergic receptors respectively. Both anesthetics have profound effects on brain dynamics and produce anesthetic states that differ appreciably from those induced by propofol. [▪1,23,62]
In general, using mathematical models to help understand how the EEG signatures of anesthetics relate to their mechanisms of action in neural circuits is important scientifically and also practically as it provides anesthesiologists with an interpretable, neurophysiologically-based approach to monitoring the brain states of patients receiving anesthesia care.
Key Points.
Propofol acts on GABAergic circuits in the cortex, thalamus and brainstem.
Propofol-induced oscillations in these circuits induce changes in arousal level.
Alpha and slow oscillations are markers of propofol-induced unconsciousness.
Burst suppression is likely due to cyclic depletion of the brain’s energy stores.
Acknowledgments
Research supported by supported by a National Institutes of Health (NIH) Director’s Pioneer Award DP1-OD003646, an NIH Director’s Transformative Research Award R01 GM104948-01 (to E.N.B.) and a Burroughs-Welcome Fund Careers at the Scientific Interface Award (to S.C.). This work was also supported by the Massachusetts General Hospital Department of Anesthesia, Critical Care, and Pain Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brown EN, Lydic R, Schiff ND. General anesthesia, sleep, and coma. N Engl J Med. 2010;363:2638–2650. doi: 10.1056/NEJMra0808281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci. 2004;5:709–720. doi: 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- 3.Raemer DB, Buschman A, Varvel JR, Philip BK, Johnson MD, Stein DA, Shafer SL. The prospective use of population pharmacokinetics in a computer-driven infusion system for alfentanil. Anesthesiology. 1990;73:66–72. doi: 10.1097/00000542-199007000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Shafer SL, Varvel JR. Pharmacokinetics, pharmacodynamics, and rational opioid selection. Anesthesiology. 1991;74:53–63. doi: 10.1097/00000542-199101000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Gambus PL, Troconiz IF. Pharmacokinetic - Pharmacodynamic modeling in anesthesia. Optimal Dose Identification. 2001;1220:89–97. [Google Scholar]
- 6▪.Ching S, Purdon PL, Vijayan S, Kopell NJ, Brown EN. A neurophysiological-metabolic model for burst suppression. Proc Natl Acad Sci U S A. 2012;109:3095–3100. doi: 10.1073/pnas.1121461109. This work provides a combined neurophysiological and metabolic model to describe burst suppression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liley DT, Walsh M. The Mesoscopic Modeling of Burst Suppression during Anesthesia. Front Comput Neurosci. 2013;7:46. doi: 10.3389/fncom.2013.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sleigh JW, Vizuete JA, Voss L, Steyn-Ross A, Steyn-Ross M, Marcuccilli CJ, Hudetz AG. The electrocortical effects of enflurane: experiment and theory. Anesth Analg. 2009;109:1253–1262. doi: 10.1213/ANE.0b013e3181add06b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCarthy MM, Brown EN, Kopell N. Potential network mechanisms mediating electroencephalographic beta rhythm changes during propofol-induced paradoxical excitation. J Neurosci. 2008;28:13488–13504. doi: 10.1523/JNEUROSCI.3536-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10▪.Ching S, Cimenser A, Purdon PL, Brown EN, Kopell NJ. Thalamocortical model for a propofol-induced alpha-rhythm associated with loss of consciousness. Proc Natl Acad Sci U S A. 2010;107:22665–22670. doi: 10.1073/pnas.1017069108. This work proposes oscillations in thalamocortical circuits as the mechanism for the spatially coherent frontal alpha oscillations observed during propofol induced unconsciousness. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steyn-Ross ML, Steyn-Ross DA, Sleigh JW. Gap junctions modulate seizures in a mean-field model of general anesthesia for the cortex. Cogn Neurodyn. 2012;6:215–225. doi: 10.1007/s11571-012-9194-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12▪.Foster BL, Bojak I, Liley DT. Understanding the effects of anesthetic agents on the EEG through neural field theory. Conf Proc IEEE Eng Med Biol Soc. 2011;2011:4709–4712. doi: 10.1109/IEMBS.2011.6091166. This work proposes a mean-field theory model to describe anesthetic actions in neural circuits. [DOI] [PubMed] [Google Scholar]
- 13.Hill S, Tononi G. Modeling sleep and wakefulness in the thalamocortical system. J Neurophysiol. 2005;93:1671–1698. doi: 10.1152/jn.00915.2004. [DOI] [PubMed] [Google Scholar]
- 14.Sheeba JH, Stefanovska A, McClintock PV. Neuronal synchrony during anesthesia: a thalamocortical model. Biophys J. 2008;95:2722–2727. doi: 10.1529/biophysj.108.134635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steyn-Ross ML, Steyn-Ross DA, Sleigh JW. Modelling general anaesthesia as a first-order phase transition in the cortex. Prog Biophys Mol Biol. 2004;85:369–385. doi: 10.1016/j.pbiomolbio.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 16.Steyn-Ross ML, Steyn-Ross DA, Sleigh JW. Interacting Turing-Hopf Instabilities Drive Symmetry-Breaking Transitions in a Mean-Field Model of the Cortex: A Mechanism for the Slow Oscillation. Physical Review X. 2013:3. [Google Scholar]
- 17.Steyn-Ross ML, Steyn-Ross DA, Sleigh JW, Wilson MT. A mechanism for ultra-slow oscillations in the cortical default network. Bull Math Biol. 2011;73:398–416. doi: 10.1007/s11538-010-9565-9. [DOI] [PubMed] [Google Scholar]
- 18.Storer KP, Reeke GN. gamma-Aminobutyric acid receptor type A receptor potentiation reduces firing of neuronal assemblies in a computational cortical model. Anesthesiology. 2012;117:780–790. doi: 10.1097/ALN.0b013e318269ba6d. [DOI] [PubMed] [Google Scholar]
- 19.Bai D, Pennefather PS, MacDonald JF, Orser BA. The general anesthetic propofol slows deactivation and desensitization of GABA(A) receptors. J Neurosci. 1999;19:10635–10646. doi: 10.1523/JNEUROSCI.19-24-10635.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang DS, Zurek AA, Lecker I, Yu J, Abramian AM, Avramescu S, Davies PA, Moss SJ, Lu WY, Orser BA. Memory deficits induced by inflammation are regulated by alpha5-subunit-containing GABAA receptors. Cell Rep. 2012;2:488–496. doi: 10.1016/j.celrep.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cimenser A, Purdon PL, Pierce ET, Walsh JL, Salazar-Gomez AF, Harrell PG, Tavares-Stoeckel C, Habeeb K, Brown EN. Tracking brain states under general anesthesia by using global coherence analysis. Proc Natl Acad Sci U S A. 2011;108:8832–8837. doi: 10.1073/pnas.1017041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22▪▪.Feshchenko VA, Veselis RA, Reinsel RA. Propofol-induced alpha rhythm. Neuropsychobiology. 2004;50:257–266. doi: 10.1159/000079981. This is one of the first reports of the association between alpha oscillations and propofol-induced unconsciousness. [DOI] [PubMed] [Google Scholar]
- 23▪.Purdon PL, Pierce ET, Mukamel EA, Prerau MJ, Walsh JL, Wong KF, Salazar-Gomez AF, Harrell PG, Sampson AL, Cimenser A, et al. Electroencephalogram signatures of loss and recovery of consciousness from propofol. Proc Natl Acad Sci U S A. 2013;110:E1142–1151. doi: 10.1073/pnas.1221180110. This paper uses detailed behavioral testing and high-density EEG recordings to give detailed descriptions of the associations between several EEG signatures and loss and recovery of consciousness induced by propofol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fulton SA, Mullen KD. Completion of upper endoscopic procedures despite paradoxical reaction to midazolam: a role for flumazenil? Am J Gastroenterol. 2000;95:809–811. doi: 10.1111/j.1572-0241.2000.01866.x. [DOI] [PubMed] [Google Scholar]
- 25.Gugino LD, Chabot RJ, Prichep LS, John ER, Formanek V, Aglio LS. Quantitative EEG changes associated with loss and return of consciousness in healthy adult volunteers anaesthetized with propofol or sevoflurane. Br J Anaesth. 2001;87:421–428. doi: 10.1093/bja/87.3.421. [DOI] [PubMed] [Google Scholar]
- 26.John ER, Prichep LS, Kox W, Valdes-Sosa P, Bosch-Bayard J, Aubert E, Tom M, di Michele F, Gugino LD. Invariant reversible QEEG effects of anesthetics. Conscious Cogn. 2001;10:165–183. doi: 10.1006/ccog.2001.0507. [DOI] [PubMed] [Google Scholar]
- 27.Borgeat A, Fuchs T, Tassonyi E. Induction characteristics of 2% propofol in children. Br J Anaesth. 1997;78:433–435. doi: 10.1093/bja/78.4.433. [DOI] [PubMed] [Google Scholar]
- 28.Cunningham MO, Whittington MA, Bibbig A, Roopun A, LeBeau FE, Vogt A, Monyer H, Buhl EH, Traub RD. A role for fast rhythmic bursting neurons in cortical gamma oscillations in vitro. Proc Natl Acad Sci U S A. 2004;101:7152–7157. doi: 10.1073/pnas.0402060101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adodra S, Hales TG. Potentiation, activation and blockade of GABAA receptors of clonal murine hypothalamic GT1-7 neurones by propofol. Br J Pharmacol. 1995;115:953–960. doi: 10.1111/j.1476-5381.1995.tb15903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jensen O, Goel P, Kopell N, Pohja M, Hari R, Ermentrout B. On the human sensorimotor-cortex beta rhythm: sources and modeling. Neuroimage. 2005;26:347–355. doi: 10.1016/j.neuroimage.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 31.McCarthy MM, Kopell N. The Effect of Propofol Anesthesia on Rebound Spiking. Siam Journal on Applied Dynamical Systems. 2012;11:1674–1697. [Google Scholar]
- 32.Hutt A, Longtin A. Effects of the anesthetic agent propofol on neural populations. Cogn Neurodyn. 2010;4:37–59. doi: 10.1007/s11571-009-9092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liley DT, Sinclair NC, Lipping T, Heyse B, Vereecke HE, Struys MM. Propofol and remifentanil differentially modulate frontal electroencephalographic activity. Anesthesiology. 2010;113:292–304. doi: 10.1097/ALN.0b013e3181e3d8a6. [DOI] [PubMed] [Google Scholar]
- 34.Hindriks R, van Putten MJ. Meanfield modeling of propofol-induced changes in spontaneous EEG rhythms. Neuroimage. 2012;60:2323–2334. doi: 10.1016/j.neuroimage.2012.02.042. [DOI] [PubMed] [Google Scholar]
- 35.Hutt A. The anesthetic propofol shifts the frequency of maximum spectral power in EEG during general anesthesia: analytical insights from a linear model. Front Comput Neurosci. 2013;7:2. doi: 10.3389/fncom.2013.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lopes da Silva FH, Hoeks A, Smits H, Zetterberg LH. Model of brain rhythmic activity. The alpha-rhythm of the thalamus. Kybernetik. 1974;15:27–37. doi: 10.1007/BF00270757. [DOI] [PubMed] [Google Scholar]
- 37.Contreras D, Destexhe A, Sejnowski TJ, Steriade M. Spatiotemporal patterns of spindle oscillations in cortex and thalamus. J Neurosci. 1997;17:1179–1196. doi: 10.1523/JNEUROSCI.17-03-01179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Contreras D, Destexhe A, Steriade M. Spindle oscillations during cortical spreading depression in naturally sleeping cats. Neuroscience. 1997;77:933–936. doi: 10.1016/s0306-4522(96)00573-8. [DOI] [PubMed] [Google Scholar]
- 39.Destexhe A, Contreras D, Sejnowski TJ, Steriade M. A model of spindle rhythmicity in the isolated thalamic reticular nucleus. J Neurophysiol. 1994;72:803–818. doi: 10.1152/jn.1994.72.2.803. [DOI] [PubMed] [Google Scholar]
- 40.Foster BL, Liley DT. Nitrous oxide paradoxically modulates slow electroencephalogram oscillations: implications for anesthesia monitoring. Anesth Analg. 2011;113:758–765. doi: 10.1213/ANE.0b013e318227b688. [DOI] [PubMed] [Google Scholar]
- 41.Foster BL, Liley DT. Effects of nitrous oxide sedation on resting electroencephalogram topography. Clin Neurophysiol. 2013;124:417–423. doi: 10.1016/j.clinph.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 42.Supp GG, Siegel M, Hipp JF, Engel AK. Cortical hypersynchrony predicts breakdown of sensory processing during loss of consciousness. Curr Biol. 2011;21:1988–1993. doi: 10.1016/j.cub.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 43.Williamson SJ, Kaufman L, Lu ZL, Wang JZ, Karron D. Study of human occipital alpha rhythm: the alphon hypothesis and alpha suppression. Int J Psychophysiol. 1997;26:63–76. doi: 10.1016/s0167-8760(97)00756-3. [DOI] [PubMed] [Google Scholar]
- 44.Murphy M, Bruno MA, Riedner BA, Boveroux P, Noirhomme Q, Landsness EC, Brichant JF, Phillips C, Massimini M, Laureys S, et al. Propofol anesthesia and sleep: a high-density EEG study. Sleep. 2011;34:283–291A. doi: 10.1093/sleep/34.3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45▪▪.Tinker JH, Sharbrough FW, Michenfelder JD. Anterior shift of the dominant EEG rhytham during anesthesia in the Java monkey: correlation with anesthetic potency. Anesthesiology. 1977;46:252–259. doi: 10.1097/00000542-197704000-00005. This work is the first report of the phenomenon of anteriorization in association with anesthesia-induced loss of consciousness. [DOI] [PubMed] [Google Scholar]
- 46▪.Vijayan S, Ching S, Purdon PL, Brown EN, Kopell NJ. Thalamocortical Mechanisms for the Anteriorization of Alpha Rhythms during Propofol-Induced Unconsciousness. J Neurosci. 2013;33:11070–11075. doi: 10.1523/JNEUROSCI.5670-12.2013. This work proposes a model to suggest that anteriorization is due to the differences in the neurophysiology and neuroanatomy of the frontal and occipital circuits in response to GABAergic inhibition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vijayan S, Kopell NJ. Thalamic model of awake alpha oscillations and implications for stimulus processing. Proc Natl Acad Sci U S A. 2012;109:18553–18558. doi: 10.1073/pnas.1215385109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Funahashi M, Higuchi H, Miyawaki T, Shimada M, Matsuo R. Propofol suppresses a hyperpolarization-activated inward current in rat hippocampal CA1 neurons. Neurosci Lett. 2001;311:177–180. doi: 10.1016/s0304-3940(01)02169-3. [DOI] [PubMed] [Google Scholar]
- 49.Ying SW, Abbas SY, Harrison NL, Goldstein PA. Propofol block of I(h) contributes to the suppression of neuronal excitability and rhythmic burst firing in thalamocortical neurons. Eur J Neurosci. 2006;23:465–480. doi: 10.1111/j.1460-9568.2005.04587.x. [DOI] [PubMed] [Google Scholar]
- 50.Amzica F. Basic physiology of burst-suppression. Epilepsia. 2009;50 (Suppl 12):38–39. doi: 10.1111/j.1528-1167.2009.02345.x. [DOI] [PubMed] [Google Scholar]
- 51.Steriade M, Amzica F, Contreras D. Cortical and thalamic cellular correlates of electroencephalographic burst-suppression. Electroencephalogr Clin Neurophysiol. 1994;90:1–16. doi: 10.1016/0013-4694(94)90108-2. [DOI] [PubMed] [Google Scholar]
- 52.Stecker MM, Cheung AT, Pochettino A, Kent GP, Patterson T, Weiss SJ, Bavaria JE. Deep hypothermic circulatory arrest: II. Changes in electroencephalogram and evoked potentials during rewarming. Ann Thorac Surg. 2001;71:22–28. doi: 10.1016/s0003-4975(00)02021-x. [DOI] [PubMed] [Google Scholar]
- 53.Young GB. The EEG in coma. J Clin Neurophysiol. 2000;17:473–485. doi: 10.1097/00004691-200009000-00006. [DOI] [PubMed] [Google Scholar]
- 54.Ohtahara S, Yamatogi Y. Epileptic encephalopathies in early infancy with suppression-burst. J Clin Neurophysiol. 2003;20:398–407. doi: 10.1097/00004691-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 55.Hirsch N, Taylor C. Pharmacological and pathological modulation of cerebral physiology. Anaesthesia & Intensive Care Medicine. 2010;11:349–354. [Google Scholar]
- 56.Cunningham MO, Pervouchine DD, Racca C, Kopell NJ, Davies CH, Jones RS, Traub RD, Whittington MA. Neuronal metabolism governs cortical network response state. Proc Natl Acad Sci U S A. 2006;103:5597–5601. doi: 10.1073/pnas.0600604103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57▪.Lewis LD, Ching S, Weiner VS, Peterfreund RA, Eskandar EN, Cash SS, Brown EN, Purdon PL. Local cortical dynamics of burst suppression in the anaesthetized brain. Brain. 2013 doi: 10.1093/brain/awt174. Propofol-induced slow oscillations are not spatially coherence, unlike propfol induced alpha oscillations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bojak I, Liley DT. Modeling the effects of anesthesia on the electroencephalogram. Phys Rev E Stat Nonlin Soft Matter Phys. 2005;71:041902. doi: 10.1103/PhysRevE.71.041902. [DOI] [PubMed] [Google Scholar]
- 59.Alkire MT, Hudetz AG, Tononi G. Consciousness and anesthesia. Science. 2008;322:876–880. doi: 10.1126/science.1149213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hudetz AG. General anesthesia and human brain connectivity. Brain Connect. 2012;2:291–302. doi: 10.1089/brain.2012.0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee U, Ku S, Noh G, Baek S, Choi B, Mashour GA. Disruption of frontal-parietal communication by ketamine, propofol, and sevoflurane. Anesthesiology. 2013;118:1264–1275. doi: 10.1097/ALN.0b013e31829103f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brown EN, Purdon PL, Van Dort CJ. General anesthesia and altered states of arousal: a systems neuroscience analysis. Annu Rev Neurosci. 2011;34:601–628. doi: 10.1146/annurev-neuro-060909-153200. [DOI] [PMC free article] [PubMed] [Google Scholar]